

Abstract
Prostate cancer (PCa) remains a principal cause of mortality in developed countries. Because no clinical interventions overcome resistance to androgen ablation therapy, management of castration resistance and metastatic disease remains largely untreatable. Metastasis is a multistep process in which tumor cells lose cell-cell contacts, egress from the primary tumor, intravasate, survive shear stress within the vasculature and extravasate into tissues to colonize ectopic sites. Tumor cells reestablish migratory behaviors employed during nonneoplastic processes such as embryonic development, leukocyte trafficking and wound healing. While mesenchymal motility is an established paradigm of dissemination, an alternate, ‘amoeboid’ phenotype is increasingly appreciated as relevant to human cancer. Here we discuss characteristics and pathways underlying the phenotype, and highlight our findings that the cytoskeletal regulator DIAPH3 governs the mesenchymal-amoeboid transition. We also describe our identification of a new class of tumor-derived microvesicles, large oncosomes, produced by amoeboid cells and with potential clinical utility in prostate and other cancers.
THE AMOEBOID PHENOTYPE
The classic mesenchymal mode of tumor cell migration has been actively investigated for many years.1 Cells migrating in this manner polymerize actin into filopodia and lamellipodia at their leading edge, where these protrusions facilitate recognition of chemotactic gradients and adhesion to underlying substrata.2,3 At sites of integrin engagement with the extracellular matrix (ECM), focal contacts form and mature into focal adhesions through recruitment and concentration of kinases (e.g., focal adhesion kinase), adaptor proteins (e.g., α-actinin) and actin binding proteins (e.g., paxillin). Following adhesion, actomyosin contractility generates traction forces enabling forward translocation of the cell body. Adhesions at the trailing edge are then released, and the process continues in a cyclic fashion as the cell migrates away from the primary tumor.2
Transition from epithelial to mesenchymal features (EMT) in cancer cells is a well- recognized mechanism of motility that is relevant to disease progression. A defining characteristic of cells having undergone EMT is dependence on pericellular proteolysis for migration. The observation that some migrating cells are relatively insensitive to protease inhibition led to the suggestion that alternative, nonproteolytic mechanisms of tumor cell escape must exist. Wolf et al.4 were the first to show that, under conditions of protease inhibition, certain tumor cells undergo a dramatic morphological, biochemical and migratory transition, converting from spindle-like to rounded morphologies, with loss of focal integrins. These tumor cells also formed constriction rings, enabling squeezing of the cell body through ECM fibers. Notably, the transition was accomplished in the absence of proteolytic matrix remodeling. Sahai and Marshall extended these observations, showing that RhoA/ROCK (Rho kinase) signaling and pliable matrices prompted tumor cells to adopt a rounded, blebbing morphology.5 Similarly to observations by Wolf and colleagues4, inhibition of proteolysis provoked elongated cells to become round, a conversion that induced sensitivity to ROCK inhibitors. Given the migratory resemblance to the amoeba Dictyostelium discoideum, this phenotype has been coined ‘amoeboid’ motility.
Following these two seminal studies, further investigation unveiled additional biochemical and cellular features inherent in amoeboid migration. Wilkinson et al.6 observed that heightened RhoA/ROCK signaling induced cortical actomyosin contractility through phosphorylation of myosin light chain (MLC2). In vivo, MLC2 localized perpendicularly to the direction of migration, providing amoeboid cells with the force necessary to deform proximal matrices and thereby push fibers from their path.7 ECM deformation appears to confer a migratory advantage, as amoeboid behavior predominates at tumor margins,8,9 with migration rates in vivo 10–30 times those observed in culture and relative to cells migrating in a mesenchymal fashion.2 Amoeboid behavior also confers greater sensitivity to chemotactic agents,10,11 potentiating intravasation,12,13 and enables cell survival during extensive shear stress within the vasculature.14 Extravasation and colonization are also promoted, as evinced by increased pulmonary metastases of amoeboid variants in murine metastasis models.8,12,15,16 Collectively, these observations suggest that a ‘mesenchymal-to-amoeboid’ transition (MAT) increases tumor cell aggressiveness relative to EMT, and thereby augments transit through the metastatic cascade.17
RELATIONSHIP OF EMT TO MAT
Both EMT and MAT are adaptive and reversible mechanisms mediating diverse aspects of the plasticity underlying dissemination. Not surprisingly these behaviors share promigratory features, however, they are also functionally and morphologically distinguishable.
