

Abstract
The mode of action of the bacterial ter cluster and TelA genes, implicated in natural resistance to tellurite and other xenobiotic toxic compounds, pore-forming colicins and several bacteriophages has remained enigmatic for almost two decades. Using comparative genomics, sequence-profile searches and structural analysis we present evidence that the ter gene products and their functional partners constitute previously underappreciated, chemical stress response and anti-viral defense systems of bacteria. Based on contextual information from conserved gene neighborhoods and domain architectures, we show that the ter gene products and TelA lie at the center of membrane-linked metal recognition complexes with regulatory ramifications encompassing phosphorylation-dependent signal transduction, RNA-dependent regulation, biosynthesis of nucleoside-like metabolites and DNA processing. Our analysis suggests that the multiple metal-binding and non-binding TerD paralogs and TerC are likely to constitute a membrane-associated complex, which might also include TerB and TerY, and feature several, distinct metal-binding sites. Versions of the TerB domain might also bind small molecule ligands and link the TerD paralog-TerC complex to biosynthetic modules comprised of phosphoribosyltransferases (PRTases), ATP grasp amidoligases, TIM-barrel carbon-carbon lyases, and HAD phosphoesterases, which are predicted to synthesize novel nucleoside-like molecules. One of the PRTases is also likely to interact with RNA by means of its Pelota/Ribosomal protein L7AE-like domain. The von Willebrand factor A domain protein, TerY, is predicted to be part of a distinct phosphorylation switch, coupling a protein kinase and a PP2C phosphatase. We show, based on the evidence from numerous conserved gene neighborhoods and domain architectures, that both the TerB and TelA domains have been linked to diverse lipid-interaction domains, such as two novel PH-like and the Coq4 domains, in different bacteria and are likely to comprise membrane-associated sensory complexes that might additionally contain periplasmic binding-protein-II and OmpA domains. The TerD and TerB domains and the TerY-associated phosphorylation system are also functionally linked to distinct DNA-processing complexes, which contain proteins with SWI2/SNF2 and RecQ-like helicases, multiple AAA+ ATPases, McrC-N-terminal domain proteins, several restriction endonuclease fold DNases, DNA-binding domains and a type-VII/Esx-like system, which is at the center of a predicted DNA transfer apparatus. These DNA-processing modules and associated genes are predicted to be involved in restriction or suicidal action in response to phages and possibly repairing xenobiotic-induced DNA damage. In some eukaryotes, certain components of the ter system appear to have recruited to function in conjunction with the ubiquitin system and calcium-signaling pathways.
INTRODUCTION
Recent studies on natural resistance to deleterious substances have emerged as a rich resource for the identification of novel pathways, mechanisms and molecules that are involved in stress response1–8. These studies have revealed that organisms have multiple complementary approaches to deal with chemical stress, which together confer both robustness and evolvability to cellular systems2, 9. They have also indicated that responses to the same substances might be channelized via distinct pathways – some of these are generic pathways that facilitate countering of diverse deleterious substances irrespective of their structure and mode of action3. These include both general efflux mechanisms such as those constituted by multi-drug resistance proteins (MDRs) as well as back-ups for various cellular systems that reinforce them against the action of chemicals, or function analogous to phenotypic capacitors by allowing the cell to withstand attacks on multiple nodes in the biological networks2, 3, 6, 7, 9. Other pathways are more specific and are deployed against particular substances or classes of substances and potentially neutralize them by acting on them directly2, 6. Furthermore, stress response might involve systems acting at various levels of cellular organization and compartmentalization3, 5, 7. For example, in eukaryotes a significant role is played by the mitochondrion in supplying energy required for certain natural resistance processes, whereas the chromatin complexes appear to aid in establishing epigenetic states that allow transcription of genes specifically related to countering stress3. Several chemogenomics studies on natural chemical resistance in model organisms have uncovered numerous uncharacterized proteins whose biochemical activities and biological functions initially appeared to be opaque6–8. However, we have previously shown that sequence analysis in combination with other forms of contextual information might considerably help in clarifying the action and biochemistry of these proteins and also yield predictions that might lead to understanding previously unexpected facets of the stress response3, 4.
The oxyanion of Tellurium, an element which is rarely found in the environment, is highly toxic to bacteria10–12. Tellurium resistance genes were initially identified in the ter operon (terZABCDEF)in pMER610 and R478 plasmids, the kilA operon (kilA, telA, telB) in the plasmids of the IncPα group and the chromosomal teh operon (tehAB) from Escherichia coli12. Tellurium resistance genes have also been identified in other bacteria, such as trgAB in Rhodobacter sphaeroides, the tmp gene in Pseudomonas syringae12 and homologs of the E.coli ter operon genes in Alcaligenes13. The products of at least some of these genes appear to have a direct role in tellurium resistance. For example, the methylase activity of tehB, an AdoMet-utilizing Rossmann fold methylase on tellurite, has a direct role in its detoxification14, 15. In line with the above-described pattern of multi-level responses to chemical stress, studies also point to a more generic role for some of the other genes initially recovered in natural tellurite resistance screens. These include TelA from the KilA operon, the deletion of whose ortholog in the firmicute Listeria monocytogenes results in reduced resistance to many cell-membrane-targeting anti-microbials, like the lantibiotics, nisin and gallidermin, the beta-lactams, cefuroxime and cefotaxime, and bacitracin16. Likewise, the E.coli ter genes, in addition to tellurite resistance, are involved in resistance to infection by several bacteriophages such as T5 and λ (the Phi phenotype) and membrane-perforating colicins (the PacB phenotype)12, 17. Clostridium acetobutylicum homologs of the Ter genes also conferred resistance to methyl methanesulfonate (MMS), mitomycin C (MC), and UV, when expressed in recA mutant strains of E. coli13. During intracellular growth Yersinia pestis was shown to express several general stress response related genes, with upregulation of the TerD and TerE genes, in addition to genes encoding other protective factors such as superoxide dismutase-A (sodA)18. In the plant pathogenic Streptomyces scabies, a TerD ortholog was found to be overexpressed when it is exposed to the protective lipidic polymer suberin found in its potato host19. Another study has shown that the deletion of tdd8, a TerD protein, in Streptomyces coelicolor, had an effect on differentiation and spore morphology, in addition to reduced tellurite resistance20. Together, these observations suggest that further analysis of the tellurite resistance operons such as the kilA and ter operons could throw light on the more general systems that help bacteria withstand both a wide range of chemical insults and other stresses, such as viral and toxin attacks.
There is some evidence for functional diversification among the different genes in the ter operon from E.coli. Disruption of TerD, TerC and TerZ reduce or abolish all stress resistance phenotypes, suggesting their general involvement across the different responses, ranging from those directed against tellurite to those against phages and colicins. Disruption of TerA only results in reduction of the phage-related Phi phenotype leaving the rest intact17. In contrast, tellurite resistance appears to specifically require terB, terC, terD and terE but apparently not the other genes21. Despite multiple studies pointing to the importance of the ter operon and the TelA gene in stress response across distant bacterial lineages, they have remained largely mysterious in terms of biochemistry and mode of action12. In terms of the proteins encoded by these genes, TerC is known to be a multi-TM protein, which is related to another E. coli membrane protein Alx that is upregulated in response to high pH22. A recent structural analysis has shown that TerD, TerE and TerZ have a common beta-sandwich fold with two calcium binding sites23. Similarly structure determination of the TerB protein has suggested that it adopts a novel alpha-helical fold24. The only directly observed effect of the action of the Te-resistance determinants has been the deposition of tellurium crystals in the vicinity of the membrane, with reduced deposition of the metal inside cells25–27. In Pseudomonas species it has been suggested that these Te crystals are released into the surrounding medium by budding off of the outer membrane vesicles containing the crystals27.
To gain further insights into this potential bacterial stress response system, we investigated the ter operon components by analyzing and synthesizing information from their sequences, structures, gene neighborhoods and domain architectures. Consequently, we show that the ter gene products and TelA lie at the center of an intricate, previously unrecognized network of functionally linked proteins with roles related to membrane-linked sensing of stress, phage restriction, DNA repair, post-transcriptional gene regulation and production of a novel nucleoside-like metabolite that might function either as a stress signal or an extremolyte.
RESULTS AND DISCUSSION
Determination of the gene-context- and domain architecture-based functional network for components of the ter operon
To better understand the ter operon components, we utilized the fact that increasing numbers of diverse newly sequenced genomes provide novel contextual information from gene neighborhoods and also allow testing the strength of previously observed connections28–30. To expand the contextual information pertaining to the ter operon products we first systematically identified all their homologs from across the three superkingdoms of life using completely sequenced genomes deposited in the non-redundant (nr) database through iterative sequence profile searches with the PSI-BLAST program. The core ter operon contains three main protein families, namely the TerD family that includes the paralogous proteins TerD, TerA, TerE, TerF and TerZ, the TerB family, and the TerC family10, 12, 23. With these, we also included in our initial focus the TerY family of von Willebrand factor A (vWA) domains, whose genes were found to be associated in conserved gene neighborhoods with the core ter genes31. Representatives of the TerD, TerB, and TerC families are found across most major bacterial lineages, with relatively infrequent examples of lateral transfers of individual families to archaea and eukaryotes (Supplementary material). In bacteria, their phyletic distribution and phylogenetic relationships suggest considerable lateral mobility, keeping with the original identification of the ter operon on mobile gammaproteobacterial plasmids10, 12, 23 (Fig. 1). Having identified members of the above four families, we systematically determined their gene neighborhoods to extract predicted operons that encode them. This established that the TerD, TerB, TerC and TerY families are indeed genomically linked in predicted operons across diverse bacteria, primarily from the actinobacterial, firmicute and gammaproteobacterial lineages, but show no such linkage in archaea (Fig. 1, Supplementary material). However, we also found that they were additionally present, independently of each other, across diverse bacteria, linked to numerous other families of proteins encoded by several conserved gene-neighborhoods. Some of these contexts also contained homologs of TelA that has been independently established as a tellurite-resistance determinant (Fig. 1, 2)10, 12, 16. Finally, we extended the linkages obtained above by transitively investigating the gene-neighborhood and domain architecture contexts of the newly recovered conserved genes that tended to widely co-occur with the initially defined set of ter genes (Fig. 2C and D, Fig 3). As a result we uncovered a vast network of contextual information in the form of several distinct types of predicted operons that linked one or more of the ter genes to a wide array of genes with a multitude of biochemical functions (Fig. 4). Given that products of genes belonging to the same operon tend to functionally interact28–30, we were able to predict the wide swath of biochemical functions that are united by the ter system. A summary of all major families of functionally linked domains uncovered in this analysis is provided in Table 1 (along with details regarding the statistical support in sequence searches for the inferred relationships, when applicable), representatives of major operons types are shown in Figure 2 and notable domain architectures are depicted in Figure 3.
Figure 1.
