

Abstract
Although implanted medical devices (IMDs) offer many benefits, they are susceptible to bacterial colonization and infections. Such infections are difficult to treat because bacteria could form biofilms on the implant surface, which reduce antibiotics penetration and generate local dormant regions with low pH and low oxygen. In addition, these infections are hard to detect early because biofilms are often localized on the surface. Herein, an optical sensor film is developed to detect local acidosis on an implanted surface. The film contains both upconverting particles (UCPs) that serve as a light source and a pH indicator that alters the luminescence spectrum. When irradiated with 980 nm light, the UCPs produce deeply penetrating red light emission, while generating negligible autofluorescence in the tissue. The basic form of the pH indicator absorbs more of upconversion luminescence at 661 nm than at 671 nm and consequently the spectral ratio indicates pH. Implanting this pH sensor film beneath 6-7 mm of porcine tissue does not substantially affect the calibration curve because the peaks are closely spaced. Furthermore, growth of Staphylococcus epidermidis on the sensor surface causes a local pH decrease that can be detected non-invasively through the tissue.
Keywords: Upconverting particles, in situ pH sensor film, S. epidermidis
1. Introduction
The aging population and advances in biomedical devices are generating increased demand for implanted medical devices (IMDs), such as prosthetic heart valves, orthopedic implants and intravascular catheters.[1] Although IMDs bring substantial benefits to patients, usually several percent become infected, depending on the types of implants and patient population.[2] These implant infections are difficult to treat because bacteria could form biofilms on the implant surface, and these biofilms reduce antibiotic penetration and contain dormant bacteria especially in hypoxic and acidic regions.[3] Treatment usually requires implant removal, antibiotic treatment of residual infection, and device replacement, although at early stages more conservative treatments such as débridement become viable.[2a, 2b] However, the bacteria are often localized near implant surfaces specifically at early stages, which brings challenges to the current detection methods. For instance, diagnosis based on symptoms (pain, swelling, and fever) are non-specific, while laboratory tests and radiography are also problematic at early stages since the biofilms are often localized to inaccessible regions on implant surfaces.[2a, 2c, 2d, 4] Magnetic resonance imaging (MRI) and computed tomography (CT) can detect bone resorption and sinus tracts but they are not helpful until later stage of infection.[2a] Invasive diagnostic methods such as specimens from periprosthetic tissue may not provide reliable information due to the limited sample area.[2a] Therefore, a local surface-sensitive and noninvasive sensor is needed to detect implant infections early and monitor bacterial growth in situ. Herein, we demonstrate proof-of-principle for a novel technique to non-invasively detect local pH changes from bacterial growth on a luminescent pH sensing film.
2. Background
Low pH on an implant surface could be an indication of bacterial infection. Indeed, acidosis is a well-known product of aerobic respiration and fermentation and was used in classic microbiology to classify bacterial species in vitro based upon what energy sources they metabolize, as determined by a change in the growth medium's pH. For example, Figure 1A shows a photograph of Staphylococcus epidermidis ATCC 12228 after overnight growth on a TSA plate supplemented with 0.0025% (w/w) phenol red dye and 1% (w/w) glucose. The phenol red turned yellow in regions where there were bacterial colonies, which indicates an acidic environment. This acidification is due to bacterial respiration and fermentation which generate weak acidic species (e.g. carbonic acid, lactic acid) as schematically illustrated in Figure 1B. Furthermore, this metabolic activity generates local acidic and hypoxic regions within biofilms, especially near the implant surface, which are believed to play a significant role in antibiotic resistance.[3b] For instance, large pH gradients in the axial direction of oral bacterial biofilms were detected by combining fluorescence lifetime imaging with two-photon excitation microscopy (TPE).[5] In addition, three dimensional pH gradients within E. coli and mixed-culture wastewater biofilms were revealed via imaging ratiometric pH nanosensors with confocal fluorescence microscopy.[6] However, these detection methods could not be used to map pH profiles within biofilms on IMDs surface in situ due to optical scattering and background fluorescence from the tissue.
Figure 1.
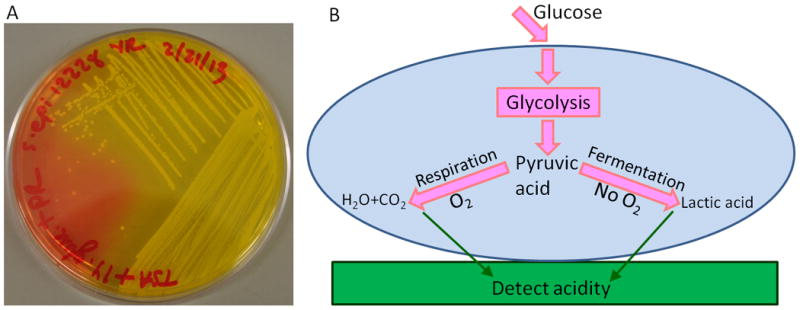
A. A photograph of pH change caused by Staphylococcus epidermidis 12228 growth on 1% glucose TSA plate supplemented with phenol red. The phenol red turns from red (neutral) to yellow (acidic) in regions with bacterial colonies; B. Schematic drawing of extracellular acidification caused by cellular glucose metabolic pathways.