Intercellular adhesion
Acquisition of a mesenchymal phenotype is characterized by loss of epithelial markers (e.g., E-cadherin) and expression of ectopic markers (e.g. N-cadherin). This ‘cadherin switch’ weakens adherens junction strength1, facilitating single-cell migration. Cells migrating in an amoeboid fashion may similarly become extricated from cell-cell constraints through reduced expression of junctional components.18
Cell-matrix adhesions
A key difference between amoeboid and mesenchymal migration modes lies in their relative dependence on integrins. Cells exhibiting a mesenchymal phenotype display focal, clustered localization and increased utilization of integrins, and differences in integrin subtypes in the front and rear of the cells. These changes strengthen anchorage to the substratum for forward translocation of the cell body.19 In contrast, amoeboid cells exhibit reduced surface expression and engagement of integrins4,20 and display uropods.21,22 MAT is associated with reduced focal adhesion kinase autophosphorylation at Y39720,23,24, can be induced by low integrin expression, integrin blocking antibodies or integrin peptidomimetics,25 and is modulated by integrin turnover and downstream signaling.26,27,28,29,30 Because an inverse correlation exists between adhesion strength and migration speed, the weaker adhesions of amoeboid cells likely contribute to their faster migration rates.25,31
Pericellular proteolysis
A defining characteristic of EMT is dependence on proteases for ‘path generation’ during migration. Co-clustering of β1-integrins and matrix metalloproteinases (MMPs)4 at sites of ECM contact allows focalized proteolysis of matrix fibers that allosterically impede migration.1 In contrast, MMPs in amoeboid cells are localized to the cytosol.4 Consistent with this distribution, MMPs are less central to amoeboid motility, which can be refractory to protease inhibition.4,5,7 MAT thus represents a mechanism by which tumor cells can migrate and invade under conditions where protease activity is suppressed.4,32
Directional and random migratory modes
In three-dimensional (3D) culture, both mesenchymal and amoeboid cells migrate directionally. This directionality is dictated by the leading edge in mesenchymal cells, yet in amoeboid cells is instead dependent on the site and direction of bleb outgrowth. Such directional migratory behaviors in 3D are in contrast to those in 2D, in which persistent migration in one direction is characteristic only of cells migrating in a mesenchymal fashion. In contrast to directional persistence in 3D, in 2D cultures amoeboid cells instead display random and faster rates of motility,25 consistent with the inverse correlation between directional persistence and migration speed.31
Morphogenesis through cytoskeletal remodeling
Morphology constitutes a fundamental distinction between amoeboid and mesenchymal phenotypes. Mesenchymal cells are characteristically elongated and polarized, with leading and trailing edges. In contrast, amoeboid cells are rounded/ellipsoid without signs of planar polarization and display prominent membrane blebs. Actin remodeling is employed by both cell types to produce these respective morphologies. Motile cells operating in a mesenchymal mode use actin polymerization to generate chemo-and substrate-sensing filopodia, and lamellipodia and stress fibers for traction and maturation of focal adhesions.3,33 In contrast, amoeboid cells display heightened actomyosin contractility, which provides the force for displacement of matrix fibers3,7,33,34 and facilitates bleb dynamics.35 Both mesenchymal and amoeboid cells are naturally occurring subpopulations within the DU145 PCa cell line (Figure 1a).
Figure 1.
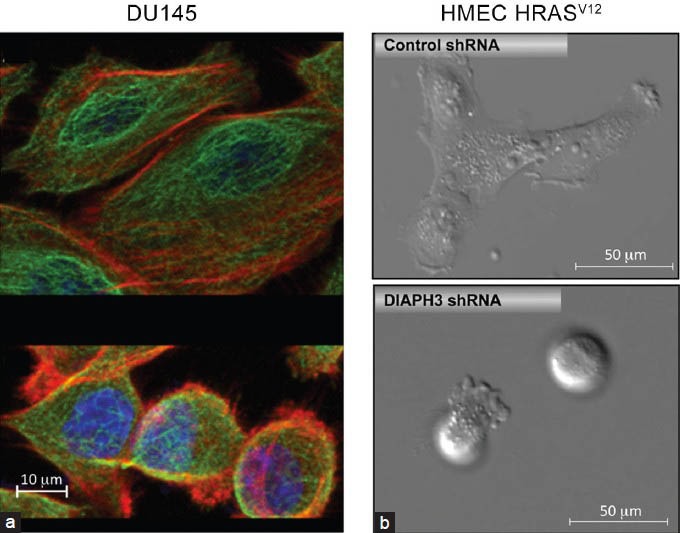
(a) Mesenchymal (top) and amoeboid (bottom) subpopulations occur naturally within DU145 PCa cells. Cells were stained with antitubulin (green) and phalloidin (red) to demonstrate differences in tubulin and actin cytoskeletal organization. The cell nucleus is blue (DAPI). Note the difference in size between mesenchymal and amoeboid cells. (b) Mesenchymal (top)-amoeboid (bottom) transition in HMEC-HRasV12-transformed HMECs upon DIAPH3 silencing.23 DAPI: 4’,6-diamidino-2-phenylindole; HMEC: human mammary epithelial cells; PCa: prostate cancer.
Though less characterized than actin contractility, microtubule (MT) dynamics also influence EMT and MAT. These long, cylindrical tubulin polymers undergo stochastic cycles of elongation and disassembly.36 This ‘dynamic instability’ underlies MT-dependent processes, perturbation of which elicits numerous features of neoplastic transformation. EMT appears to render the MT cytoskeleton less stable than in differentiated epithelia.37 Functional loss of tumor suppressor proteins can cause MT disruption, which cooperates with the pleiotropic events underlying EMT.37 Regulators of MT dynamics also affect EMT.38,39,40 While relevant to EMT, MT instability may contribute even more substantially to MAT. Inhibiting MT polymerization with vincristine promotes amoeboid invasion through hyperactivation of GEFH1, an activator of RhoA/ROCK41. Overexpression,42 downregulation of inhibitory phosphorylation,42 or loss of sequestration by p27kip1,12,43 of the MT depolymerizer stathmin also induces MAT, in part through disruption of endocytic trafficking.44 Such events are detected in human tumors,42 including PCa.45,46
Consistent with a role for the cytoskeleton in triggering amoeboid behavior, our laboratory has identified the diaphanous-related formin-3, DIAPH3, as potentially a pivotal regulator of MAT in prostate cancer and possibly other tumor types.23 DIAPH3 belongs to the formin family that shares tandem FH1 and FH2 domains, which nucleate, elongate and bundle linear actin filaments and/or stabilize MT.47 DIAPH3 silencing causes redistribution of actin structures (stress fiber loss and cortical MLC2 relocalization) and reduces MT stability, alterations associated with transition to an ellipsoid, blebbing and amoeboid phenotype23 (Figure 1b). In 3D matrices, invading DIAPH3-silenced cells assume rounded morphologies, while controls are elongated. Consistent with these cytoskeletal defects and the amoeboid characteristics above, DIAPH3 silencing disrupts endocytic trafficking, suppresses focal adhesions and promotes migration, invasion and metastatic colonization.23 Enforced DIAPH3 expression instead induces mesenchymal characteristics, including N-cadherin upregulation, suppression of membrane blebbing and increased stress fiber formation,23 phenotypes modulated by the phosphorylation state of DIAPH3 at S624. Our studies situate DIAPH3 as a node controlling mesenchymal and amoeboid behaviors.