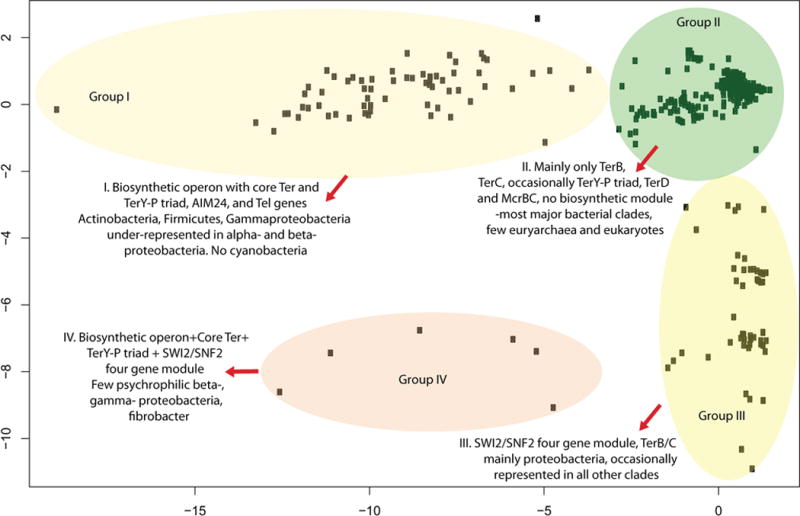
Principal Component Analysis of the phyletic patterns of ter and associated genes in 1348 genomes. Plotting of PC1 vs PC2 resolved the organisms into four major groups, which reflect the presence of the biosynthetic module in combination with the TerBCD and TerY-P triad gene modules, the presence of primarily TerBC without associated biosynthetic operons, the presence of the SWI2/SNF2 four-gene module, and the combined presence of all these modules. The underlying phyletic pattern for the PCA is available in the Supplementary material.
Figure 2.
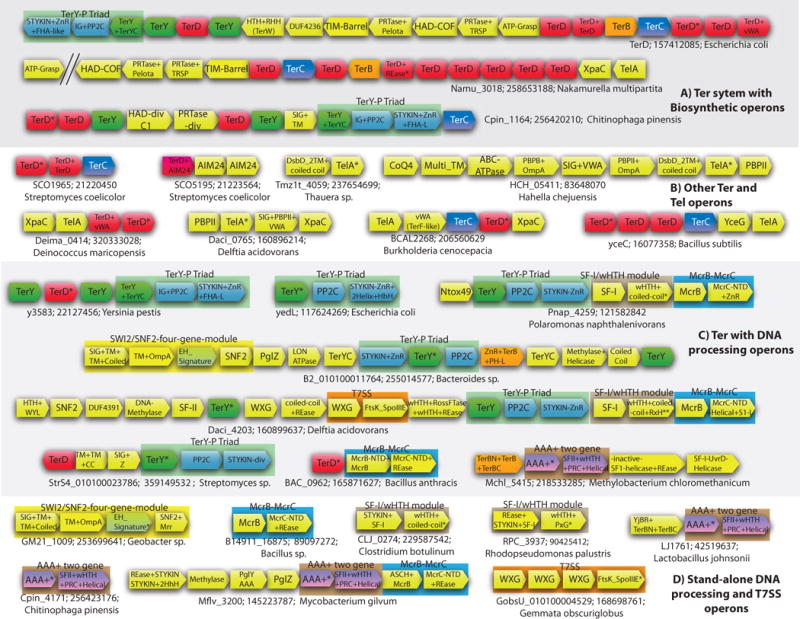
Operons of the ter system and associated systems. The gene neighborhood data for some of the genes encoding Ter system proteins is depicted using arrows. The direction of the arrow is the direction of transcription of the gene. The gene name, Genbank identifier (gi), and the species name of the starred gene is shown next to the operon. The multi-gene modules that always occur together have been boxed. The genes are cartoon representation and not to scale. The depicted operons are representative of many operons spanning a range of diverse organisms. The complete list is provided in the Supplementary material. (A) Ter system genes found with the novel biosynthetic operons. (B) Other operons of Ter and TelA genes. (C) DNA-processing related operons. In the T7SS containing operon of Bordetella the gene with two stars (**) has not been translated in Genbank. The protein and the gene can be found in the Supplementary materials. (D) Standalone versions of DNA processing and Type 7 Secretion System. Abbreviations of domain names are as in Table 1. Other abbreviations: SF-1 – SF-I Helicase; SF-II – SF-II Helicase; SNF2 – SNF2 Helicase;CC – coiled coil; Z -uncharacterized globular region.
Figure 3.
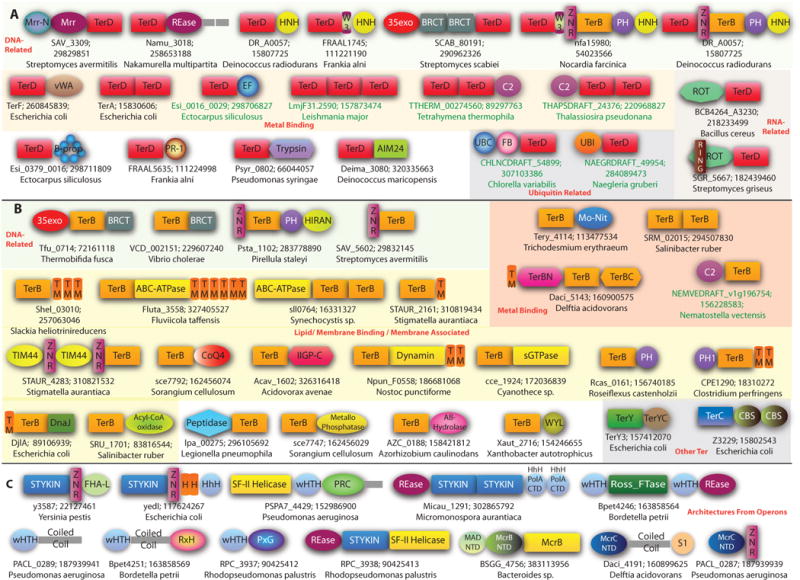
Domain architectures of proteins found in the ter system and associated operons. The domains are not to scale. (A) Domain Architectures of the TerD domain. (B) Domain architectures of TerB, TerY and TerC domain. (C) Domain Architectures of proteins found in the ter system operons. Domain architectures are labeled with a representative gene name, the Genbank identifier (GI) number, and the species name separated by semicolons. The eukaryotes are shown in green. The functional categories are shown in red letters. Domain abbreviations are in the Table 1. Coiled coil regions are shown with a grey rectangle. Other abbreviations: TM – Transmembrane Helix; SIG –Signal peptide; H – Hydrophobic Helix.
Figure 4.
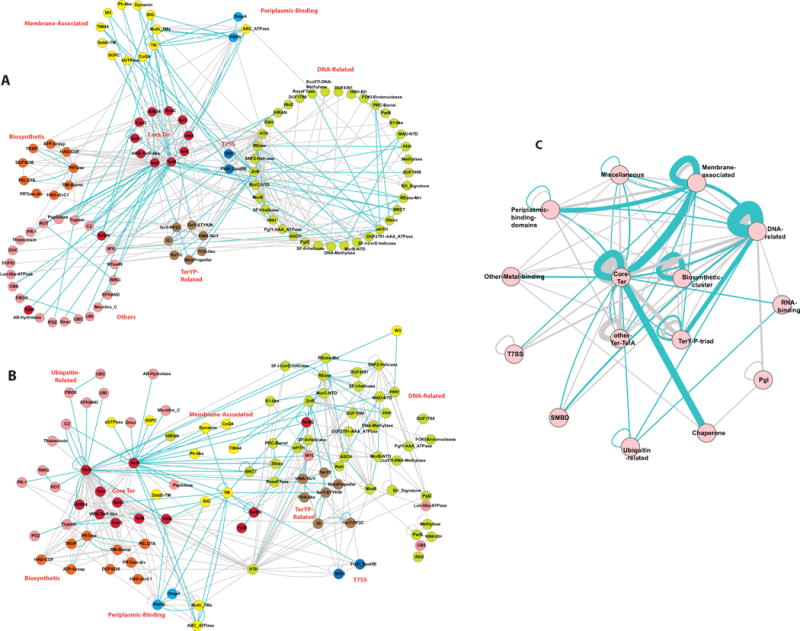
Domain network graph of the Ter system proteins and the proteins encoded in their gene neighborhood. The graphs were rendered using the Cytoscape program. The network is an ordered graph with the blue edges representing the connection between adjacent domains combined in the same polypeptides and the grey edges representing the context in the gene neighborhood. (A) The nodes of the network arranged by function. (B) The “force-directed” network was derived using spring-embedded layout utilizing the Kamada-Kawai algorithm, which works well for graphs with 50–100 nodes. The natural clustering of the functional categories has been pointed out. (C) Condensed network, where the domain belonging to a given functional category have been collapsed in to that category name.
Table 1.