Several methods are availble to measure tissue pH, however, they all have limitations, which hinder their ability to monitor pH variation on IMDs surface in vivo. For instance, microelectrodes could be used to measure tissue pH. For example, in patients with total hip replacement prosthesis, intraoperative pH measurements with microelectrodes observed a pH of 5.8 on the implant surface due to septic loosening, which is significantly lower than the nearby muscle pH 7.4.[7] However, non-invasive monitoring of a large surface on an implant through tissue would require large flexible electrodes, a power source and telemetry, which would bring manufacturing challenges.[8] MRI can measure pH using 31P or 13C-carbonate contrast agents but the resolution and sensitivity are poor and it is not surface specific.[9] Positron emission tomography (PET) can measure pH based upon accumulation of 11C- dimethyloxazolidinedione in acidic regions.[10] However, the technique is not surface-specific and has poor spatial resolution and sensitivity for this application. pH sensors based on optical methods have several advantages such as non-invasiveness, high specificity and better portability, which make them suitable for detecting pH in real time and in situ.[11] However, there are several requirements that would need to be fulfilled before they could detect pH on IMD surfaces through tissue. First, the excitation and emission signal must penetrate through the tissue and generate minimal autofluorescence background. Second, ratiometric pH sensors are necessary to account for variation in total signal intensity due to light attenuation in tissue. Third, these ratiometric pH sensors should be designed to minimize spectral distortion and calibration error caused by tissue absorption and scattering.
To address these issues, we designed a pH sensor with two components: an upconversion luminescence layer serving as a light source, and a layer of pH-sensitive dye that modulates the upconversion luminescence spectrum. We chose upconverting particles (UCPs) as the light source because they could be excited by absorbing multiple near infrared photons with low incident intensity, which allows deep optical penetration through tissue and generates bright luminescence essentially free from autofluorescence.[12] Additionally, UCPs have high photostability and low toxicity, which makes them useful for long time intracellular tracking in vitro and tumor imaging in animals in vivo.[13] UCP-based sensors have been designed to detect biological molecules, metal ions, and pH.[14] For instance, Wolfbeis' group has designed several sensor films based on the different absorbance of the pH indicators on the green and red luminescence of UCPs for detecting carbon dioxide, ammonia and pH.[14c, 15] However, these sensors would not be applicable for measurements through tissue because the signal used to calculate pH (i.e. the ratio of green to red light intensity) would be strongly affected by absorption and scattering in the tissue.[16] In addition, the ratio of green to red upconversion luminescence also depends upon the incident light intensity which varies with tissue thickness.[17] To minimize these spectral distortions, our pH sensor films measure the absorbance of a pH indicator bromocresol green (BG) at two closely-spaced red luminescence peaks of UCPs (inner filter effect). The ratio between these closely spaced peaks is not dependent on the illumination power and the tissue attenuates both wavelengths similarly.
3. Results and Discussion
Our design employs two layers: a layer of UCPs in polydimethylsiloxane (PDMS) that serves as a background-free red light source and a sensing layer consisting of the colorimetric pH indicator, BG, encapsulated in silica. Upon irradiaiton with 980 nm light, the UCPs emit red luminescence with a spectrum that is modulated by the pH-dependent absorption from the BG-loaded sensing-layer. The luminescence layer is fabricated separately from the sensing layer in order to simplify the fabrication process and increase the flexibility of the design: for example, new types of sensors could be designed by changing the indicators using the same luminescent film. Below, we characterize the luminescent upconversion layer and describe the pH sensor design (section 3.1), we then study the sensor film's reversibility, stability, and pH calibrations curves with and without a layer of tissue (section 3.2), and finally we apply the film for real time measurements of pH during bacterial growth (section 3.3).
3.1. UCPs characterization and pH sensor design
Our goal is to show the proof-of-principle for non-invasively detecting pH variation on IMDs in real time in order to monitor bacterial infection on IMDs. Figure 2 shows the concept of our pH sensor film. The pH sensor film comprises of a BG-doped silica layer and a film of UCPs encapsulated in a layer of PDMS as schematically drawn in Figure 2A. The size of the UCPs ranges from 200 nm to 300 nm with some degree of aggregation as shown in the TEM images in Figure S1 (Supporting information). Although the size and morphology of the UCPs is not uniform, in this application, the particles are embedded within a polymer film and the shape does not directly affect the performance of the pH sensor. UCPs with brighter red luminescence emission bands will be used for chemical measurements through deeper tissue in the future.[18] The XRD patterns shown in Figure S2A (Supporting information) match with the standard powder positions of Y2O2S hexagonal phase (JCPDS Card No. 24-1422) displayed in Figure S2B (Supporting information), which confirms the composition and structure of the host materials. All the luminescence spectra were obtained with the setup shown in Figure 2B. The red line in Figure 2C represents the luminescence spectrum of UCPs dispersed by a grating with 150 lines/mm. The inset shows the fine structure of red emission peaks of the UCPs resolved by a grating with 1200 lines/mm. These red emission peaks are assigned to the 4F9/2→4I15/2 transition of Er3+. The fine structure has been observed before and is attributed to 4f-electron crystal-field interaction.[17, 19] The luminescence generated by the UCPs passes through the sensing layer which alters the spectrum in a pH-dependent manner. In order to compare the upconversion luminescence with the BG dye absorption, Figure 2C shows both the UCP luminescence spectrum (solid red line, plotted on the left axis and inset) and the absorption spectra of the BG film in standard pH buffer 3 and 7 (orange and green lines, respectively, plotted against the right axis). At high pH, the absorption spectra of the BG-doped silica film overlap with the red luminescence of UCPs. Figure S3A (Supporting Information) shows the absorption spectra in response to a larger range of standard pH buffers (from 3 to 10), and the presence of a clear isobestic point at ∼500 nm indicates that there are only two forms of the dye, protonated and deprotonated, with pH-dependent concentration ratios. The 613 nm deprotonated absorption peak intensity increases with increasing pH (Figure S3B, Supporting Information). More importantly, deprotonated BG absorbes more light at 661 nm than that at 671 nm. This effect is more clearly observed in Figure 2D, which shows the normalized luminescence spectra of the pH sensor film in response to standard buffer 4, 7 and 9. The shorter wavelength of red upconversion luminescence decreased more than the longer wavelength when pH increases. Therefore, a ratiometric pH sensor could be designed by taking the ratio of these two peaks. In addition, we note that without the BG sensor film, the upconversion luminescence spectrum of the UCPs is not pH dependent, especially after encapsulation in PDMS. For example, the normalized emission spectra of UCPs before (solid black line) and after (dotted red line) incubation in 1 M sulfuric acid for 5 days overlap almost perfectly as shown in Figure S4 (Supporting information), which indicates that the UCPs encapsulated in the PDMS layer are not sensitive to external pH.