Non-apoptotic membrane blebbing
While mesenchymal cells display lamellipodia and filopodial protrusions, a defining characteristic of amoeboid cells is protrusion of bulky, non-apoptotic and dynamic membrane blebs from the cell surface. Cortical tension,48 substrate adhesion strength,49 and relative RhoA and Rac1 activities2 influence the choice to bleb or form lamellipodia.50 Dissociation of the actin cortex and plasma membrane or alternatively cortical ruptures,35,51 initiates bleb formation. Hydrostatic pressure inflates these structures, whose growth is reverted by cortex regeneration.52 Blebs contribute to amoeboid migration, and their release can modify the tumor microenvironment (TME, see below).
Our group recently demonstrated that membrane blebs formed from amoeboid cells can be shed into the extracellular space.53,54 Such extracellular vesicle (EV) shedding produces atypically large (1–10 μm) EV that can condition the TME and reach the circulation. We named this type of poorly-characterized particle a ‘large oncosome’, employing the term ‘oncosome’ used previously by Janusz Rak and colleagues as a tumor-derived microvesicle that carries tumor biomarkers and can transfer signaling complexes to recipient cells.55 Oncosomes horizontally transfer proteins, mRNA, miRNA and metabolites to neighboring tumor and stromal cells,55 in a manner distinct from paracrine signaling by soluble factors. The realization that the size range of large oncosomes far exceeds that of other EV (e.g., exosomes, ≤70 nm) identified a significant advantage for their visualization and isolation. Thus, while detection of smaller EV requires electron microscopy, large oncosomes are of sufficient size to be visualized by light microscopy or immunofluorescence methods. These tools can be applied in human tissues, raising the possibility for detection of amoeboid cells in situ, a significant advance for clinicopathologic detection of this malignant phenotype in tumor biopsies.54 In addition to detection techniques in tissues, we have also developed a method for large oncosome identification using size beads and immunoflow cytometry,54 and more recently, a filtration-based system enabling exclusion of smaller EV and selective enrichment for large oncosomes.56 With these approaches, we have identified large oncosomes shed from cultured cells and in biological fluids from mice and humans with PCa.
Using a quantitative blebbing assay, we observed that the oncogene Akt1 and mitogens of the EGF family promote large oncosome genesis, a process attenuated by epidermal growth factor receptor (EGFR) inhibition with gefinitib.53 Proteomic analysis of microvesicles shed from cells overexpressing activated Akt1 revealed the presence of numerous signaling mediators, including Akt1, Src and a biomarker for metastatic PCa, caveolin-1.53 More recently, analyses of large oncosomes from tumorigenic RWPE-2 or non-tumorigenic RWPE-1 isogenic prostate cells revealed abundant, differentially-expressed miRNAs.56 Large oncosomes display gelatinase activity,53 promote gene expression in recipient fibroblasts54 and stimulate migration in cancer and endothelial cells.54 Together, these disparate bioactivities suggest that large oncosomes contribute to cancer progression. In support of this notion and MAT induction by DIAPH3 loss,23 DIAPH3 silencing stimulates formation and shedding of large oncosomes,23,53,54 which promote proliferation and motility in recipient cells.53 Similarly, DIAPH3 silencing enhances the secretion of smaller EV, which stimulate proliferation in recipient prostate and heterologous cancer cells (Kim et al. in press). Our data suggest that EV secretion is upregulated in amoeboid cells, and that the diverse biological activities of these microvesicles contribute to the malignancy and greater invasiveness of such tumor cell variants.
PATHWAYS MEDIATING THE AMOEBOID PHENOTYPE
Networks underlying MAT remain poorly defined, though their elucidation would greatly facilitate therapeutic strategies with which to impact metastasis. Below, we summarize pathways modulating this transition (Figure 2).
Figure 2.
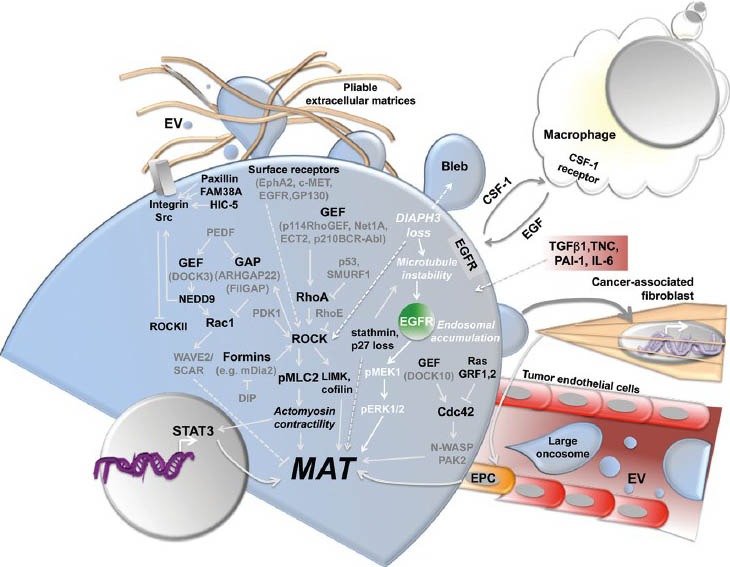
Signal transduction mechanisms and cell processes that regulate or are associated with the amoeboid phenotype. CSF-1: Colony stimulating factor 1; EGFR: epidermal growth factor receptor; EPC: endothelial progenitor cell; EV: extracellular vesicle; GAP: GTPase-activating protein; GEF: guanine nucleotide exchange factor; IL-6: interleukin-6; MAT: mesenchymal-to-amoeboid transition; PAI: plasminogen activator inhibitor type-1; TGFβ1: transforming growth factor beta 1; TNC: Tenascin C.