| Domain | Fold | Search details | Comments |
|---|---|---|---|
| Ter components | |||
| TerD | β -sandwich fold | E. coli TerD protein GI: 157412085 | In Pfam 26.0 TerD (PF02342) has been erroneously classified under the vWA clan due to the vWA fusion in TerF |
| vWA | α/β Rossmanoid fold | 1) TerY. GI: 157412073 2) TerFC-like: GI: 260845839 (fused to TerD); GI: 226312577 (solo) 3) generic vWA |
1) some proteobacteria and flavobacteria have a C terminal TerYC 2) some TerFC exist as solos |
| TerB | α helical | 1) TerB. Z1612 GI: 15801100 2) DUF533 GI: 15802259 |
|
| TerBN | Predominantly α helical | GI: 160900575 residues 127.333 | Overlaps with PFAM DUF4016 |
| TerBC | Predominantly α helical | GI: 160900575 residues 651.798 | C terminal of TerB in DUF4016-like TerBN containing proteins |
| TerY-C | Metal-chelated by 8 conserved C and H | C term of TerY3 GI: 157412070 residues: 219.335 |
Metal binding. Also occurs as solo eg. CLOBAR_01126 gi: 164687485 |
| TerY-P triad components | |||
| TerY-STYKIN | kinase | GI: 117624267 Residues 1.303. PSI-Blast. 2nd iteration E values > 10−37 | Forms the TerY-P triad |
| TerY-PP2C | GI: 15802552 | Forms the TerY-P triad Fused to IG in some instances |
|
| Betapropeller | GI: 158335667 | Fused to TerY-STYkin and ZnR | |
| IG | GI: 157412069 Residues 140.280 PSI-BLAST. recovers IG in 2nd iteration e value > 2×10−06 | Fused to TerY-PP2C | |
| FHA-Like | GI: 121583391 Residues 404.498 | Fuseed to Tery-STYKIN | |
| Ter-TelA other components | |||
| AIM24 | DSBH | GI: 320335663 | Fused to TerD and as solos in operon with TerD |
| YjbR | CyaY-like | 1) overlaps with DUF419. GI: 104774241 | In operon with AAA ATPase and SF-II helicase dyad along with TerBN |
| yceG | α+β | GI: 16077362 | |
| XpaC | α helical | 1) XpaC GI: 321313695 2) XpaC-divergent GI: 320333025, 334343691, 308067731 |
|
| TelA | α helical | GI: 16077363 | |
| Biosynthetic gene cluster | |||
| Pelota (PF01248) | α+β Chorimate Mutase-like fold | C terminus of PRTase (DUF2983) GI: 15800688, residues 267–371. PSI-Blast: recovers the pelota domain of Neurospora crassa protein GI: 85102905 of the NHP2/L7aE family; 2nd iteration with an e value of 2e−04 | RNA binding. In PFAM 26.0 both the PRTase and Pelota domains are combined as a single model DUF2983 |
| Phosphoribosyltransferase | α/β. PRTase | 1) Z1167 from E.coli (N terminus of DUF2983 GI: 15800688, residues 1–267). PSI-BLAST: purine and pyrimidine PRTases in the 3rd iteration with e-values<10−3. HH-pred: P-value 3.4×10−6, probability 94.1%, PDB: 2jbhA hypoxanthine-guanine PRTase 2) Z1169 from E.coli (GI: 15800690, residues 1–220; N terminal to DUF3706). PSI-BLAST Recovers orotate PRTase in the 2nd iteration e <10−5. HH-Pred. P-value 7.9×10−24, probability 99.8, OPRTase pdb: 1o57 3) comF-like Cpin_1168 GI: 256420214. PSI-BLAST. Recovers comF proteins in 2nd iteration e=8×10−04 |
In a few bacteria the PRTases of the Z1167 and Z1169 families are fused (gi: 332668740) |
| ATP-GRASP ligase | ATP-GRASP | E.coli Z1170 GI: 15800691. PSI-BLAST: CarB family ATP-GRASP 2nd iteration, e value 10−31 | Most closely related to the carbamoyl phosphate synthetase |
| HAD phosphoesterase | HAD | 1) HAD-COF E.coli Z1168 GI: 15800689. PSI-BLAST. HAD 2nd iteration, e value 10−12. HH-PRED. Prob. 100% P-value 6.5×10−36 pdb: 1nrwA 2) HAD-div Cpin_1167 GI: 256420213 3) HAD-C1 Psyr_0807 GI: 66044062 |
1) Cof-like proteins belong to “C2” cap-containing assemblage 2) tetra-helical ‘C1’ cap assemblage |
| C-C bond lyase | TIM-Barrel | E.coli Z1166 GI: 15800687 | Most closely related to enzymes like citrate lyase |
| TRSP | GI: 15800690, residues 230–380; overlaps with PFAM DUF3706 | In PFAM 26.0 DUF3706 has been imperfectly defined. Has conserved GXXE and TRSP signatures |
|
| DNA related | |||
| wHTH | wHTH | 1) wHTH (fused to RossFTase and REase) GI: 163858564. Residues 1.212. PSI-BLAST. Recovers wHTH in the 1st iteration (PFAM RTCR – not classified as HTH) 2) Distorted wHTH (fused to RossFTase and REase) GI: 163858564. Residues 362.439 3) wHTH (fused to SF-II-Helicase and PRC-Barrel). GI: 152986900 residues 400.513. HH-pred Prob. 97.3%. P-value 3×10−08 1oyw(405.505) 4) wHTH (fused to coiled-coil; PACL_0289-like): GI: 187939941 Residues 50.165. PSI-Blast recovers HTH after 6th iteration with e> 10−03. HHpred Probability 97.3% P-value 2.4×10−08 pdb:2hr3 |
DNA-binding The distorted wHTH overlaps with the improperly defined DUF1887 |
| RXH | α-helical | GI:163858569 Residues 390.646 | Fused to wHTH (the version fused to coiledcoil; PACL_0289-like) proteins in TerY-P associated gene clusters |
| PxG | GI: 344191412 Residues 140.474 | Fused to wHTH (PACL_0289-like). These proteins do not have the coiled-coil region. In operon with REase+STYKIN+SF-I-helicase | |
| BRCT | α/β three layered sandwich | 1) BRCT fused to TerB GI: 72161118. Residues 335.411 2) BRCT fused to fused to TerD GI: 290962326. Residues 217.295 & 343.421 |
Two BRCT domains are fused to TerD and 35exo, while one BRCT is fused to TerB and 35exo |
| HhH | Bi-helical hairpin | 1) TerY-STYKIN fused HhH. GI: 117624267. Residues 509.649. HH-pred Probability 98.76%. P-value 4.9×10−13 pdb 1wcnA 2) RNA_pol_A_CTD. GI: 302865792 Residues 774.843 |
|
| ZnR | Zn ribbon | 1) TerY-STYKIN fused ZNR. Gi. 117624267 residues 304.338 2) fused to TerB. Gi. 117624267 residues 1.40 |
Fused to the STYKIN of the TerY-P triad |
| S1-like | OB fold | GI: 325267434. residues 300.367 | S1-like fused to McrC-NTD |
| HIRAN | GI: 283778890. Residues 417.473 | Fused to ZnR, TerB and PH-like domain | |
| ASCH | PUA | GI: 333897583 | Fused to McrB and REase |
| Abi2 | helical | GI: 21224934. Residues 250.324 | In operon with AAA-ATPase (DUF2791) |
| PRC-Barrel | β-barrel | GI: 152986900 residues 515.567 | fused to SF-II-Helicase and wHTH |
| McrB-NTD | 1) DUF3578. GI: 229137733 (fused to McrB) | Potential nucleotide binding. Found fused to McrB and REases | |
| MAD-NTD | GI: 374384750. Residues 3.88 | Potential nucleotide binding. Found N terminal to McrB-NTD in McrB containing proteins and also fused to HNH | |
| MCRC-NTD | 1) DUF2357. GI: 239826784 (with REase- McrC catalytic domain) 2) GI: 325267434 (fused to S1-like) 3) GI: 115376453 (fused to ZnR) 4) GI: 325680706 (fused to Peptidase) |
Stimulatory domain for the activity of the McrB NTPase; partly maps to PFAM DUF2357 Fused to S1-like, ZnR, McrC-REase, and peptidase |
|
| SF-I Helicase | Two tandem P-loop NTPase domains | 1) GI: 383754211; GI: 359414882 (fused to STYKIN) 2) GI: 217958516 (in operon with TerD) 3) GI: 218533283 (inactive SF-I-Helicase+REase) 4) GI: 344336707 |
1) forms the dyad with wHTH+coiled-coil 2) Related to the UvrD helicase; 3) in operon with (DUF2791-AAA_ATPase) 4) In operon with TerY-P triad and McrC-NTD+S1 |
| SF-II Helicase | Two tandem P-loop NTPase domains | 1) GI: 152986900 residues 33.395 2) GI: 160899638 |
1) Fused to wHTH, PRC-Barrel and a C terminal domain. Part of the AAA_ATPase (DUF2791) dyad 2) In operon with T7SS and with McrB/McrC |
| SF-II: SNF2-Helicase | Two tandem P-loop NTPase domains | 1) GI: 157155470 ; GI: 15641763 (fused to Rease-Mrr) 2) GI: 163858552 (in operon with T7SS) 3) GI: 302865791 |
1) In operon with EH-Signature protein |
| EH_Signature | α-helical domain | GI: 253699641 GM21_1009 | In the four gene operon with SIG+TM+TM+C-term, SIG+OmpA, and SNF2-Helicase |
| RossFTase | Rossmann fold | GI: 163858564. Residues 213.361. | Overlaps with the improperly defined DUF1887 |
| AAA+ NTPase | P-Loop-NTPase | 1) DUF2791 GI: 152987173 2) McrB 3) PglY; GI: 296123320 |
PglY, a component of the Pgl systems |
| DUF1788 | GI: 220916261 | In operon with PglZ and AAA_ATPase (DUF2791) | |
| DUF4391 | GI: 163858553 | In operon with TerY | |
| DUF1998 | GI: 89893419 (fused to Helicase and ZnR) | In operon with AAA_ATPase (DUF2791) | |
| PglZ | Alkaline phosphatase | GI: 134101638 | |
| HNH | HNH | 1) GI: 15807725 2) HNH-EH: DUF1524-nuclease GI: 21224935 |
Fused to proteins with TerD and TerB |
| REase | Restriction Endonuclase Fold (REase) | 1) REase-Mrr: fused to TerD GI: 290960911 2) DUF3883 GI: 383790095 (fused to McrB-NTD) 3) DUF524 (McrC catalytic domain). GI: 239826784 4) C term of GI: 160899632 4) NERD. GI: 302865792 residues 13.127 5) REase(fused to RossFTase and wHTH): GI: 163858564 residues 431.587 |
5) The REase fused to RossFTase overlaps with the improperly defined DUF1887. |
| 3′–5′ exonuclease | RNAseH | GI: 72161118 | |
| ParB | DUF262; 1) GI: 21224935 2) GI: 254228175 |
Fused to HNH-EH 1) In operon with AAA_ATPase (DUF2791) 2) In operon with EH-Signature protein |
|
| FOKI-Endonuclease | REase | DUF1819 GI: 89893421 | |
| DNA-Methylase | Rossmann | 1) GI: 262194453 2) GI: 356451333 |
1) In operon with DUF2791-AAA_ATPase and PglZ 2) In operon with EH-signature protein |
| Eco571-DNA-Methylase | Rossmann | GI: 262193922 | In operon with DUF2791-AAA_ATPase and PglZ |
| STYKIN | Serine/threonine/tyrosine Kinase fold | 1) GI: 359414882 Fused to SF-I Helicase | In the operon with wHTH+coiled-coil that do not have the TerYP-Triad |
| Mrr-N | Novel α+β | 2) GI: 290960911 | Found fused to DUF4236 and Mrr family REase fold domains with C-terminal TerD domains. Predicted DNA-binding domain |
| DUF4236 | Predominantly β | GI: 15800686. residues 1.59 | Found in certain ter operons with biosynthetic module; usually fused to the Mrr-N domain. Could be a novel DNA-binding domain |
| Type 7 Secretion System | |||
| FtsK_SpoIIIE | P-Loop-NTPase | GI: 160899633 | |
| WXG | 4 helical bundle | GI: 160899636 | |
| Other Metal Binding | |||
| C2 | β -sandwich fold | GI: 89297763 | Fused to TerD in eukaryotes |
| Mo-Nit | Conserved cysteines | GI: 113954696 residues 160.240 | Fused to TerB in cyanobacteria (Pfam Mo_nitro_C) |
| EF-HAND | α-helical | GI: 298706827 | Fused to TerD in eukaryotes |
| Lipid/Membrane binding or Membrane associated | |||
| TerC | Membrane-spanning helices | 1) TerC 2) DUF475. DR_2226 GI: 15807218 |
1) 7 TM helices 2) two extra TM helices inserted between the helices 3 and 4 |
| Multi_TM permease | Membrane-spanning helices | GI: 83648076 | Multi transmembrane Transporter. In operon with TelA, ABC-ATPase and others (see Fig 1 and 4) |
| DSBD 2 TM | Membrane-spanning helices | GI: 237654700 | In operon with TelA |
| GTPase | P-Loop-NTPase | 1) Dynamin GI: 172039480 2) TRAFAC GTPase related to Hsr1 GI: 172036839 |
Both fused to TerB |
| PH-Like | β-barrel | 1) GI: 113971539 Residues 16.145. HHpred -Probability 95.94% P-value 3.2×10−06 pdb:3hsa 2) GI: 148654659. Residues 146.267 HHPred-Probability 90% P-value P-value=10–5 GLUE domain PDB: 2cay |
1) This PH domain is fused to TerB and 2TMs (called PH1 in Fig 2B) 2) These PH-like domains along with a TerB are fused to a variety of domains like TerD, HIRAN, HNH |
| IIGP-C C terminal domain of interferon-inducible GTPase |
α helical fold | GI: 326316418. Residues 156.342. PDB: 1tq4; p=10−6 iteration 4 in a PSIBLAST search with Acav_1602 from Acidovorax; | Fused to TerB in proteobacteria. (overlaps with PFAM DUF697) |
| W3 | GI: 54023566 residues 400.443 | A small domain with three conserved tryptophans (W3), which is only found in actinobacteria and their viruses. overlaps with the N-terminal portion of the PFAM model for DUF2510 | |
| TIM44 | NTF2-like | GI: 310821532. Residues 121.258 & 290.430 | bind lipids and localize to the inner face of membranes |
| CoQ4 | α helical | 1) fused to TerB. GI: 148239309 2) GI: 83648078 In operon with TelA, PBPII and others (see Fig 1 & 4) |
Potential an isoprenoid phosphoesterase |
| AcylCoA-Oxidase | α helical | GI: 83816544 | Fused to TerB |
| Periplasmic-Binding domains | |||
| OmpA | 1) GI: 253699640 (SIG+OmpA) 2) GI: 83648074 (fused to PBPII) |
1) In the four gene operon with SIG+TM+TM+C-term, EH-Signature, and SNF2-Helicase 2) In operon with TelA, ABC-ATPase and others (see Fig 1 and 4) |
|
| PBPII | 1) GI: 317133199 (fused to vWA) 2) GI: 317133197 3) GI: 83648074 (fused to OmpA) 4) GI: 93005589, GI: 169825919 |