Figure 2.
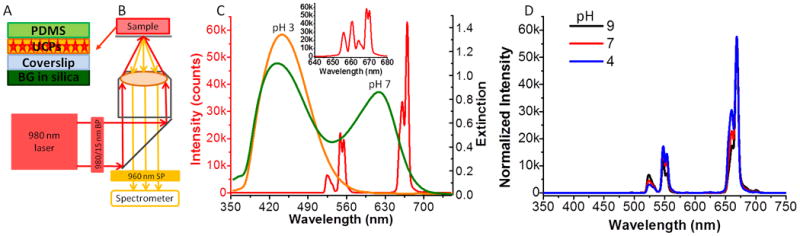
A. Schematic drawing of the side view of the pH sensor film; B. Schematic diagram of the setup for spectra acquisition; C. The overlap between the luminescence spectrum of UCPs (red line, plotted against the left y-axis) and the extinction spectra of BG-doped silica films in response to buffer 3.0 (orange line, right y-axis) and buffer 7.0 (green line, right y-axis). Inset: the red luminescence of UCPs dispersed with the higher resolution grating (1200 lines/mm); D. The luminescence spectra of UCPs dispersed with a lower resolution grating (150 lines/mm) in response to different standard buffers. The spectra were normalized to the peak intensity at 668 nm.
In order to minimize spectral distortion due to wavelength-dependent optical attenuation in the tissue, and also to minimize the power dependence of the UCP luminescence ratio, we generated a pH calibration curve based upon the ratio of two closely-spaced red emission peaks at 671 nm and 661 nm. To better resolve the spectrum at these wavelengths, we used a high-resolution grating with 1200 lines/mm. To test if these two closely-spaced red emission peaks had the same power dependence, the intensity of the two peaks was recorded as a function of laser intensity on a logarithmic scale as shown in Figure 3A. The two linear fit lines had the same slope, indicating that these two emission peaks had the same power dependence. The slope was 1.35, attributed to a two photon process that begins to saturate at high power intensities.[17] In contrast, Figure 3B shows the intensities of the green and red emission peaks as a function of laser power on a logarithmic scale (in order to see both peaks in the same spectrum, we used a grating with 150 grooves mm-1). The green and red peaks had different slopes, indicating that their ratio depends upon the incident light intensity. Specifically, the intensity ratio of green emission over red emission changed by 46 % when the excitation power changed by a factor of 8, while the intensity ratio of the two closely-spaced red emission peaks was unaffected (<1%) within experimental uncertainty. Therefore, the pH calibration curve based upon the two closely-spaced red emission peaks would not be affected by variations in excitation power intensity, while calibrations based on the red and green emission peaks would be strongly affected.
Figure 3.
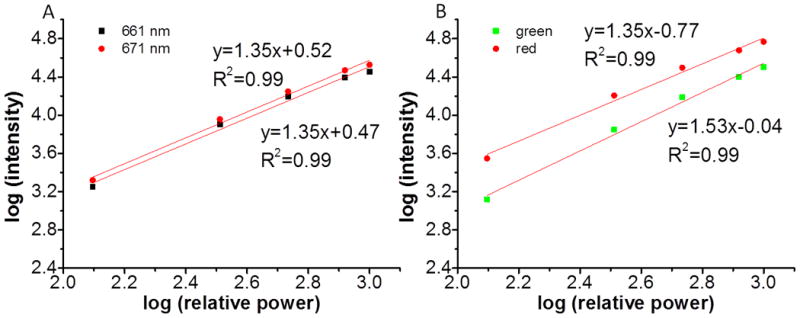
A. Power dependence of the two closely-spaced red emission peaks, 661 nm (black squares) and 671 nm (red dots) of the UCPs. B. Power dependence of the green and red emission of the UCPs in green squares and red dots, respectively.
3.2. pH sensing layer reversibility, stability, and calibration curves with/without tissue
To apply these sensors for monitoring pH variation on IMDs, the sensor layer must be reversible with a response time shorter than the expected for pH changes due to bacterial growth. Figure 4A shows the real time response of the films to alternate PBS (pH 7.4) and standard buffer 5, repetitively. Nearly the same response was obtained for 6 repetitions with no evidence of degradation. This indicates the BG-doped silica films have good reversibility. The response time was about 10 min for a 90% change, which is longer than the result reported from literature.[20] The response was relatively slow because our BG-doped silica films were spin-coated twice. Samples with a single coating responded within 1 min, however, double coatings increase the amount of BG and thus increase the sensitivity. Since a 10 min response time is still rapid compared to the time expected for pH changes due to bacterial growth, we used the double coatings in all the experiments.
Figure 4.
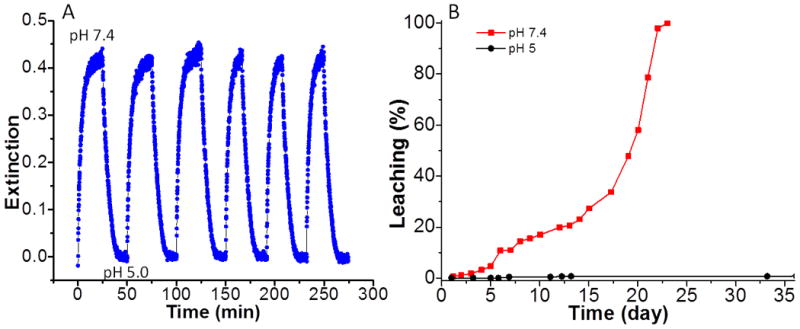
A. Real-time reversibility of BG-doped silica film absorbance in response to alternate immersion in pH 7.4 and pH 5.0 buffers; B. Leaching study of BG-doped silica film in pH 7.4 (red squares) and pH 5.0 (black dots).