Rho family GTPases, their regulators and downstream targets
The canonical GTPases Rac1, Cdc42 and RhoA, play critical roles in specifying migration strategies.5,8,57 Like all GTPases, the Rho family is cyclically activated by guanine nucleotide exchange factors (GEFs) and deactivated by GTPase-activating proteins (GAPs).58 RhoA and Rac1 control the transition between amoeboid and mesenchymal phenotypes, respectively, and display an inter-relationship also with Cdc42. While inducing filopodia during mesenchymal motility, Cdc42 also paradoxically promotes amoeboid migration; activation by DOCK10 induces MAT,59 while this Cdc42-mediated transition is suppressed by RasGRF1/2.15 In contrast to Cdc42, Rac1 functions as an antagonist of the amoeboid phenotype. Hyperactivation of Rac1 by the GEF DOCK3 promotes mesenchymal behavior,8 while inactivation by the GAPs ARHGEF228 or FilGAP60 promotes MAT. Arguably, the most characterized GTPase mediator of the amoeboid transition is RhoA, which is activated by disparate upstream GEFs.27,61,62,63,64 Signaling through its effector kinase ROCK, RhoA promotes actomyosin contractility, and thereby enables ECM deformation and ‘path finding’ through tissue matrices. RhoA/ROCK signaling is positively regulated by PDK116 and p53 loss,33 and negatively regulated by RhoE/Rnd316,65 and the ubiquitin E3 ligase SMURF1.34 Consistent with the requirement of RhoA/ROCK for MAT, we observed that DIAPH3 silencing promotes ROCK activation, as evinced by enhanced MLC2 and MYPT-1 phosphorylation.23
Receptor tyrosine kinases
Chemotactic sensitivity is heightened by transition to an amoeboid phenotype. In agreement with their contribution to PCa progression and metastasis,66,67 the hepatocyte growth factor receptor, c-MET,68 and Ephrin A1 receptor, EphA2,69,70 are both implicated in MAT. Extensive work by the Condeelis laboratory demonstrates that amoeboid migration is also highly EGFR-responsive, with amoeboid cells hypersensitized to EGF, a soluble factor that facilitates chemotaxis and promotes metastasis.14,71 Concordantly, we observed elevated EGFR activation and EGF-responsive membrane blebbing, migration and invasion in DIAPH3-deficient PCa cells.23,53
The TME
Mitogenic and morphogenic components within the TME participate in conversion of tumor cells to an amoeboid phenotype. Secretion of tenascin C72 or plasminogen activator inhibitor type-1 (PAI-1)73 by tumor cells promotes MAT, a transition similarly induced by the proinflammatory cytokines IL-674 and transforming growth factor β1.62 Crosstalk with stroma also potentiates amoeboid migration.75 Paracrine interactions, through reciprocal ligand secretion and complementary receptor expression, promote co-migration of amoeboid cells and macrophages.13 Carcinoma cell migration is further mediated by activation of cancer-associated fibroblasts (CAFs) by the pro-inflammatory cytokine OSM, which promotes CAF contractility and in turn ECM remodeling.74 While CAFs induce EMT in PCa cells, in cooperation with the tumor cells they recruit endothelial progenitor cells that stimulate further transition to an amoeboid phenotype.76 Adhesion of PCa cells to endothelial progenitor cells increases following MAT, potentially promoting neovascularization and intravasation.76
Amoeboid-derived large oncosomes have the potential to pleiotropically condition the TME. Enriched in numerous oncogenic biomolecules, large oncosomes stimulate signal transduction, proliferation and migration in recipient tumor cells,53,54 and gene expression in CAFs.56 Large oncosomes upregulate pro-metastatic factors in fibroblasts, enhance migration in tumor endothelial cells and activate stromal myofibroblasts to enhance PCa cell migration.53,54 EV shed by amoeboid cells also suppress immune cell proliferation (Kim et al. in press). It is tempting to speculate that amoeboid-derived EV contribute to the ‘relay system’ proposed by Wyckoff and colleagues,13 by transmitting promigratory signals from the minority of highly chemotactic amoeboid cells to the remainder of the tumor mass.
Cytoskeletal remodeling and formins
Cytoskeletal rearrangements are fundamental to the reprogramming underlying motile behaviors in tumor cells. Alterations in actin and MT networks thus contribute to both EMT and MAT. Formins regulate cytoskeletal dynamics downstream of Rho GTPases77 and are thereby poised to contribute to both mesenchymal and amoeboid morphogenesis. Indeed, several formins and their upstream regulators induce transition to an amoeboid phenotype. Overexpression of the Formin-like protein 2 evokes MAT,78 and FHOD1 promotes membrane blebbing in conjunction with ROCK1 and Src79, while Dia1 promotes bleb-based cell motility through the GEF LARG.80 Eisenmann and colleagues demonstrated that inhibition of mDia2, through binding to DIA-interacting protein (DIP), similarly induces an amoeboid transition.81,82 Interestingly, the DIAPH3 locus encodes multiple splice variants that differentially impact actin dynamics and membrane blebbing.83 Thus, formins both positively and negatively participate in the amoeboid transition.