1) &2) in operon with TelA 3) in operon with TelA and ABC-ATPase 4) in operon with TerD and the Biosynthetic operon |
|
| Ubiquitin related | |||
| Ubiquitin (UBI) | GI: 284089473 (fused to TerD) | Fused to TerD in eukaryotes | |
| RING | GI: 21219375 | Inserted into the ROT domain fused to TerD | |
| UBC | GI: 307103386 | fused to FBOX and TerD | |
| FBOX | GI: 307103386 | fused to E2 domain and TerD | |
| SMBD | |||
| CBS | GI: 21224934. Residues 1.200 | ||
| WYL | GI: 154246655 Residues 40.150 (fused to TerB) GI: 160899642 fused to HTH (in operon with T7SS) |
||
| Other NTPases | |||
| ABC ATPase | P-Loop-ATPase | 1) GI: 83648075 2) GI: 220907668, 327405527 (fused to TerB) 3) GI:188586145 |
1) In operon with TelA, PBP+OmpA and others (see Fig 1 and 4) 3) SMC-like ABC ATPase fused to PHP domain |
| AAA+: Lon-like-ATPase | P-Loop-ATPase | GI: 255014574 | In operon with TerY-P triad and EH-Signature |
| RNA related | |||
| ROT/TROVE | α-helical repeats | GI: 218233499 | RNA binding domain |
| Mut7-C | PIN domain-like Rossmannoid |
GI: 115345561 | DUF82; RNase; In operon with AAA+ two gene module and TerBN+TerB+TerBC |
| Ntox49 | BECR fold nuclease | GI:121582837 | Distantly related to barnase, colicin E5 and RelE |
| Miscellaneous | |||
| Calcineurin-like phosphoeserase | 4-layered sandwich | 1) fused to TerB: gi 262193394 residues 270.479 2) GI: 104774242 (in operon with DUF2791-AAA_ATPase and SF-II – helicase dyad) |
Could function as potential nucleases or phosphoesterases |
| Trypsin | B barrel | GI: 28868154 (fused to TerD and YceG) | peptidase |
| Thioredoxin | GI: 21220837 | In operon with TerD | |
| α/β-Hydrolase | α/β 3-layer sandwich | GI: 158421812 | DUF3141; Fused to TerB |
| Peptidase | GI: 148358342 residues 166.379 | fused to TerB | |
| PR-1 | α+β | GI: 111224998 | Fused to TerD in actinobateria |
| DnaJ | All α | GI: 89106939 | Fused to TerB; cochaperone for Hsp70 |
| DOC | α+β | GI; 89893424 | Protein nucleotidyltransferase toxin; In operon with -AAA +ATPase two gene module and PglZ |
Footnotes: All PFAM references are to version 26.0. Statistical support are provided for the domains that are not identified by PFAM, and for those previously assigned DUF names by PFAM. Statistical support is given in the form of e-values from PSI-BLAST searches or from HH-PRED searches. In the case of HH-PRED searches the probability and pvalue of a hit to a HMM of the domain derived from given PDB is shown.
Major gene-neighborhood and domain architecture themes of the ter components
Our analysis revealed several distinct themes among the gene-neighborhoods of the ter and associated genes and domain architectures of their products. These can be visualized as being modularly structured, with some components of the original ter operon forming a single unit, which might be connected, all together or singly, either in the same extended operon or in one polypeptide, to various other distinct modules comprised of a diverse set of protein domains. Thus, this subset of ter components from the original ter operon might be seen as a functional hub that connects to various ensembles of domains, each with a distinct function (illustrated as a network in Fig. 4). Below, we briefly summarize these major contextual themes:
-
(i)
Contexts comprised of TerD, TerC and some additional linked genes. Operons combining homologs of TerD and TerC from the original ter operon are found in most major bacterial lineages (Fig. 1, 2). These operons are typically characterized by the presence of more than one paralog belonging to the TerD family encoded by tandem genes (Fig. 2). The minimal versions of these are comprised merely of a tandem array of TerD paralogs, while the more complex versions also contain one or more additional genes encoding members the TerC, TelA, and TerF C-terminal (TerF-C) vWA domain families. Three other poorly characterized families of proteins namely the YceG, XpaC and Aim24 (a distinct version of the double stranded β-helix fold) domains might also be encoded by these operons (Fig. 2B).
-
(ii)
Contexts with TerY and a potential phosphorylation-dependent signaling switch. In this thematic group of operons the core set of ter genes, i.e. those coding for the multiple TerD paralogs, and in some cases additionally TerC and the other families mentioned in the above group, are combined with a further three gene module. The three genes in this module encode the vWA domain protein TerY, a protein phosphatase of the PP2C superfamily and a serine/threonine/tyrosine (STY) protein kinase and are henceforth called the TerY-phosphorylation (TerY-P) triad. This TerY-P triad might also occur as a standalone module independently of the ter components described in the previous group. TerY-P triad might also occur combined with some other distinct modules either by itself or along with some of the ter and associated genes described in the above group (Fig. 2A and C; See below).
-
(iii)
Contexts combining ter gene and components of a novel biosynthetic module. In this group of operons one or more paralogous TerD genes, TerC, and in some cases additional genes, such as TelA, YceG, XpaC or the TerY-P triad, are combined with a further distinctive module comprised of a set of genes encoding a group of predicted biosynthetic enzymes. We hereinafter refer to this module as the “biosynthetic module”, which minimally codes for a predicted amide bond synthetase with an ATP-grasp catalytic domain, a TIM barrel enzyme, at least two phosphoribosyltransferases, and a phosphatase of the HAD superfamily. In some cases the biosynthetic module might occur as a distinct gene cluster potentially sharing a promoter region with the rest of ter genes, but on the opposite strands in the head-to-head orientation (Fig. 2A). In the gene neighborhoods, which combine the ter genes with the biosynthetic module, the ter cluster is often characterized by the presence of an additional component, TerB. The biosynthetic module rarely occurs independently of the remaining ter components, and on those occasions (e.g. certain Bacillus species) there is always a ter gene cluster elsewhere in the genome (Supplementary Material). A second, more reduced biosynthetic module, coding for just a predicted phosphoribosyltransferase and a HAD superfamily phosphatase, is found embedded within a longer operon with the core ter components, the TerY-P triad and TelA in a small set of organisms (Fig. 2A e.g. Chitinophaga pinensis).
-
(iv)
Contexts combining ter components to diverse DNA repair/recombination or phage restriction related modules. The TerD domain was originally reported as being fused to a HNH/EndoVII DNase domain32. In this study we found several additional combinations of both the TerD and TerB domains in the same polypeptide to other domains that are specifically found in DNA-processing and recombination proteins (Fig. 3A and B). Further, TerD, TerB and the TerY-P triad are combined in conserved operons with genes for several DNA-manipulating enzymes, some of which are also found in the previously reported phage restriction operon, namely the Phage Growth Limitation (Pgl) system (Fig. 2C)33, 34. In these cases, the TerY-P triad is also combined with a multi-gene module that encodes a novel, mobile Type VII/Esx secretory system gene cluster (Fig. 2C).
-
(v)
Miscellaneous contexts with various ter components and contexts centered on TelA. Further TerD and TerB domains occur in both prokaryotic and eukaryotic proteins coupled with some other domains diagnostic of a variety of distinct functions (Fig. 3; See below). The TelA gene also occurs, independently of the ter genes, with other conserved genes in distinct operons primarily found in firmicutes but also less frequently in other lineages such as proteobacteria, deinococci and spirochaetes (Fig. 2B).
These contextual themes indicate that the TerD, TerB, TerC, TerY and TelA families appear to act like central units that connect and potentially bring together various other functional modules with biochemical activities as disparate as signal transduction, soluble metabolite biosynthesis, DNA repair and RNA-based regulation (Fig. 3). In the next section of this article we analyze in detail, using sequence and structure analysis, the major individual components of each of these thematic groups and explore their functional significance.
The TerD, TerC, and TerB, family proteins potentially constitute novel membrane-associated complexes with multiple independent metal-binding sites
Functional diversification of the TerD paralogs
Given that the core of the ter gene clusters code for multiple paralogous TerD domain proteins (Fig. 2A)12, 23, we first investigated the significance of this pattern. In bacteria, multiple paralogous genes in a single operon are an infrequent occurrence: it either implies that the multiple paralogous proteins function as alternative versions of a single protein or that they constitute a multi-subunit complex with functional diversification of the individual subunits35, 36. To differentiate between these alternatives, we analyzed the conservation pattern of the TerD superfamily in light of the published structure of the TerD protein (PDB:2KXV)23. The structure of the TerD domain reveals two calcium-binding sites formed by conserved residues mapping to the inter-strand loops of its β–sandwich fold (Fig. 5A). In Klebsiella pneumonia TerD, these sites are respectively constituted by D12, D67 and D70 (site 1) and D22, D24, D60, and E71 (site 2), with W11 acting as a hydrophobic structural bridge between them, possibly facilitating cooperative metal-binding by the two sites23 (Fig. 5A). Superimposing the conservation pattern of the TerD family on the structure reveals three versions of the TerD domain (Fig. 5; Supplementary material): 1) Those with all Ca2+-binding sites intact, which are predicted to bind two Ca2+ ions; 2) those in which only one Ca2+-binding site is conserved, which might at best bind a single Ca2+ ion; 3) those with poor conservation of both Ca2+-binding sites which might not bind Ca2+ ions. Combining the operonic organization of the TerD paralogs with a phylogenetic analysis of the family (Supplementary information) we observed that most operons code for at least two TerD paralogs, belonging to two distinct clades in the tree, with both Ca2+-binding sites intact. Additionally, they code for at least one more TerD paralog, which invariably has only a single Ca2+-binding site or has entirely lost it, and belongs to clades of the TerD tree that are distinct from those containing two intact Ca2+-binding sites. This suggests that the different TerD paralogs of the ter operon had largely diversified even in the ancestral operon, before its dissemination across bacteria. These observations, taken together with the fact that most ter operons code for 3–5 TerD domains, suggest that the different TerD paralogs are likely to form a multi-subunit complex of fixed stoichiometry (possibly torroidal in shape; Fig. 6), with the individual subunits being functionally distinct in terms of Ca2+-binding and possibly other properties as well. This inference is also supported by the presence of multiple tandem TerD domains in several eukaryotic TerD homologs (Fig. 3A). Given that the TerF-like TerD paralogs display a fusion to a C-terminal vWA domain, the complexes with a TerF-like protein are predicted to contain an additional metal-binding site, equivalent to the Mg2+ and Co2+ binding sites seen in other vWA domains37. While the TerD family is poorly understood, the evidence from the eukaryotic homologs of TerD suggests that at least some versions of this domain might bind soluble ligands, such as cAMP38. We observed that five residues showing strong conservation across the entire TerD family map to a surface with a shallow cavity located away from the part of the molecule containing the Ca2+-binding sites (Fig. 5A). This site could possibly mediate the interactions of TerD with other components of the system or a diffusible metabolite (see below).