We next measured sensor stability agaist leaching. The leaching curves for the BG-doped silica films in PBS (pH 7.4) and standard buffer 5 are shown in red squares and black dots in Figure 4B, respectively. In buffer 5, there was negligible BG leaching for more than a month. By comparision, in PBS, there was no significant BG leaching for about 5 days, followed by increasing BG leaching over the next 20 days. This leaching effect is probably due to the dissolution of silica at slightly basic pH 7.4. Indeed, the degradation of silica-based HPLC column due to silica dissolution in phosphate buffer pH 7.0 has been observed.[21] The stability of the BG-doped film is sufficient for testing the concept of principle here. In the future, we will covalently immobilize pH indicators in a poly(ethylene glycol) hydrogel to improve the stability and biocompatibility of the pH sensor.[22]
To test if the presence of tissue would compromise the performance of our pH sensor film, pH calibration curves were acquired with the sample either embedded between two slices of porcine muscle tissue, or with no tissue. Figure 5A shows the luminescence spectra of the pH sensor film in standard buffers ranging from pH 3-10, while Figure 5B shows the luminescence spectra of the same pH sensor film in response to different standard buffers with the excitation and emission light passing through the porcine tissue. The corresponding normalized spectra are shown in Figure 5C and Figure 5D, respectively. As we can see from Figure 5A and Figure 5B, all the luminescence spectra are background free, which is attributed to the lack of tissue autofluorescence generated by the 980 nm excitation source. It can also be seen from Figure 5C and Figure 5D that increasing the pH causes the luminescence intensity at 661 nm to decrease relative to the 671 nm intensity. This spectral change is expected because at higher pH, more of the BG indicator is deprotonated, and deprotonated BG absorbs more light at 661 nm than at 671 nm, as shown in Figure 2C and Figure S3 (Supporting Information). Figure 5E shows the effective extinction spectrum of the porcine tissue, defined as log10 (Ino tissue (λ)/Itissue(λ)), where I is the upconversion luminescence intensity at wavelength λ. Although the luminescence intensity decreases by a factor ranging from 30 to 300 after the light passes through the porcine tissue, the effective extinction spectra are quite flat, which means that the porcine tissue did not significantly distort the shape of the spectral in this region. Figure 5F shows the intensity ratio of upconversion luminescence at 671 nm over 661 nm as a function of pH for an external sensor film and a sensor film placed between two pieces of porcine tissue, with the top slice about 8 mm thick and the bottom one 6-7 mm thick. It is shown that the pH sensor film is responsive to different pH range from 5 to 10. The pKa of the pH sensor film is 7.0, while the pKa of free BG is around 4.8. This pKa shift agrees with what was reported in the literature and it is mainly attributed to the hydrophobic nature of the hybrid silica film.[20] More importantly, the two calibration curves are in good agreement, which means that the presence of thick porcine tissue does not substantially affect the pH calibration curve. Hence, it is possible to monitor pH variation through up to 6-7 mm tissue by referring to the pH calibration curve generated without passing through tissue. This calibration robustness will be useful for in vivo pH measurement and this ability may provide useful information for early diagnosis and treatment of bacterial infection on IMDs.
Figure 5.
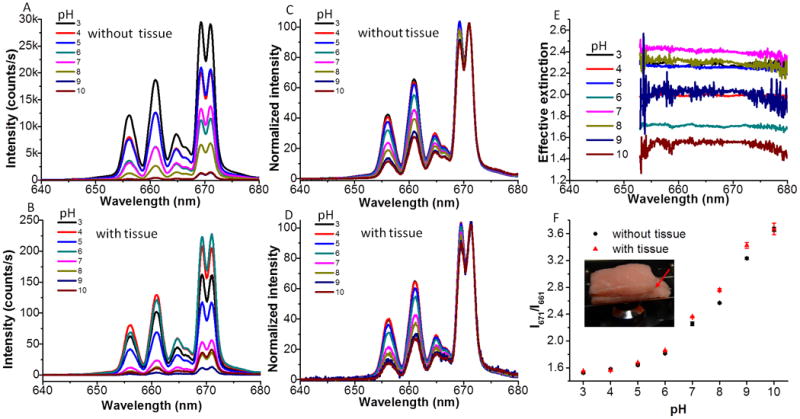
A. Upconversion luminescence spectra of the pH sensor film in response to different standard buffers range from pH 3.0 to pH 10. B. Upconversion luminescence spectra of pH sensor film in response to different standard buffers after passing through 6-7 mm porcine tissue. C and D are the spectra from A and B normalized to the peak at 671 nm, respectively. E. The effective extinction spectra of UCPs due to the presence of porcine tissue. F. pH calibration curves of the pH sensor film without (black dots) and with passing through porcine tissue (red triangles). Inset: Photograph of the pH sensor film sandwiched between two pieces of porcine tissue on the microscope stage for generating pH calibration curve through porcine tissue. The red arrow indicates that the pH sensor film was sandwiched between the two pieces of porcine tissue.