Genomic deletion of the formin DIAPH3
Our laboratory has demonstrated a relationship between DIAPH3 and the amoeboid phenotype in PCa.23 We identified the DIAPH3 locus within a small frequent focal deletion on chromosome 13q. Copy number variation analyses revealed a significant correlation between DIAPH3 loss and disease progression. Genomic DIAPH3 deletions increased in frequency with Gleason grade, most frequent in metastatic PCa.23 A 100K SNP array analysis of primary and metastatic tumors yielded a similar conclusion, one supported by fluorescent in situ hybridization on an independent cohort.53 In tissues from PCa patients, DIAPH3 protein levels were diminished in metastases relative to primary tumor or benign prostate epithelia. DIAPH3 loss was also more prevalent in disseminated tumor cells from patients with advanced disease, relative to those with organ-confined disease or solid tumors.23
In agreement with association with metastasis, targeting DIAPH3 by RNAi in cultured cells evoked an amoeboid transition, characterized by membrane blebbing, cell rounding, hyperactivation of ROCK/MLC2 and fast, random migration23. Echoing the above examples of enhanced extravasation/colonization of amoeboid cells,8,12,15,16 DIAPH3 silencing potentiated metastases in mice. Molecularly, DIAPH3 loss destabilized MT, as evinced by diminished tubulin acetylation, a posttranslational modification accumulating on MT with slow turnover rates (stable MT).84 The cytoskeletal defect was associated with disrupted endocytic trafficking of EGFR,23 whose activation promotes blebbing in PCa cells.53 EGFR accumulated in early endosomes, with transport to lysosomes and membrane recycling both mitigated. Consequently, attenuation of receptor activity was delayed, evoking downstream ERK1/2 hyperactivation. Sustained MEK1/ERK1/2 signaling was essential for maintenance of MAT. These findings suggest that assessing DIAPH3 lesions in PCa patients may have prognostic utility.
CONCLUSIONS
The studies described in this article highlight the importance of tumor cell plasticity in patient stratification and attempts to discover new, clinically informative biomarkers. Methods for identifying amoeboid cells in human tumors are emerging, including detection of large oncosome features in tumor tissue or in the blood. The role of the amoeboid phenotype in tumor progression is still poorly understood and much fascinating tumor biology lies ahead, waiting discovery.
COMPETING FINANCIAL INTERESTS
Nothing to declare.
ACKNOWLEDGEMENTS
Grant support: NCI R01 CA143777, CA112303, NIDDK R3747556, P50 DK65298 and DAMD17-03-2-0033 (M.R.F.); Steven Spielberg Discovery Fund in Prostate Cancer Research (B.K. and M.R.F.); NIH R00 CA131472 (D.D.V.); Avon Foundation Fund 02-2013-043 (D.D.V.), Ladies Auxiliary VFW Cancer Research Postdoctoral Fellowship (S.M.) and US Department of Defense Prostate Cancer Research Program W81XWH-07-1-0148 and AUAF/GlaxoSmithKline (M.H.H.).
REFERENCES
- 1.Kalluri R, Weinberg RA. The basics of epithelial-mesenchymal transition. J Clin Invest. 2009;119:1420–8. doi: 10.1172/JCI39104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74. doi: 10.1038/nrc1075. [DOI] [PubMed] [Google Scholar]
- 3.Wolf K, Friedl P. Extracellular matrix determinants of proteolytic and non-proteolytic cell migration. Trends Cell Biol. 2011;21:736–44. doi: 10.1016/j.tcb.2011.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, et al. Compensation mechanism in tumor cell migration: mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J Cell Biol. 2003;160:267–77. doi: 10.1083/jcb.200209006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sahai E, Marshall CJ. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol. 2003;5:711–9. doi: 10.1038/ncb1019. [DOI] [PubMed] [Google Scholar]
- 6.Wilkinson S, Paterson HF, Marshall CJ. Cdc42-MRCK and Rho-ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol. 2005;7:255–61. doi: 10.1038/ncb1230. [DOI] [PubMed] [Google Scholar]
- 7.Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS, Sahai E. ROCK-and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr Biol. 2006;16:1515–23. doi: 10.1016/j.cub.2006.05.065. [DOI] [PubMed] [Google Scholar]
- 8.Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–23. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- 9.Tozluoglu M, Tournier AL, Jenkins RP, Hooper S, Bates PA, et al. Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol. 2013;15:751–62. doi: 10.1038/ncb2775. [DOI] [PubMed] [Google Scholar]
- 10.Wang W, Goswami S, Lapidus K, Wells AL, Wyckoff JB, et al. Identification and testing of a gene expression signature of invasive carcinoma cells within primary mammary tumors. Cancer Res. 2004;64:8585–94. doi: 10.1158/0008-5472.CAN-04-1136. [DOI] [PubMed] [Google Scholar]
- 11.Wyckoff JB, Segall JE, Condeelis JS. The collection of the motile population of cells from a living tumor. Cancer Res. 2000;60:5401–4. [PubMed] [Google Scholar]
- 12.Berton S, Belletti B, Wolf K, Canzonieri V, Lovat F, et al. The tumor suppressor functions of p27(kip1) include control of the mesenchymal/amoeboid transition. Mol Cell Biol. 2009;29:5031–45. doi: 10.1128/MCB.00144-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004;64:7022–9. doi: 10.1158/0008-5472.CAN-04-1449. [DOI] [PubMed] [Google Scholar]