Figure 5.
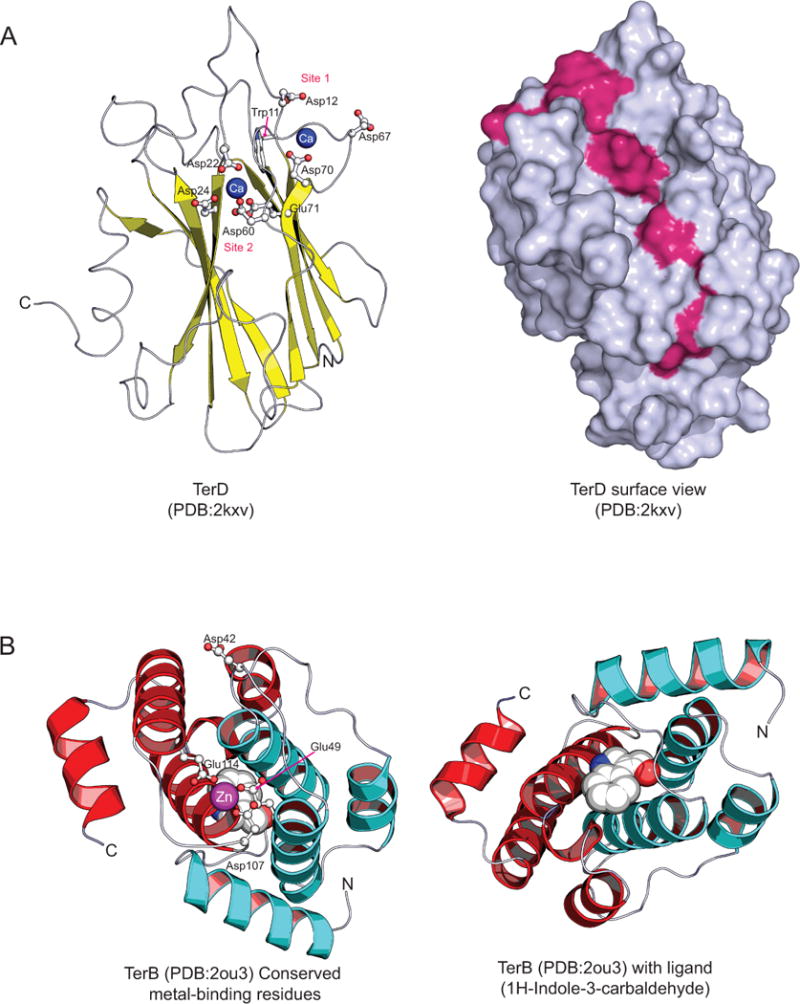
Cartoon structures illustrating (A) TerD with its calcium-binding residues and conserved shallow cavity, and (B) TerB and its metal-binding residues and ligand binding region. Helices in the two individual repeat units of TerB are colored red and blue to highlight the repeat structure. In TerD, the conserved surface residues that comprise the shallow cavity are colored in magenta.
Figure 6.
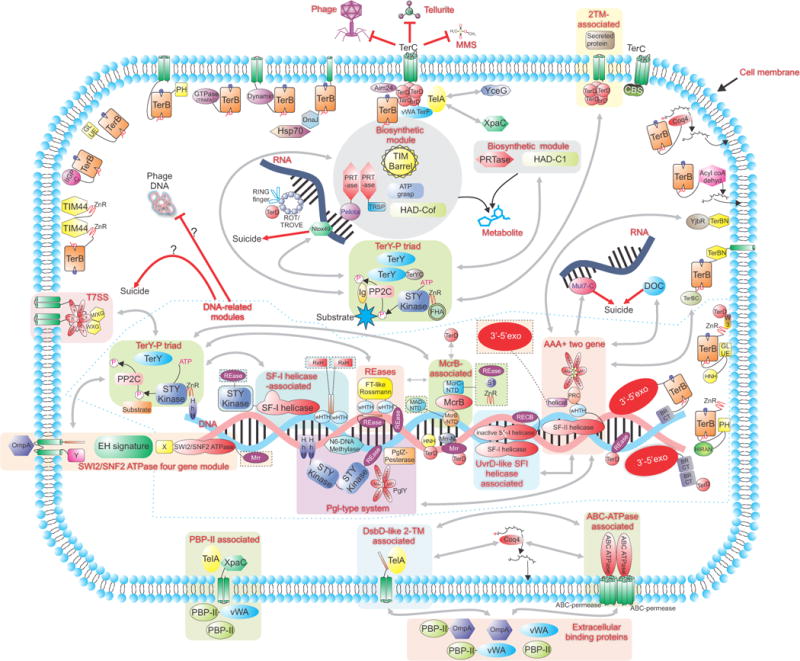
A schematic showing a virtual bacterial cell that summarizes the connections between proteins and systems described in this study along with their predicted sub-cellular localization. Predicted complexes comprised of multiple proteins are enclosed in shaded boxes. Domains that are, on occasion, fused to proteins of the core modules of the predicted complexes are shown in shaded boxes outlined by dashed lines. Bidirectional grey arrows denote connections between: individual modules of complexes or different complexes, proteins and particular complexes or between different proteins, as deciphered by conserved gene neighborhood/domain architecture analysis. These connections are only present in species where these modules are clustered in the same gene neighborhood and can be retrieved from Figures 1, 2 and the Supplementary material. Red arrows illustrate the biological consequence of the action of various complexes. Standard names are used for domain abbreviations.
TerC is a membrane protein with potential intra-membrane metal-binding sites
TerC is the one component encoded by the ter gene clusters, which is most frequently associated with the multiple TerD paralogs (Fig. 2A, B; Supplementary material). This strongly suggests that the complex formed by the TerD paralogs associates with a transmembrane unit formed by TerC (Fig. 6). In course of our analysis, we extended the TerC family to include multiple previously unrecognized members including one defined as a “domain of unknown function”, DUF475 in the Pfam database (Table 1). A multiple alignment of the extended TerC family showed that all the TerC domains share a common core of 7 TM helices. The subset previously termed DUF475 are typified by two extra transmembrane helices inserted between the helices 3 and 4 of the conserved core, and an N-terminal hydrophobic helix, which might constitute either a further TM segment or a signal peptide (Supplementary material). In the conserved 7TM core, there are several unusual charged positions embedded within the TM segments, such as a conserved DN signature in the first TM helix, a conserved R in the second helix, and a conserved DxxxxxD signature in fourth Helix 4. They also display a charged loop between Helix 3 and 4 (Supplementary material). One distinct subfamily of TerC domains has two extra N-terminal hydrophobic helices, with a conserved D on the first of these additional helices (Supplementary material). The multiple conserved acidic sites in the TerC domain suggest that it could form a structure with an intra-membrane anionic surface, which could either function as conduit or a binding site for metals. It is possible that in the TerC domains occurring independently of the TerD paralogs, where they are combined with C-terminal CBS domains39 (Fig. 3B), the latter domains could recognize a soluble adenosine-containing ligand, as has been reported for other CBS domains40, 41. Thus, as in the case of chloride channels, the CBS domain might regulate the ionic interactions of the TerC multi-TM domain42. It is possible that the overexpression of the TerC paralog alx in response to high pH22 is related to its ability to counter the adverse effects of increased alkalinity by interacting with metal ions at the membrane or by regulating transmembrane permeability.
Structural analysis of the TerB domain and identification of a potential metal-binding site in it
Though TerB is primarily present in the ter clusters only when they are combined with the biosynthetic module, TerB does not exhibit characteristics typical of a catalytic domain. Further, in terms of physical location within the gene cluster, it tends to be proximal to the TerD and TerC family genes rather than the biosynthetic module genes (Fig. 2A), suggesting that it functions primarily in conjunction with the TerD and TerC and might have a role in linking them either to the enzymes encoded by the biosynthetic module or to the diffusible product synthesized by them (Fig. 6). Our analysis of previously published structures of the TerB protein (PDB: 2jxu and 2ou3)24 indicated the presence of an internal repeat of two tetra-helical units (Fig. 5B) that are appressed against each other to constitute a 8 helical unit. The two repeats of the tetra-helical units are recognizable at the sequence level and each of them is typified by a DsxhxxxE motif (where s is small, and h is hydrophobic amino acid residue; Fig. 5B and Supplementary material) present in the hairpin between the central two helices. Hence, this tetra-helical repeat unit appears to be the ancestral structural element of the TerB domain. The conserved acidic residues from the two repeats form an asymmetric metal-binding site that is occupied by Zn2+ cations in the crystal structure of Nostoc TerB (2ou3, Fig. 5B). Additionally, this structure reveals that the Nostoc TerB binds a soluble ligand, indole-3-carbaldehyde, in a pocket opposite to the metal-binding site (Fig. 5B). We speculate that this pocket is likely to represent a site for binding the soluble ligand synthesized by the enzymes of the biochemical module, which is combined with the ter gene cluster (See below).
It is possible that the TerB product encoded by the ter gene cluster assembles into the same above-proposed membrane-associated complex as that formed by TerC and the TerD paralogs (Fig. 6). A role for the TerB family in close proximity to the membrane is also supported by our observations regarding the domain architectures of the TerB family members that occur outside of the ter operons (Fig. 3B and 5):
-
(i)
At least two TerB subfamilies combine the TerB domain with conserved TM regions. Representatives of one of the largest subfamilies of the TerB outside of the ter operons combine, distinct from the previous two, have a conserved N-terminal TM helix. These proteins additionally possess a DnaJ domain43 C-terminal to the TerB domain (Fig. 3B), suggesting that they might couple recruitment of the Hsp70 family chaperones via the DnaJ domain with sensing of metal ions proximal to the membrane by the TerB domains (Fig. 6).
-
(ii)
Two distinct TerB subfamilies combine the TerB domain respectively with a dynamin-like GTPase and a novel GTPase of the TRAFAC-clade; both of subfamilies additionally have conserved TM segments (Fig. 3B).