3.3. Real time pH monitoring through tissue
Having made and characterized the pH sensor film, we next tested if our pH sensor film could monitor the pH variation due to bacterial growth on a tryptic soy agar (TSA) plate through thick tissue. We chose S. epidermidis as the model bacterium because it is a common inhabitant of human skin and has been notorious for nosocomial infections.[1] The pH variation due to bacterial growth at the interface of the TSA plate and the pH sensor film was detected in real time through porcine tissue with a thickness of 6-7 mm. The red dots in Figure 6A represent the real time pH variation caused by S. epidermidis growth on a TSA plate supplemented with 1 % glucose as a function of time. The outliers were due to the cosmic spikes during the real time acquisition. The plot shows that the pH gradually decreased soon after the bacteria were inoculated onto the TSA plate. After the experiment concluded, the porcine tissue was removed and the TSA plate with the pH sensor film was photographed as shown in Figure 6B. S. epidermidis colonies were seen growing on the edges of the pH sensor film and the color of the BG-doped silica layer had changed from green to yellow indicating acidosis. This indicates that the presence of bacteria indeed induces pH variation, and this pH variation could be detected through thick porcine tissue with our pH sensor films. The black squares in Figure 6A are the control results by adding the same volume of PBS instead of bacterial culture. The ratios of the two chosen peaks are stable during the whole acquisition period, which indicates the pH was constant. This could be further verified through the photograph of the pH sensor film on the TSA plate taken by the end of the acquisition as shown in Figure 6C. The TSA plate was free of bacteria and the color of the pH sensor film remained green at the end of the acquisition, which indicates neutral pH. Based on the pH calibration curve through tissue with the same thickness, the pH due to S. epidermidis growth was calculated to be around 5.7 by the end of acquisition, while the pH of the control was around 7.4. This result is similar to that of Wang and co-workers who used fluorescent pH sensitive nanosensors to measure pH during E. coli growth on agarose, and observed a pH decrease from 7.8 to 6.5 over the course of 9 hr.[23] However, the use of fluorescein as the pH indicator limits their ability to detect pH through tissue. Here our design allows pH detection through biological tissue and the results show a great potential for our pH sensor films to detect real time bacterial infection on IMDs in situ.
Figure 6.
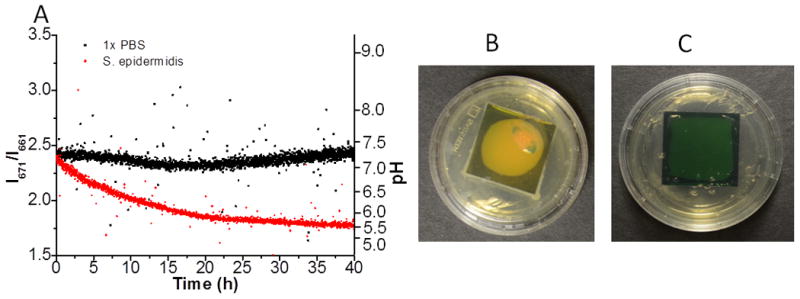
Real time pH detection with the pH sensor films passing through 6-7 mm porcine tissue. The red dots show the pH variation due to the growth of S. epidermidis inoculated onto the TSA plate; the black squares show the pH variation of the control sample, by adding the same volume of PBS.The right axis is the corresponding pH calculated based on the calibration curve through 6-7 mm porcine tissue; B. Photograph of the pH sensor film by the end of acquisition innoculated with S. epidermidis; C. Photograph of the control sample by the end of acquisition. The UCPs are coated in an approximately circular region below the film.
4. Conclusion
In conclusion, we constructed pH sensor films based on the inner filter effect between the BG-doped silica film and the two closely-spaced red emission peaks of the UCPs. The upconversion luminescence spectrum is essentially background-free when applied in biological tissues. Selecting two closely-spaced red emission peaks of the UCPs minimizes spectral distortion of the emission light caused by tissue absorption and scattering, and also minimizes the power dependence of the UCPs. Finally, the pH decrease due to the growth of S. epidermidis on the interface between the TSA plate and pH sensor film was monitored through thick porcine tissue in real time. These proof-of-principle experiments demonstrate the great potential of detecting bacterial infection on IMDs in vivo in biomedical field. In the future, we will improve the long term stability of the BG-doped silica films by changing the matrix to a more stable polymer and covalently binding the pH indicators. X-ray scintillator particles will also be introduced as an X-ray addressable in situ light source to construct images with higher spatial resolution.[24] Alternatively, photoacoustic spectroscopy could be used to detect pH through up to thick tissue with our BG-doped silica film.[25] We will also try to coat the pH sensor films to the medical implants such as fracture fixators and apply them for in vivo pH monitoring in a rabbit model.
5. Experimental Section
Materials
All chemicals were used as received without any further purification. Y2O3, Yb2O3, Er2O3, bromocresol green (BG), tetramethoxyl orthosilicate (TMOS), methyltrimethoxysilane (MTMOS), sodium fluoride (NaF) were purchased from Alfa Aesar (Ward Hill, MA). All the rare-earth nitrates were prepared by dissolving the corresponding oxides with an appropriate amount of nitric acid (6N) at elevated temperature. Carboxymethylcellulose (CMC) was purchased from Fisher Scientific (Pittsburgh, PA). D (+) Glucose was purchased from Sigma-Aldrich (Saint Louis, MO). PDMS base and its curing agent were purchased from Dow corning (Midland, MI). Hydrochloric acid (2 N), nitric acid (6 N), standard pH buffers and sulfuric acid (6 M) were all purchased from BDH (West Chester, PA). Reagent alcohol, tryptic soy broth (TSB) and tryptic soy agar (TSA) were ordered from EMD (Gibbstown, MJ). Urea was purchased from Amresco (Solon, OH). Staphylococcus epidermidis 35984 and Staphylococcus epidermidis 12228 were purchased from ATCC (Manasssas, VA).