- 14.Soon L, Braet F, Condeelis J. Moving in the right direction-nanoimaging in cancer cell motility and metastasis. Microsc Res Tech. 2007;70:252–7. doi: 10.1002/jemt.20411. [DOI] [PubMed] [Google Scholar]
- 15.Calvo F, Sanz-Moreno V, Agudo-Ibanez L, Wallberg F, Sahai E, et al. RasGRF suppresses Cdc42-mediated tumour cell movement, cytoskeletal dynamics and transformation. Nat Cell Biol. 2011;13:819–26. doi: 10.1038/ncb2271. [DOI] [PubMed] [Google Scholar]
- 16.Pinner S, Sahai E. PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol. 2008;10:127–37. doi: 10.1038/ncb1675. [DOI] [PubMed] [Google Scholar]
- 17.Klein CA. Cancer. The metastasis cascade. Science. 2008;321:1785–7. doi: 10.1126/science.1164853. [DOI] [PubMed] [Google Scholar]
- 18.Friedl P, Wolf K. Plasticity of cell migration: A multiscale tuning model. J Cell Biol. 2009;188:11–9. doi: 10.1083/jcb.200909003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huttenlocher A, Horwitz AR. Integrins in cell migration. Cold Spring Harb Perspect Biol. 2011;3:a005074. doi: 10.1101/cshperspect.a005074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carragher NO, Walker SM, Scott Carragher LA, Harris F, Sawyer TK, et al. Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene. 2006;25:5726–40. doi: 10.1038/sj.onc.1209582. [DOI] [PubMed] [Google Scholar]
- 21.Lorentzen A, Bamber J, Sadok A, Elson-Schwab I, Marshall CJ. An ezrin-rich, rigid uropod-like structure directs movement of amoeboid blebbing cells. J Cell Sci. 2011;124:1256–67. doi: 10.1242/jcs.074849. [DOI] [PubMed] [Google Scholar]
- 22.Poincloux R, Collin O, Lizarraga F, Romao M, Debray M, et al. Contractility of the cell rear drives invasion of breast tumor cells in 3D Matrigel. Proc Natl Acad Sci U S A. 2011;108:1943–8. doi: 10.1073/pnas.1010396108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hager MH, Morley S, Bielenberg DR, Gao S, Morello M, et al. DIAPH3 governs the cellular transition to the amoeboid tumour phenotype. EMBO Mol Med. 2012;4:743–60. doi: 10.1002/emmm.201200242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oppel F, Muller N, Schackert G, Hendruschk S, Martin D, et al. SOX2-RNAi attenuates S-phase entry and induces RhoA-dependent switch to protease-independent amoeboid migration in human glioma cells. Mol Cancer. 2011;10:137. doi: 10.1186/1476-4598-10-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedl P. Prespecification and plasticity: Shifting mechanisms of cell migration. Curr Opin Cell Biol. 2004;16:14–23. doi: 10.1016/j.ceb.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Ahn J, Sanz-Moreno V, Marshall CJ. The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci. 2012;125:1814–26. doi: 10.1242/jcs.101444. [DOI] [PubMed] [Google Scholar]
- 27.Carr HS, Zuo Y, Oh W, Frost JA. Regulation of focal adhesion kinase activation, breast cancer cell motility, and amoeboid invasion by the RhoA guanine nucleotide exchange factor Net1. Mol Cell Biol. 2013;33:2773–86. doi: 10.1128/MCB.00175-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deakin NO, Turner CE. Distinct roles for paxillin and Hic-5 in regulating breast cancer cell morphology, invasion, and metastasis. Mol Biol Cell. 2011;22:327–41. doi: 10.1091/mbc.e10-09-0790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McHugh BJ, Murdoch A, Haslett C, Sethi T. Loss of the integrin-activating transmembrane protein Fam38A (Piezo1) promotes a switch to a reduced integrin-dependent mode of cell migration. PLoS One. 2012;7:e40346. doi: 10.1371/journal.pone.0040346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noguchi F, Inui S, Nakajima T, Itami S. Hic-5 affects proliferation, migration and invasion of B16 murine melanoma cells. Pigment Cell Melanoma Res. 2012;25:773–82. doi: 10.1111/pcmr.12005. [DOI] [PubMed] [Google Scholar]
- 31.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84:359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 32.Wolf K, Muller R, Borgmann S, Brocker EB, Friedl P. Amoeboid shape change and contact guidance: T-lymphocyte crawling through fibrillar collagen is independent of matrix remodeling by MMPs and other proteases. Blood. 2003;102:3262–9. doi: 10.1182/blood-2002-12-3791. [DOI] [PubMed] [Google Scholar]
- 33.Gadea G, de Toledo M, Anguille C, Roux P. Loss of p53 promotes RhoA-ROCK-dependent cell migration and invasion in 3D matrices. J Cell Biol. 2007;178:23–30. doi: 10.1083/jcb.200701120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sahai E, Garcia-Medina R, Pouyssegur J, Vial E. Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol. 2007;176:35–42. doi: 10.1083/jcb.200605135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Charras GT. A short history of blebbing. J Microsc. 2008;231:466–78. doi: 10.1111/j.1365-2818.2008.02059.x. [DOI] [PubMed] [Google Scholar]
- 36.Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–42. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- 37.Hernandez P, Tirnauer JS. Tumor suppressor interactions with microtubules: keeping cell polarity and cell division on track. Dis Model Mech. 2010;3:304–15. doi: 10.1242/dmm.004507. [DOI] [PubMed] [Google Scholar]