-
(iii)
Four further TerB subfamilies are characterized by fusions to globular domains, three of which were previously uncharacterized, with potential membrane-binding functions (Fig. 3B, Table 1). Using profile-profile comparisons with the HHpred program we were able to unify the uncharacterized domain found in the first of these subfamilies with PH-like domains of the GLUE family (P-value=10−5; probability 90% hit to a HMM of the GLUE domain derived from PDB: 2cay) known to bind lipids and their derivatives in eukaryotes44. In the second subfamily too the TerB domain is flanked at the N-terminus by a previously uncharacterized family of PH-like domains (distinct from the above PH-like domains; Fig. 3B and Supplementary material), and at the C-terminus by a 2TM domain. The novel PH-like domain could potentially associate with the membrane as has been proposed for other domains with a PH-like fold (Fig. 6)45. Sequence profile searches with the globular domain found in the third of these subfamilies recovered the C-terminal α–helical domain of the vertebrate interferon induced GTPases (e.g. IIGP1; PDB: 1tq4; p=10−6 iteration 4 in a PSIBLAST search with the cognate regions of the TerB family protein Acav_1602 from Acidovorax; GI: 326316418) (Fig. 3B, Table 1). This domain maps to the region required for the interaction of the interferon-induced GTPases with the cell membrane46, suggesting a similar role for the equivalent domain in the bacterial TerB family proteins. The fourth of these subfamilies features one or more copies of a N-terminal Tim44 domain (Fig. 3B, Table 1), which is known to bind lipids and localize to the inner face of membranes (e.g. the mitochondrial inner membrane)47.
-
(iv)
A further TerB subfamily from several distinct bacterial lineages (Supplementary material) combines an N-terminal TerB domain with a C-terminal fatty acyl coA oxidase module (Fig. 3B, Table 1), which binds fatty acyl coA precursors of lipids to catalyze their oxidation using a FAD cofactor to trans-2 enoyl unsaturated versions48.
-
(v)
Finally, one TerB subfamily links the TerB domains to the Coq4 domain (Fig. 3B, Table 1), which has been shown to associate with the inner surface of membranes (e.g. the mitochondrial inner membranes)49. The Coq4 domain, originally identified as a potential enzyme for the lipid-soluble coenzyme Q biosynthesis, is entirely α-helical, with long hydrophobic helices, and a conserved HDxxHx10–13E signature that binds a metal ion required for its biological activity49; however, its exact function has not been established. Examination of a known crystal structure of this domain, determined as part of the structural genomics effort (PDB: 3KB4, Northeastern Structural genomics program), revealed that it binds a geranylgeranyl monophosphate moiety between the long hydrophobic helices with the residues from the above-mentioned signature chelating a metal in the proximity of the phosphate head group. Based on this, we propose that at least some of Coq4 domains might function as an isoprenoid phosphoesterase that potentially cleaves off the isoprenyl phosphate from the pyrophosphate form.
In conclusion, our above analysis suggests that the ter components TerD, TerC and TerB form a multi-site metal-binding multi-protein complex that associates with the cell membrane (Fig. 6). Another key component of this complex is likely to be the double stranded β-helix fold protein AIM24, which is frequently found in these neighborhoods and might also be fused to TerD in several organisms (Fig. 3A and Table 1). This proposal for a membrane associated ter complex is consistent with its observed role in membrane-proximal tellurium deposition27. This complex might also participate in altering membrane potential or permeability, a role consistent with its role in resistance to pore-forming colicins. Furthermore, when the TerB and TerC domains occur independently of the ter gene cluster, they might still have membrane-associated metal-binding capabilities, which feed into a variety of other functions; these might include recruitment of other stress response proteins, like the Hsp70 family chaperones, or alteration membrane structure (e.g. via the dynamin-like GTPase domains). Further, fusion of TerB domains to at least two distinct enzymatic domains with a role in lipid metabolism suggests that TerB domain proteins could potentially alter the membrane bilayer composition, such as via isoprenoid insertion or lipid oxidation, perhaps in response to a signal sensed by the TerB domain (Fig. 6).
Components of the TerY-phosphorylation triad
Like the above-described ter components the vWA domain of TerY has a metal-binding site37. However, its near-obligate neighborhood association with genes for a PP2C phosphatase and a STY-kinase indicates that this metal-binding activity is likely to function primarily in relationship with these enzymes, rather than the other ter components. Moreover, in the case of the TerY-P triad the gene-order is strictly preserved with the TerY gene being the first gene, followed by those for the PP2C phosphatase and STY-kinase (Fig. 2A and C). Most kinases from the TerY-P triad also usually contain a Zn-ribbon domain C-terminal to the kinase catalytic domain (Fig. 3C). These observations hint that the TerY vWA and kinase Zn-ribbon domains might function as metal-binding or metal-dependent sensors that regulate a protein-phosphorylation switch mediated by the action of the kinase and PP2C catalytic domains. The kinases from different versions of the TerY-P triad show some diversity in domain architecture C-terminal to the conserved Zn-ribbon domain (Fig. 3C). A subset of them is fused to helix-hairpin-helix domains (HhH; e.g. GI: 91211359; Fig. 3C), which are known to bind DNA in a variety of DNA-repair proteins such as RecA50. Other kinases instead show different superstructure-forming domains, either WD40 β-propellers (e.g. gi: 158335667) or tetratricopeptide repeats (e.g. gi: 17230316) at their extreme C-termini (Supplementary Material). Yet another group of these kinases (e.g. GI: 113460777), displays a C-terminal β–sandwich domain, which recovers the FHA domain (p=10−5; probability 80%) in profile-profile searches with the HHpred program (Fig 2C and Table 1). One divergent group of these kinases (e.g. GI: 269125743) lack the Zn-ribbon but instead have a novel C-terminal Immunoglobulin (Ig) domain that is also found in the phosphoesterase protein PglZ of the Pgl phage restriction system (Table 1 and Supplementary Material).
When TerY genes are combined with the above-described ter genes (TerD paralogs, TerC, and infrequently TerB) into a single long gene-neighborhood, then there are several notable additions to the basic TerY-P triad (Fig. 2A and 2B): i) In these gene clusters the TerY genes invariably undergo one or more duplications to result in multiple TerY paralogs. ii) At least one of these TerY paralogs is distinguished from the remaining ones by a distinct C-terminal domain (the TerY-C domain), which has 8 conserved metal chelating cysteines or histidines (Supplementary material) that is never found in the standalone TerY-P triads.iii) The PP2C phosphatases in these gene neighborhoods are typified by a previously unrecognized Ig domain (Fig. 2A).iv) The kinases from these TerY-P triads are those that contain the above-mentioned FHA-related domain. These observations suggest that, when the TerY-P triad is combined with the complex formed by the core ter components, the subunit stoichiometry in terms of the number of TerY domains might change, possibly due to the multiple TerY paralogs interacting with the multiple TerD subunits. Additionally, the TerY-C and Ig domains might mediate specific interactions between the phosphatase and TerY or with the other ter components. A second set of gene-neighborhoods, thus far seen only in actinobacteria and chloroflexi, have a single TerD family gene that apparently shares a common promoter with the TerY-P triad genes but always runs on the opposite strand (Fig. 2C). These operons lack TerC, but are characterized by two additional genes, respectively encoding a membrane protein with two TM segments (e.g. gi: 359149534) and a secreted protein (e.g. gi: 359149533). In this system a distinct membrane-associated complex might be assembled by the above membrane protein along with the TerD family protein and the TerY-P triad (Fig 1C; Fig. 6).
Together the above observations suggest that independently or in conjunction with the complex of other ter components the TerY-P triad constitutes a signaling unit that might respond to stresses. The diversity in C-terminal domain architectures of the kinases in TerY-P triad point to a potential diversity of targets on which the phosphorylation switch operates. The fusion to the HhH domain observed in a diverse group of bacteria suggests that one possible target is a DNA-associated protein, which could couple the stress sensing with DNA-processing or restriction of bacteriophages (Fig. 6). The associations with the WD40, TPR, Ig and FHA-like might help the enzymes of the TerY-P triad interact with specific protein substrates directly recognized by these domains. In particular, the TerY-P triad could be an important element that transmits signals between the membrane-associated complex with the core ter components and intracellular targets.
The biosynthetic module: prediction of a novel nucleotide-like metabolite and RNA-based regulation
In an attempt to decipher the nature of the molecule generated by the ter-associated biosynthetic module (Fig. 2A), we undertook a detailed analysis of the individual enzymes encoded by it and endeavored to piece together a tentative pathway based on their predicted activity. We discuss below the core enzymes of this module and their predicted activities.
The phosphoribosyltransferases
One of the most striking features of the biosynthetic operon is the presence of two distinct enzymes belonging to the phosphoribosyltransferase (PRTase) family. All characterized members of the PRTase family utilize 5-phospho-α-D-ribose 1-diphosphate (PRPP) as a substrate and typically replace the diphosphate with purine, pyrimidine or a NH2 group along with anomeric inversion of the ribose ring51, 52. These reactions are a key step in nucleotide and amino acid biosynthesis51, 53. The presence of these enzymes immediately suggested that the ter-associated biosynthetic module is involved in synthesis of a β–N-riboside monophosphate derivative. The first of the predicted PRTase family members in the biosynthetic module (e.g. Z1169 from E.coli) shows a relatively straightforward relationship to characterized members of the PRTase superfamily: PSI-BLAST searches with the N-terminal globular domain (e.g. GI: 15800690, residues 1–220) recovers the orotate PRTases (OPRTases; PDB: 1oro)53 in the 2nd iteration with e-value <10−5. Indeed, these PRTases specifically share several sequence features with the OPRTases to the exclusion of other PRTases suggesting that they might utilize orotate as a substrate (Supplementary material). Consistent with this, they conserve all key features associated with binding of the diphosphate moiety of PRPP53. However, these PRTases from the biosynthetic module are distinguished from the conventional OPRTases involved in pyrimidine biosynthesis by the presence of a distinct C-terminal domain, which overlaps with the imperfectly defined DUF3706 domain in the Pfam database, and contains highly conserved GXXE and TRSP signatures (the TRSP domain in Table 1 and Fig. 3).
The second PRTase protein (e.g. Z1167 from E.coli) from the biosynthetic operons overlaps with an alignment termed DUF2983 in the Pfam database. However our analysis revealed that it contains two distinct globular domains. PSI-BLAST searches initiated with the N-terminal globular domain (e.g. GI: 15800688; range 1–267) recovered significant hits to purine and pyrimidine PRTases in the 3rd iteration with e-values<10−3. However, this PRTase domain was far more divergent than any of the previously characterized base PRTase domains and showed no specific similarities to any particular PRTase family (Supplementary material and Table 1). Its sequence conservation pattern revealed that it possesses a conserved PRPP-binding site, suggesting that it indeed recognizes the basic substrate used by other members of this superfamily51, 53. Our sequence searches showed that the C-terminal globular domain conserved in these proteins is a pelota domain (e.g. PSIBLAST search with GI: 15800688, residues 267–371, recovers the pelota domain in ribosomal protein L7AE in the 2nd iteration with e-values<10−4). The pelota domain is a RNA-binding domain found in several RNA-binding proteins such as pelota, eRF1, SECIS and ribosomal proteins like S12, L7AE and L30AE54. Interestingly, certain PRTase family members, such as PyrR, have been shown to function as nucleotide sensors that regulate transcriptional attenuation of the pyrimidine biosynthesis operon by specifically binding the pyr mRNA55–58. As the pelota domain is known to bind specific RNA secondary structures59, we propose that the second PRTase protein in the ter-associated biosynthetic module might function as a RNA-binding regulator, which might regulate the expression of this module based on the sensing of a nucleotide-like molecule by the PRTase domain. However, as there is nothing to preclude this protein from functioning as a catalytically active PRTase, it is possible that it also functions with the above OPRTase-like enzyme as a heterodimer.