Synthesis and characterization of UCPs
Upconverting particles Y2O2S: Yb3+, Er3+ (UCPs) were synthesized according to literature with some modifications.[26] First, precursors were prepared using a homogeneous precipitation method. For a typical reaction, urea (6 g) was added to 100 mL solution, containing Y(NO3)3 (10 mM), Yb(NO3)3 (0.65 mM ) and Er(NO3)3 (0.13 mM). Precipitates were generated by heating the mixture to 80 °C for 2 h. The precipitates were centrifuged for three times with distilled water to remove the excess reagents. Then a slight amount of NaF was added to the particles before they were calcinated at 800 °C for 2 h. The sulfuration reaction was performed in a quartz tube furnace with sulfur powder at 700 °C for 1 h under argon protection. To acquire transmission electron microscopy (TEM) images, a drop of UCP solution was added to a TEM copper grid and dried at room temperature. The images were taken with an H-7600T (Hitachi) at an acceleration voltage of 120 kV. The composition and structure of the UCPs were confirmed by powder XRD patterns, which were obtained on a Rigaku diffractometer at 40 kV and 40 mA. The upconversion luminescence spectra of the UCPs were obtained with the setup displayed in Figure 2B. A 500 mW 980 nm diode-pumped solid state laser (Changchun New Industries Optoelectronics Technology, Changchun, China), filtered with a 980/15 nm band pass filter (Chroma Technology Corp, Bellows Falls, VT), was directed to a 5x objective lens and focused to the sample on the stage. The upconversion luminescence spectrum was collected with the same objective and passed through a 960 nm short pass emission filter (Chroma Technology Corp, Bellows Falls, VT) before being focused to a spectrometer (DNS 300, DeltaNu, Laramie, WY). The upconversion luminescence was spatially dispersed by a grating with 1200 lines/mm otherwise indicated and collected with a cooled CCD camera (iDUS-DU420A-BV, Andor, South Windsor, CT), set to 100 kHz transfer such that each digital count corresponded to 15 photoelectrons. The spectra were recorded with Andor's Solis software. For studying the spectral variation of UCPs in response to sulfuric acid, the coverslip deposited with only the UCPs, which were encapsulated in a layer of PDMS, was immersed to sulfuric acid (1 M) for five days. The luminescence spectra were obtained before and after being incubated in sulfuric acid.
Preparation and characterization of BG-doped silica film
Organically modified silica films doped with BG were prepared according to literature with some small modifications.[20] The hybrid silica sol was prepared by combining TMOS with MTMOS in a molar ratio of 2:1. In a typical preparation, TMOS (320 μL) and MTMOS (138 μL) were mixed in reagent alcohol (228 μL), an appropriate amount of water was added to maintain the molar ratio of silane over H2O to be 1:4, then HCl (166 μL, 0.1 M) was added to initiate the polymerization. Then, BG (30 mg) was added to the mixture. After being stirred for 24 h at room temperature, an aliquot of the sol (40 μL) was spread onto a precleaned microscopic coverslip (18 mm x 18 mm) for 30 s and then spun cast at 2500 rpm for 20 s to form a transparent, uniform silica film. These films were dried at room temperature under a relative humidity environment of ca. 40 % for 1 day. Then another layer was coated on the top of the first layer with another fresh sol, which was prepared with the same protocol as the first layer. The prepared films were dried at room temperature for at least 3 days before further experiments. To measure the absorbance spectra of BG-doped silica films in response to different pH, different standard pH buffers (20 μL) were added to the surface of a BG-doped film for 15 min. Then, the film was attached to a movable stage and UV-vis absorbance spectra were taken with a UV-2101pc spectrometer (Shimadzu, Torrance, CA). To study the reversibility of the BG-doped silica films, a BG-doped film was fixed to a Petri-dish bottom and placed onto the microscope stage. Absorbance variations in response to alternate pH 5.0 and PBS (pH 7.4) were recorded in real time with the microscope head lamp (tungsten filament) as the light source. The spectra were recorded with an acquisition time of 0.1 s in every 30 s for 20 min. For studying the leaching property of the BG-doped silica films, the films were incubated in 20 mL buffer 5 (pH 5.0) or PBS, respectively. 3 mL of the solutions were taken out at indicated time for UV-vis absorbance measurements with the corresponding buffers as references. After measurements, the solutions were put back to the samples. Standard calibration curves for free BG in buffer 5 and PBS (pH 7.4) were generated to quantify the BG leached out from the doped silica films.
pH sensor film preparation
pH sensor films were prepared according to the following procedures. First, UCPs were dispersed in 0.5 % CMC with a concentration of 3 mg/mL. Then 150 μL of the solution was added to the other side of the BG-doped silica films, a film of UCPs was formed after being dried at room temperature overnight. Then PDMS with a base to curing agent ratio of 10:1 was spread onto the surface of UCP film to keep the UCPs in position. The PDMS layer was cured at room temperature for 3 days.
pH calibration curves without and with porcine tissue
To generate calibration curves, a pH sensor film was immersed into a series of standard buffers, ranging from pH 3 to pH 10 for 15 min. The pH sensor film was sealed with plastic wrap before obtaining upconversion luminescence spectra with the setup described above. To generate the calibration curve without passing through porcine tissue, the film was sandwiched between two pieces of white paper and the acquisition time was 2 s. To generate the calibration curve with tissue, the film was placed between two pieces of porcine tissue and the acquisition time was 20 s. pH calibration curves were obtained by plotting the peak intensity ratio of 661 nm over 671 nm as a function of time.