- 38.Ha GH, Kim JL, Breuer EK. TACC3 is essential for EGF-mediated EMT in cervical cancer. PLoS One. 2013;8:e70353. doi: 10.1371/journal.pone.0070353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li N, Jiang P, Du W, Wu Z, Li C, et al. Siva1 suppresses epithelial-mesenchymal transition and metastasis of tumor cells by inhibiting stathmin and stabilizing microtubules. Proc Natl Acad Sci U S A. 2011;108:12851–6. doi: 10.1073/pnas.1017372108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shan B, Yao TP, Nguyen HT, Zhuo Y, Levy DR, et al. Requirement of HDAC6 for transforming growth factor-beta1-induced epithelial-mesenchymal transition. J Biol Chem. 2008;283:21065–73. doi: 10.1074/jbc.M802786200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eitaki M, Yamamori T, Meike S, Yasui H, Inanami O. Vincristine enhances amoeboid-like motility via GEF-H1/RhoA/ROCK/Myosin light chain signaling in MKN45 cells. BMC Cancer. 2012;12:469. doi: 10.1186/1471-2407-12-469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belletti B, Nicoloso MS, Schiappacassi M, Berton S, Lovat F, et al. Stathmin activity influences sarcoma cell shape, motility, and metastatic potential. Mol Biol Cell. 2008;19:2003–13. doi: 10.1091/mbc.E07-09-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 44.Belletti B, Pellizzari I, Berton S, Fabris L, Wolf K, et al. p27kip1 controls cell morphology and motility by regulating microtubule-dependent lipid raft recycling. Mol Cell Biol. 2010;30:2229–40. doi: 10.1128/MCB.00723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mistry SJ, Bank A, Atweh GF. Targeting stathmin in prostate cancer. Mol Cancer Ther. 2005;4:1821–9. doi: 10.1158/1535-7163.MCT-05-0215. [DOI] [PubMed] [Google Scholar]
- 46.Tradonsky A, Rubin T, Beck R, Ring B, Seitz R, et al. A search for reliable molecular markers of prognosis in prostate cancer: a study of 240 cases. Am J Clin Pathol. 2005;137:918–30. doi: 10.1309/AJCPF3QWIG8FWXIH. [DOI] [PubMed] [Google Scholar]
- 47.Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- 48.Tinevez JY, Schulze U, Salbreux G, Roensch J, Joanny JF, et al. Role of cortical tension in bleb growth. Proc Natl Acad Sci U S A. 2009;106:18581–6. doi: 10.1073/pnas.0903353106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lammermann T, Sixt M. Mechanical modes of ‘amoeboid’ cell migration. Curr Opin Cell Biol. 2009;21:636–44. doi: 10.1016/j.ceb.2009.05.003. [DOI] [PubMed] [Google Scholar]
- 50.Bergert M, Chandradoss SD, Desai RA, Paluch E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci U S A. 2012;109:14434–9. doi: 10.1073/pnas.1207968109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fackler OT, Grosse R. Cell motility through plasma membrane blebbing. J Cell Biol. 2008;181:879–84. doi: 10.1083/jcb.200802081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Charras GT, Hu CK, Coughlin M, Mitchison TJ. Reassembly of contractile actin cortex in cell blebs. J Cell Biol. 2006;175:477–90. doi: 10.1083/jcb.200602085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Vizio D, Kim J, Hager MH, Morello M, Yang W, et al. Oncosome formation in prostate cancer: association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009;69:5601–9. doi: 10.1158/0008-5472.CAN-08-3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Di Vizio D, Morello M, Dudley AC, Schow PW, Adam RM, et al. Large oncosomes in human prostate cancer tissues and in the circulation of mice with metastatic disease. Am J Pathol. 2012;181:1573–84. doi: 10.1016/j.ajpath.2012.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Al-Nedawi K, Meehan B, Micallef J, Lhotak V, May L, et al. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat Cell Biol. 2008;10:619–24. doi: 10.1038/ncb1725. [DOI] [PubMed] [Google Scholar]
- 56.Morello M, Minciacchi VR, de Candia P, Yang J, Posadas E, et al. Large oncosomes mediate intercellular transfer of functional microRNA. Cell Cycle. 2013;12:1–11. doi: 10.4161/cc.26539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parri M, Chiarugi P. Rac and Rho GTPases in cancer cell motility control. Cell Commun Signal. 2010;8:23. doi: 10.1186/1478-811X-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–80. doi: 10.1038/nrm1587. [DOI] [PubMed] [Google Scholar]
- 59.Gadea G, Sanz-Moreno V, Self A, Godi A, Marshall CJ. DOCK10-mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr Biol. 2008;18:1456–65. doi: 10.1016/j.cub.2008.08.053. [DOI] [PubMed] [Google Scholar]
- 60.Saito K, Ozawa Y, Hibino K, Ohta Y. FilGAP, a Rho/Rho-associated protein kinase-regulated GTPase-activating protein for Rac, controls tumor cell migration. Mol Biol Cell. 2012;23:4739–50. doi: 10.1091/mbc.E12-04-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Daubon T, Chasseriau J, El Ali A, Rivet J, Kitzis A, et al. Differential motility of p190bcr-abl-and p210bcr-abl-expressing cells: respective roles of Vav and Bcr-Abl GEFs. Oncogene. 2008;27:2673–85. doi: 10.1038/sj.onc.1210933. [DOI] [PubMed] [Google Scholar]