The ATP-grasp amide bond synthetase
Identification of a protein with an ATP-grasp structure (e.g. E.coli Z1170; GI: 15800691) in the biosynthetic module pointed to a nucleotide-dependent ligase activity. We compared this ATP-grasp domain to a comprehensive alignment of diverse ATP-grasp domains that we had published earlier60. This indicated that these ATP-grasp domains were closely related to the carbamoylphosphate synthetase type ATP-grasp domains, and shared with them a synapomorphic, active-site associated signature NxQ in the penultimate strand of the core ATP-grasp fold (Supplementary material). Carbamoylphosphate synthetase catalyzes the ligation of ammonia to carbon dioxide61, suggesting that the ATP-grasp enzyme found in the biosynthetic module is also likely to function as an amide-bond- forming enzyme60, probably catalyzing the ligation of the amino group of an amino acid to a carboxylate group, such as that found in the above-proposed orotate moiety (Supplementary material).
The TIM barrel enzyme
Our profile-profile comparisons with the TIM barrel enzyme encoded by the biosynthetic operon (e.g. Z1166; GI: 15800687) indicated that it is most closely related to the carbon-carbon bond lyases, such as the citrate lyase (β-subunit), 4-hydroxy-2-oxo-heptane-1,7-dioate aldolase and malate synthase, and the macrophomate synthase62–65. It shares with these enzymes key active site motifs, namely an ED signature in the 2nd, an arginine in the 3rd, a glutamate in the 5th, and an asparate in the 6th β-α unit of the TIM barrel domain, suggesting that it catalyzes a comparable reaction as these enzymes (Supplementary material). While most enzymes of this family are conventional carbon-carbon bond lyases, the macrophomate synthase catalyzes a more complex version of this reaction. Strikingly, it is simultaneously a natural catalyst of the Diels-Alder cyclo-addition reaction of an enolate (electron-rich dienophile) and a pyrone (electron-deficient diene) reaction, as well as a decarboxylase of oxaloacetate62. Based on these reactions we propose that the TIM barrel enzyme from the biosynthetic module might act as a C-C bond lyase on the amino acid moiety ligated by the above-mentioned ATP-grasp enzyme along with its decarboxylation to produce a potential orotate derivative (See supplement for a speculative rendering of the possible reaction catalyzed by this enzyme). This step might be compared in part to the action of the C-C bond lyase, orotidine-5′-phosphate decarboxylase66, in pyrimidine biosynthesis.
The HAD superfamily enzyme
The HAD superfamily enzyme found in these operons (e.g. E.coli Z1168; GI: 15800689) is related to the Cof-like proteins, which belong to the “C2” cap-containing assemblage of HAD enzymes (here the cap structural element is inserted between the 3rd strand and 4th helix of core HAD fold)67. Its conserved active site residues strongly support it being a phosphoesterase (Supplementary material). Accordingly, we propose that this HAD enzyme functions as a phosphatase that cleaves the 5-phosphate moiety of β–N-riboside monophosphate derivative synthesized by the action of the above PRTases. This proposal is supported by a comparable phosphatase step seen in pyrimidine and purine biosynthesis in which the 5′-nucleotidase converts pyrimidine or purine 5′ monophosphates to the cognate ribonucleoside61. A few proteobacteria contain a gene for a second HAD phosphoesterase embedded within the biosynthetic gene cluster (e.g. P. syringae Psyr_0807 GI: 66044062). This HAD enzyme is distinct from the above enzymes in having a tetrahelical “C1” cap (here the cap structure is inserted into the characteristic β–hairpin after the 1st strand of the HAD fold)67.
The above analysis shows that the general biochemical activities of the individual enzymes of the biosynthetic module might be predicted with considerable confidence. On the whole their activities resemble related enzymes involved in nucleotide biosynthesis. This, coupled with the relationship of the first PRTase to the orotate PRTase, and the presence of the HAD phosphoesterase, strongly suggests that the biosynthetic module generates a pyrimidine-derived ribonucleoside.
Enzymes of the second smaller biosynthetic module also point to a ribonucleoside derivative
The more reduced biosynthetic modules that are embedded with a smaller group of ter gene clusters (Fig. 2A) encode just two enzymes, namely a PRTase and a HAD superfamily enzyme. This PRTase (e.g. Chitinophaga pinensis Cpin_1168; GI: 256420214) is distinct from those described above and is instead related the ComF-like PRTases that are required for DNA uptake during transformation of competent bacteria68. Likewise, the HAD superfamily enzyme (e.g. Chitinophaga pinensis Cpin_1167; GI: 256420213) found in these operons forms a clade distinct from those described above, and has a tetra-helical ‘C1’ cap67. These observations indicate that both this PRTase and HAD phosphatase have been combined with the ter components for the second time, independently of those described above. These two enzymes are predicted to catalyze respectively, the formation of a β–N-riboside monophosphate using a pyrimidine or purine and its cognate ribonucleoside through phosphatase action. In this case, the absence of the ATP-grasp and TIM barrel enzymes suggests that this system might use a pre-available base, such as orotate, rather than further modifying it. Nevertheless, the independent constitution of comparable biosynthetic operons on two occasions in conjunction with the ter system suggests that deployment of a ribonucleoside derivative is an important aspect of the action of this system.
The ter components are linked to a variety of DNA-processing systems
Architectures of TerD and TerB domains reveal multiple connections to DNA processing and repair
We observed in this study that the previously reported proteins with TerD and the HNH nuclease domains additionally contain Zn-ribbon, TerB, and PH-like (related to the GLUE domain; see above) domains between the originally identified TerD and HNH domains32 (Fig. 3A). The actinobacterial versions of these proteins have, in addition to the PH-like domain, a small domain with three conserved tryptophans (W3), which is only found in actinobacteria and their viruses (overlaps with the N-terminal portion of the Pfam model for DUF2510) in a wide variety of predicted membrane proteins (Table 1; Supplementary material). Based on the domain architectures of these proteins we propose that the W3 domain might provide a further membrane-interaction interface. We also found that both the TerB and TerD domain are fused the BRCT domain and a 3′–5′ DNase domain of the RNaseH fold similar to those found in DNA polymerases50 (Fig. 3A and B). However, their architectures display an interesting difference: While the TerD domain is to the C-terminus of the 3′–5′ DNase and two BRCT domains, the TerB domains are inserted between those two domains (Fig. 3A and B). The TerD domain is also linked to two distinct nuclease domains with the restriction endonuclease (REase) fold (also called PD-(D/E) XK in the Pfam database; Fig. 3A)69:twice independently to nuclease domains belonging to the Mrr-like DNase family and once to a nuclease belonging to a previously uncharacterized family with the REase fold (Supplementary material). Other than nuclease domains, the TerB domain is also fused to the HIRAN domain and the BRCT domain (distinct from the above-mentioned versions with 3′–5′ DNase; Fig. 3B). A number of lines of evidence suggest that the HIRAN domain is a DNA-binding domain with a specialized role in DNA-repair, and is found fused to a number of DNA repair and recombination-related domains70. In eukaryotes, several BRCT domains have been implicated in binding phosphopeptides and polyADP ribose to coordinate DNA repair71. However, there is currently no evidence for the bacterial BRCT domains binding phosphopeptides in DNA repair or chromatin-linked polyADP ribose. Instead, the BRCT domains fused to bacterial NAD-dependent ligases are implicated in binding DNA-termini in the context of strand ligation71, 72. The versions of the BRCT domain fused to the TerB domain are specifically related to those found in the NAD-dependent DNA ligases and are accordingly predicted to be involved in recognition of DNA breaks.
The above architectures indicate that the TerD and TerB domains have been repeatedly combined with different DNA processing-related domains on independent occasions. These observations point to a possible role for components of the ter system in linking the sensing of xenobiotic-induced and other stresses directly to DNA repair. The presence of PH-like and actinobacterial W3 domains in the TerD-TerB-HNH domains proteins suggests that such proteins could potentially localize to membranes and operate on DNA in association with sensing of a stress-related signal at the cell membrane (Fig. 6). The proteins combining the TerB domain with a BRCT domain are usually found in genomic contexts indicative of a degenerate, integrated prophage, and are found on at least one active virus, the Vibrio phage K139 (Supplementary material). The prophage-encoded versions might be potentially utilized by the cells as a defensive mechanism to restrict invading phages as has been previously reported for certain prophages73. Indeed, more generally, the previously reported phage resistance role associated with the ter system might relate to the activation of functionally linked DNA –processing systems.
The TerY-P triad might be combined with two distinct DNA-processing modules
In addition to a subset of kinases from the TerY-P triad, which are fused to DNA-binding HhH domains and have a potential role in DNA-repair (see above), we found that the TerY-P triad displays two other notable connections to DNA-processing-related proteins. In both cases three genes of the core TerY-P triad are combined with distinct multi-gene modules, components of which also occur independently in several bacteria.
Multiple distinct components combine to form the first TerY-P associated DNA-processing module
The more prevalent multi-gene DNA processing module, which is combined with the TerY-P triad, minimally encodes the following components (Fig. 2C, Fig 3, Table 1):
-
(i)
A giant superfamily-I (SF-I) DNA helicase protein whose helicase region is characterized by a unique large insert just upstream of the Walker B motif (GI: 383754211; Table 1; Supplementary material).
-
(ii)
A winged helix-turn-helix (wHTH) domain protein that is likely to bind DNA74. These proteins contain a N-terminal wHTH domain fused to either C-terminal coiled-coil regions, which probably aid their dimerization, or to another conserved globular domain with a conserved PxG signature. All those associated with the TerY-P triad contain the coiled-coil region. A subset of these has a further C-terminal α-helical globular domain with a conserved histidine followed by a RxH signature (Fig. 3C, Fig. 6, Table 1).
The genes coding for the above SF-I helicase and wHTH proteins occur independently of the TerY-P triad in several organisms as a standalone two-gene element (Fig. 2D; labeled the SF-I/wHTH module). In most of these cases, the SF-I helicase proteins shows a further N-terminal fusion to a STY-kinase domain. It is possible that in these cases this fused kinase domain performs a DNA-associated phosphorylation function comparable to that of the kinase from the TerY-P triad. Other than the above two proteins, some of these stand-alone elements might also encode an additional REase-fold DNase domain protein, which in several cases is also fused to the N-terminus of the kinase-SF-I helicase protein (e.g. RPC_3937; gi: 90425412; from Rhodopseudomonas palustris; Fig. 2D). Standalone versions of this two gene element most frequently encode a version of the wHTH protein with the C-terminal globular domain with the conserved PxG signature instead of the coiled coil region (Fig. 3D).