Bacterial culture and real time pH detection through tissue
Fresh culture of Staphylococcus epidermidis ATCC 35984 was prepared by inoculating sterile TSB (5 mL) with a single colony from a TSA plate streaked for less than one week. Bacteria were grown to the stationary phase under shaking conditions at 37 °C for 16-18 h. The number of bacteria was quantified by measuring the optical density at 600 nm (Smartspec 3000, Bio-Rad). The culture was diluted to 108 CFU/mL with sterile PBS. Afterwards, 2 μL of this diluted culture was added to the center of a 1 % glucose TSA plate (35 mm in diameter). The pH sensor film immersed in PBS for 12 h was then laid on top of the TSA plate after the spotted bacterial culture evaporated. Then porcine tissue with a thickness of 6-7 mm was used to wrap the plate. The tissue-wrapped TSA plate was put into a larger Petri dish, which was placed onto the microscope stage. Upconversion luminescence spectra were obtained every 1 min with an acquisition time of 20 s for about 40 h. The porcine tissue was exposed to the 980 nm laser intermittently when data was being acquired with the introduction of a shutter in front of the laser. Sterilized PBS (2 μL) was added to the TSA plate for the control experiment. Spectra were also recorded in real time with the same parameters. Peak ratio variations as a function of time were obtained by analyzing the raw spectra without further normalization or smoothing. Images of the pH sensor films in the TSA plates were taken after the acquisition was terminated. For determining the real time pH variation, the calibration curve through 6-7 mm porcine tissue obtained above was fitted to a fourth power polymonimal curve.
Supplementary Material
Acknowledgments
This research was supported in part by SCBiomat Center of Biomedical Research Excellence (COBRE) through NIH award 1R15EB014560. Electron microscopy images were acquired at the Clemson University Electron Microscope Facility and use of this facility was funded by The Center of Biomaterials for Tissue Regeneration (CBTR) funded under NIH grants 5P20RR021949 and 8P20GM103444.
Footnotes
Supporting Information: Supporting Information, including the TEM images of UCPs, XRD pattern of UCPs, UV-vis absorbance spectra of the BG-doped silica film and luminescence spectra change of UCPs embedded in PDMS in response to sulfuric acid, is available online from Wiley InterScience or from the author.
References
- 1.Bryers JD. Biotechnol Bioeng. 2008;100:1. doi: 10.1002/bit.21838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Zimmerli W. Best Pract Res Cl Rh. 2006;20:1045. doi: 10.1016/j.berh.2006.08.003. [DOI] [PubMed] [Google Scholar]; b) Hetrick EM, Schoenfisch MH. Chem Soc Rev. 2006;35:780. doi: 10.1039/b515219b. [DOI] [PubMed] [Google Scholar]; c) Mody RM, Zapor M, Hartzell JD, Robben PM, Waterman P, Wood-Morris R, Trotta R, Andersen RC, Wortmann G. J Trauma Acute Care Surg. 2009;67:758. doi: 10.1097/TA.0b013e3181af6aa6. [DOI] [PubMed] [Google Scholar]; d) Ehrlich GD, Stoodley P, Kathju S, Zhao Y, McLeod BR, Balaban N, Hu FZ, Sotereanos NG, Costerton JW, Stewart PS, Post JC, Lin Q. Clin Orthop Relat Res. 2005:59. doi: 10.1097/00003086-200508000-00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Vertes A, Hitchins V, Phillips KS. Anal Chem. 2012 doi: 10.1021/ac2029997. [DOI] [PubMed] [Google Scholar]; b) Stewart PS, Franklin MJ. Nat Rev Micro. 2008;6:199. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]; c) Costerton JW, Stewart PS, Greenberg EP. Science. 1999;284:1318. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.a) Trampuz A, Piper KE, Jacobson MJ, Hanssen AD, Unni KK, Osmon DR, Mandrekar JN, Cockerill FR, Steckelberg JM, Greenleaf JF, Patel R. N Engl J Med. 2007;357:654. doi: 10.1056/NEJMoa061588. [DOI] [PubMed] [Google Scholar]; b) Esposito S, Leone S. Int J Antimicrob Ag. 2008;32:287. doi: 10.1016/j.ijantimicag.2008.03.010. [DOI] [PubMed] [Google Scholar]
- 5.Vroom JM, De Grauw KJ, Gerritsen HC, Bradshaw DJ, Marsh PD, Watson GK, Birmingham JJ, Allison C. Appl Environ Microbiol. 1999;65:3502. doi: 10.1128/aem.65.8.3502-3511.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hidalgo G, Burns A, Herz E, Hay AG, Houston PL, Wiesner U, Lion LW. Appl Environ Microbiol. 2009;75:7426. doi: 10.1128/AEM.01220-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Konttinen YT, Takagi M, Mandelin J, Lassus J, Salo J, Ainola M, Li TF, Virtanen I, Liljeström M, Sakai H, Kobayashi Y, Sorsa T, Lappalainen R, Demulder A, Santavirta S. J Bone Miner Res. 2001;16:1780. doi: 10.1359/jbmr.2001.16.10.1780. [DOI] [PubMed] [Google Scholar]