- 62.Giampieri S, Manning C, Hooper S, Jones L, Hill CS, et al. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol. 2009;11:1287–96. doi: 10.1038/ncb1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Terry SJ, Elbediwy A, Zihni C, Harris AR, Bailly M, et al. Stimulation of cortical myosin phosphorylation by p114RhoGEF drives cell migration and tumor cell invasion. PLoS One. 2012;7:e50188. doi: 10.1371/journal.pone.0050188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weeks A, Okolowsky N, Golbourn B, Ivanchuk S, Smith C, et al. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am J Pathol. 2012;181:662–74. doi: 10.1016/j.ajpath.2012.04.011. [DOI] [PubMed] [Google Scholar]
- 65.Belgiovine C, Frapolli R, Bonezzi K, Chiodi I, Favero F, et al. Reduced expression of the ROCK inhibitor Rnd3 is associated with increased invasiveness and metastatic potential in mesenchymal tumor cells. PLoS One. 2010;5:e14154. doi: 10.1371/journal.pone.0014154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Knudsen BS, Edlund M. Prostate cancer and the met hepatocyte growth factor receptor. Adv Cancer Res. 2004;91:31–67. doi: 10.1016/S0065-230X(04)91002-0. [DOI] [PubMed] [Google Scholar]
- 67.Walker-Daniels J, Coffman K, Azimi M, Rhim JS, Bostwick DG, et al. Overexpression of the EphA2 tyrosine kinase in prostate cancer. Prostate. 1999;41:275–80. doi: 10.1002/(sici)1097-0045(19991201)41:4<275::aid-pros8>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 68.Laser-Azogui A, Diamant-Levi T, Israeli S, Roytman Y, Tsarfaty I. Met-induced membrane blebbing leads to amoeboid cell motility and invasion. Oncogene. 2013 doi: 10.1038/onc.2013.138. [DOI] [PubMed] [Google Scholar]
- 69.Taddei ML, Parri M, Angelucci A, Bianchini F, Marconi C, et al. EphA2 induces metastatic growth regulating amoeboid motility and clonogenic potential in prostate carcinoma cells. Mol Cancer Res. 2011;9:149–60. doi: 10.1158/1541-7786.MCR-10-0298. [DOI] [PubMed] [Google Scholar]
- 70.Parri M, Taddei ML, Bianchini F, Calorini L, Chiarugi P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009;69:2072–81. doi: 10.1158/0008-5472.CAN-08-1845. [DOI] [PubMed] [Google Scholar]
- 71.Wang W, Wyckoff JB, Frohlich VC, Oleynikov Y, Huttelmaier S, et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 2002;62:6278–88. [PubMed] [Google Scholar]
- 72.Grahovac J, Becker D, Wells A. Melanoma cell invasiveness is promoted at least in part by the epidermal growth factor-like repeats of tenascin-C. J Invest Dermatol. 2013;133:210–20. doi: 10.1038/jid.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cartier-Michaud A, Malo M, Charriere-Bertrand C, Gadea G, Anguille C, et al. Matrix-bound PAI-1 supports cell blebbing via RhoA/ROCK1 signaling. PLoS One. 2012;7:e32204. doi: 10.1371/journal.pone.0032204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sanz-Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, et al. ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell. 2011;20:229–45. doi: 10.1016/j.ccr.2011.06.018. [DOI] [PubMed] [Google Scholar]
- 75.Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009. doi: 10.1016/j.cell.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 76.Giannoni E, Taddei ML, Parri M, Bianchini F, Santosuosso M, et al. EphA2-mediated mesenchymal-amoeboid transition induced by endothelial progenitor cells enhances metastatic spread due to cancer-associated fibroblasts. J Mol Med (Berl) 2013;91:103–15. doi: 10.1007/s00109-012-0941-9. [DOI] [PubMed] [Google Scholar]
- 77.Chesarone MA, DuPage AG, Goode BL. Unleashing formins to remodel the actin and microtubule cytoskeletons. Nat Rev Mol Cell Biol. 2010;11:62–74. doi: 10.1038/nrm2816. [DOI] [PubMed] [Google Scholar]
- 78.Kitzing TM, Wang Y, Pertz O, Copeland JW, Grosse R. Formin-like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene. 2010;29:2441–8. doi: 10.1038/onc.2009.515. [DOI] [PubMed] [Google Scholar]
- 79.Hannemann S, Madrid R, Stastna J, Kitzing T, Gasteier J, et al. The Diaphanous-related Formin FHOD1 associates with ROCK1 and promotes Src-dependent plasma membrane blebbing. J Biol Chem. 2008;283:27891–903. doi: 10.1074/jbc.M801800200. [DOI] [PubMed] [Google Scholar]
- 80.Kitzing TM, Sahadevan AS, Brandt DT, Knieling H, Hannemann S, et al. Positive feedback between Dia1, LARG, and RhoA regulates cell morphology and invasion. Genes Dev. 2007;21:1478–83. doi: 10.1101/gad.424807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Eisenmann KM, Harris ES, Kitchen SM, Holman HA, Higgs HN, et al. Dia-interacting protein modulates formin-mediated actin assembly at the cell cortex. Curr Biol. 2007;17:579–91. doi: 10.1016/j.cub.2007.03.024. [DOI] [PubMed] [Google Scholar]
- 82.Wyse MM, Lei J, Nestor-Kalinoski AL, Eisenmann KM. Dia-interacting protein (DIP) imposes migratory plasticity in mDia2-dependent tumor cells in three-dimensional matrices. Plos One. 2012;7:e45085. doi: 10.1371/journal.pone.0045085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stastna J, Pan X, Wang H, Kollmannsperger A, Kutscheidt S, et al. Differing and isoform-specific roles for the formin DIAPH3 in plasma membrane blebbing and filopodia formation. Cell Res. 2012;22:728–45. doi: 10.1038/cr.2011.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Verhey KJ, Gaertig J. The tubulin code. Cell Cycle. 2007;6:2152–60. doi: 10.4161/cc.6.17.4633. [DOI] [PubMed] [Google Scholar]