- 8.Wike-Hooley JL, Haveman J, Reinhold HS. Radiother Oncol. 1984;2:343. doi: 10.1016/s0167-8140(84)80077-8. [DOI] [PubMed] [Google Scholar]
- 9.Gillies RJ, Raghunand N, Garcia-Martin ML, Gatenby RA. IEEE Eng Med Biol. 2004;23:57. doi: 10.1109/memb.2004.1360409. [DOI] [PubMed] [Google Scholar]
- 10.Rottenberg DA, Ginos JZ, Kearfott KG, Junck L, Dhawan V, Jarden JO. Ann Neurol. 1985;17:70. doi: 10.1002/ana.410170116. [DOI] [PubMed] [Google Scholar]
- 11.a) Gu B, Yin MJ, Zhang AP, Qian JW, He S. Opt Express. 2009;17:22296. doi: 10.1364/OE.17.022296. [DOI] [PubMed] [Google Scholar]; b) Wang F, Widejko RG, Yang Z, Nguyen KT, Chen H, Fernando LP, Christensen KA, Anker JN. Anal Chem. 2012;84:8013. doi: 10.1021/ac3018179. [DOI] [PubMed] [Google Scholar]
- 12.a) Haase M, Schäfer H. Angew Chem Int Ed. 2011;50:5808. doi: 10.1002/anie.201005159. [DOI] [PubMed] [Google Scholar]; b) Wang F, Banerjee D, Liu Y, Chen X, Liu X. Analyst. 2010;135:1839. doi: 10.1039/c0an00144a. [DOI] [PubMed] [Google Scholar]; c) Ong LC, Gnanasammandhan MK, Nagarajan S, Zhang Y. Luminescence. 2010;25:290. doi: 10.1002/bio.1229. [DOI] [PubMed] [Google Scholar]; d) Wang C, Tao H, Cheng L, Liu Z. Biomaterials. 2011;32:6145. doi: 10.1016/j.biomaterials.2011.05.007. [DOI] [PubMed] [Google Scholar]
- 13.a) Nam SH, Bae YM, Park YI, Kim JH, Kim HM, Choi JS, Lee KT, Hyeon T, Suh YD. Angew Chem. 2011;123:6217. doi: 10.1002/anie.201007979. [DOI] [PubMed] [Google Scholar]; b) Chatterjee DK, Gnanasammandhan MK, Zhang Y. Small. 2010;6:2781. doi: 10.1002/smll.201000418. [DOI] [PubMed] [Google Scholar]; c) Salthouse C, Hilderbrand S, Weissleder R, Mahmood U. Opt Express. 2008;16:21731. doi: 10.1364/oe.16.021731. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Yang Y, Shao Q, Deng R, Wang C, Teng X, Cheng K, Cheng Z, Huang L, Liu Z, Liu X, Xing B. Angew Chem Int Ed. 2012;51:3125. doi: 10.1002/anie.201107919. [DOI] [PubMed] [Google Scholar]
- 14.a) Liu Q, Peng J, Sun L, Li F. ACS Nano. 2011;5:8040. doi: 10.1021/nn202620u. [DOI] [PubMed] [Google Scholar]; b) Deng R, Xie X, Vendrell M, Chang YT, Liu X. J Am Chem Soc. 2011;133:20168. doi: 10.1021/ja2100774. [DOI] [PubMed] [Google Scholar]; c) Sun LN, Peng H, Stich MIJ, Achatz D, Wolfbeis OS. Chem Commun. 2009;0:5000. doi: 10.1039/b907822c. [DOI] [PubMed] [Google Scholar]; d) Zhang P, Rogelj S, Nguyen K, Wheeler D. J Am Chem Soc. 2006;128:12410. doi: 10.1021/ja0644024. [DOI] [PubMed] [Google Scholar]; e) Wang L, Yan R, Huo Z, Wang L, Zeng J, Bao J, Wang X, Peng Q, Li Y. Angew Chem. 2005;117:6208. doi: 10.1002/anie.200501907. [DOI] [PubMed] [Google Scholar]
- 15.a) Ali R, Saleh SM, Meier RJ, Azab HA, Abdelgawad II, Wolfbeis OS. Sensor Actuat B-Chem. 2010;150:126. [Google Scholar]; b) Mader HS, Wolfbeis OS. Anal Chem. 2010;82:5002. doi: 10.1021/ac1007283. [DOI] [PubMed] [Google Scholar]
- 16.Yu X, Li M, Xie M, Chen L, Li Y, Wang Q. Nano Research. 2010;3:51. [Google Scholar]
- 17.Kumar GA, Pokhrel M, Martinez A, Dennis RC, Villegas IL, Sardar DK. J Alloys Compd. 2012;513:559. [Google Scholar]
- 18.a) Wang J, Wang F, Wang C, Liu Z, Liu X. Angew Chem Int Ed. 2011;50:10369. doi: 10.1002/anie.201104192. [DOI] [PubMed] [Google Scholar]; b) Wang F, Deng R, Wang J, Wang Q, Han Y, Zhu H, Chen X, Liu X. Nat Mater. 2011;10:968. doi: 10.1038/nmat3149. [DOI] [PubMed] [Google Scholar]
- 19.Gruber JB, Quagliano JR, Reid MF, Richardson FS, Hills ME, Seltzer MD, Stevens SB, Morrison CA, Allik TH. Phys Rev B. 1993;48:15561. doi: 10.1103/physrevb.48.15561. [DOI] [PubMed] [Google Scholar]
- 20.Makote R, Collinson MM. Anal Chim Acta. 1999;394:195. [Google Scholar]
- 21.Kirkland JJ, Henderson JW, DeStefano JJ, van Straten MA, Claessens HA. J Chromatogr A. 1997;762:97. doi: 10.1016/s0021-9673(96)00945-4. [DOI] [PubMed] [Google Scholar]
- 22.O'Neal DP, Meledeo MA, Davis JR, Ibey BL, Gant VA, Pishko MV, Cote GL. IEEE Sens J. 2004;4:728. [Google Scholar]
- 23.Wang Xd, Meier RJ, Wolfbeis OS. Angew Chem Int Ed. 2013;52:406. doi: 10.1002/anie.201205715. [DOI] [PubMed] [Google Scholar]
- 24.Chen H, Patrick AL, Yang Z, VanDerveer DG, Anker JN. Anal Chem. 2011;83:5045. doi: 10.1021/ac200054v. [DOI] [PubMed] [Google Scholar]
- 25.a) Wilson K, Homan K, Emelianov S. Nat Commun. 2012;3:618. doi: 10.1038/ncomms1627. [DOI] [PubMed] [Google Scholar]; b) Ray A, Yoon HK, Koo Lee YE, Kopelman R, Wang X. Analyst. 2013;138:3126. doi: 10.1039/c3an00093a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xing M, Cao W, Pang T, Ling X. Solid State Commun. 2009;149:911. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.