-
(iii)
A McrB clade AAA+ NTPase. At least some versions of this AAA+ domain have been shown to utilize GTP instead of ATP and are subunits of the McrBC restriction endonuclease complex. In this complex, the McrB ATPase forms heptameric rings around DNA and translocates the DNA through the central channel using the free-energy of GTP hydrolysis75, 76. In the MrcBC system, two McrB heptameric torroids that are bound at different sites cause the translocation of DNA with resultant looping of the DNA, which brings the two torroids together with subsequent cleavage of DNA by the McrC subunit75, 76. Typically, these McrB AAA+ domains are usually combined with an N-terminal globular domain, the McrB-N-terminal domain (McrB-NTD; partly matched by the model of the domain unknown function DUF3578 in the Pfam database), which based on the evidence from McrB is predicted to function as a potential site-specific DNA-binding domain77, 78. In support of this proposal, the McrB-NTD domain is also found fused to REase domains in other proteins (Supplementary material). In the gene-neighborhoods where this gene cluster is combined with the TerY-P triad, the encoded McrB type AAA+ protein is distinguished by a long N-terminal coiled-coil segment (Fig, 2C and D, Fig. 3C and 6).
-
(iv)
A protein with a McrC-N-terminal domain (McrC-NTD). We observed that in these gene-neighborhoods the gene for the McrB AAA+ NTPase is always immediately adjacent to a gene encoding a protein with an uncharacterized conserved domain (e.g. gi: 325267434; Table 1). Using sequence profile searches, we were able to unify this domain with the domain found N-terminal to the McrC endonuclease domain (e.g. PSI-BLAST searches initiated with a profile of this domain recovered the N-terminal domain of McrC; e-value=10−4; iteration 13; Supplementary material). Secondary structure prediction revealed that this domain is a predominantly α-helical domain. It displays a conserved ENR signature and a further C-terminal acidic residue; the asparagine in the former motif is nearly absolutely conserved (Supplementary material). Studies on the McrBC system have indicated that McrC stimulates the intrinsic GTPase activity of McrB by 30-fold and is required to dimerize two McrB torroids75, 79. Based on this observation and the strict gene-neighborhood association, which we recovered between the genes for the McrB and McrC-NTD family proteins, we propose that the McrC-NTD specifically functions as stimulatory domain for the activity of the McrB NTPase. It is conceivable that the conserved signatures associated with this domain help it function analogous to GTPase activating proteins such as the RGS domain by specifically interacting with McrB80. In proteins encoded by the gene neighborhoods containing the TerY-P triad, the McrC-NTD proteins are fused to different types of DNA-binding domains (Fig. 3C; Table 1). One subset is fused to a S1-like OB fold domain, similar to those found in the cold shock proteins, and is predicted to bind single-stranded nucleic acids54, whereas the other subset is fused to a Zn-ribbon domain.
McrB AAA+ and McrC-NTD are also encoded by two gene elements independently of the TerY-P triad in numerous bacteria (Fig. 2D; labeled McrB-McrC). In these cases the McrC-NTD is typically fused to a C-terminal REase domain. Such a two gene element is also combined with a single TerD gene in certain bacteria like Bacillus anthracis (e.g. gi: 165871627; Fig. 2C). Several of the McrB proteins from these elements display a further, previously unknown globular domain N-terminal to the McrB-NTD and AAA+ domains. This globular domain is additionally found in restriction endonucleases fused to HNH DNase domains (Fig. 3C, Table 1, Supplementary material). Hence, we term it the MAD-NTD for McrB ATPase-DNase N-terminal domain. The architectures suggest that the MAD-NTD is also likely to function as DNA-binding domain.
-
(v)
The gene neighborhoods coupling the TerY-P triad to this DNA-processing complex typically also encode at least two distinct REase domains. The first of these is more widespread, and is a previously unrecognized clade of REase domains, with a unique glycine-rich insert between the D and the ExK signatures of the REase active site (GI:383790095, Spiaf_0871; DUF3883 in PFAM). The second, less frequently found REase protein, is typified by a N-terminal region containing a domain structurally similar to the Rossmanoid domain found in formyltransferases, flanked by two wHTH domains on either side (giI:160899632 ; Daci_4198) (Fig 2C). The Rossmannoid domain, the second distorted wHTH domain and the REase domain also occur in the Csa3-like proteins linked to certain CRISPR systems81.
Some of these TerY-P triad gene neighborhoods with the above DNA-processing module are further extended via a combination with a novel gene cluster encoding a mobile Type VII/Esx secretion system (T7SS) gene cluster (Fig. 2C). The T7SS system was initially found to be a unique secretion system of the Gram-positive bacteria clade (i.e. actinobacteria, firmicutes, chloroflexi clade), which is dependent upon an ATPase pump of the FtsK-HerA superfamily, with 3 tandem ATPase domains82, 83. Proteins secreted via this system are characterized by a “leader domain” in the form of a four-helical bundle termed the ESAT-6 or WxG domain, which might either occur singly or combined with several other C-terminal domains in a larger polypeptide84, 85. The novel T7SS genes, which we found combined with the TerY-P DNA-repair/recombination neighborhoods, represent the first examples of the T7SS outside of the Gram-positive clade of bacteria. In addition to a few firmicutes, it is found across a wide range of bacterial lineages, such as the planctomycetes, verrucomicrobia, lentisphaerae, proteobacteria, cyanobacteria, and fusobacteria. The phyletic pattern of this novel T7SS, with a sporadic presence in distantly related bacterial lineages is strongly suggestive of it being a mobile unit. Furthermore, it is distinguished from the classical version by the architecture of its ATPase pump, which has only one active ATPase domain, the remaining two being catalytically inactive (Table 1). The gene neighborhoods encoding this novel T7SS addition to the gene for the ATPase also always contain at least two distinct genes for standalone WXG domain proteins, which are likely to be the targets for export by this T7SS (Fig. 2C and 5). This T7SS gene-cluster is combined with the TerY-P triad and the above DNA recombination/repair gene-cluster in firmicutes, several proteobacterial lineages and fusobacteria, whereas it occurs independently of them in cyanobacteria and the planctomycete-verrucomicrobia clade (Fig. 2D). This points to a serial process of “operon accretion”, in which the originally independent TerY-P triad and the above DNA-processing module combined to give rise to a composite binary element, which was followed by addition of the T7SS gene neighborhood to result in an even larger ternary element (Fig. 2C). It should also be noted that the stand alone versions of these novel T7SS appear to represent the minimal versions of this kind of secretory apparatus, with just the ATPase pump and the WXG secretion targets. Hence, it is conceivable that they are indeed close to the ancestral versions that gave rise to the more developed T7SS that are typical of the Gram-positive bacterial clade.
Based on the components encoded by these gene-neighborhoods, we propose that in these cases signaling via the phosphorylation switch mediated by the TerY-P triad is combined with DNA processing (Fig. 6). The presence of the McrB NTPase and the McrC-NTD indicates that this DNA processing module might mediate a DNA-looping event upon recognition of specific sites as proposed for the classical McrBC system75. The REase domain proteins point to cleavage of the DNA similar to that mediated by restriction systems. This could potentially happen in the context of DNA repair in response to a xenobiotic stress recognized by the TerY-P triad. Several Csa3-related proteins from CRISPR systems share the wHTH and formyltransferase-like Rossmanoid domains found fused to one of the REase domain proteins encoded by this neighborhood81. This suggests that this DNA-processing complex being deployed in response to phage DNA as a potential phage-restriction system, which either cleaves phage DNA or limits phage infection by suicidal action on self DNA. In some cases suicidal or translation limiting action might also be initiated by RNases encoded by linked genes, such as the recently described NTox49 RNAse domain86, which is found in a subset of these neighborhoods (e.g. gi: 121582837 from Polaromonas naphthalenivorans; Fig. 2C). Based on the apparent mobility of the Esx/T7SS reported here, and the role of FtsK-HerA superfamily ATPases in the cell-to-cell mobility of conjugative transposons and T4SS-dependent plasmids87, we propose that these systems might aid in the cell-to-cell transfer of the DNA elements that encode it. In support of this proposal, we found that versions of this element found in relatively distant proteobacteria such as Delftia acidovorans, Bordetella petrii, Methylomonas methanica and Stenotrophomonas sp. SKA14 showed high levels of identity at the nucleotide level (Supplementary material). Furthermore, the element is absent in the genomes of closely related sister species of Delftia and Bordetella, supporting the idea of its recent lateral mobility between distant species.
The TerY-P triad might associate with a SWI2/SNF2 ATPase-dependent DNA-processing system that communicates with cell surface
In a few cases, for example in the bacterium Bacteroides sp. 2_1_7, the TerY-P triad, together with a gene coding for a protein containing a TerB domain, are combined in a large, predicted operon with a further four-gene module coding for a SWI2/SNF2-type superfamily-II (SF-II) DNA helicase and two other uncharacterized genes. Our investigation revealed that the latter four-gene module (Fig. 2C and D; labeled the SWI2/SNF2-four gene module) also occurs independently across a wide phyletic range of bacteria. Typically, it is found in bacteria that lack the biosynthetic module, although in a small subset of organisms, particularly psychrophiles, both modules might occur in the same genome (Fig. 1). Additional sequence analysis of the proteins encoded by this SNF2-four gene module provided interesting functional leads:
-
(i)
The SWI2/SNF2 ATPase encoded by these gene neighborhoods is related to the prokaryotic SWI2/SNF2 ATPases encoded by several restriction-modification operons, but are distinguished from all other SWI2/SNF2 ATPases by the presence of a novel, previously uncharacterized N-terminal domain (Fig. 3C & D, Table 1, Supplementary material). A subset of these SWI2/SNF2 ATPases is fused to a C-terminal REase fold domain related to the Mrr endonuclease. By analogy to the related ATPases found in the restriction-modification operons it is possible that these might function as DNA helicases88.
-
(ii)
In these gene-neighborhoods, the SWI2/SNF2 ATPase is always preceded by a gene coding for a protein with a large conserved domain, which has a strongly conserved glutamate at the N-terminus and a histidine at the C-terminus (called EH_Signature; Fig. 2D, Table 1, Supplementary material). While it could not be unified with any known catalytic domain, this pattern of conserved charged residues is not incompatible with enzymatic function, such as nuclease activity. Its strict-neighborhood association with SWI2/SNF2 ATPase strongly suggests a function in conjunction with it.
-
(iii)
The third gene in these clusters encodes a protein with an N-terminal predicted signal peptide, and an OmpA domain (Fig. 2D). This suggests that it is a secreted protein, which is consistent with the function of the OmpA domain in binding peptidoglycan89, 90.
-
(iv)
The gene coding for the OmpA domain protein is always preceded in these neighborhoods by a gene coding for fast-evolving membrane protein with 3 TM regions and a C-terminal cytoplasmic tail with a coiled-coil segment (Fig. 2D). The first of the two membrane-associated segments of this protein has a peculiar sequence signature similar to that observed in several voltage-gated ion channels and might have a role related to membrane permeability91.
Taken together, the evidence from the above analysis indicates that the products of this four gene module have the hallmarks of potentially constituting a complex with both cell-surface/membrane-linked and cytoplasmic elements. The OmpA domain interacting with extracellular peptidoglycan and the TM protein could constitute the former which communicates via the cell membrane with the latter comprised of the SWI2/SNF2 ATPase and the protein with the conserved glutamate and histidine (Fig. 6). Such a system could link extracellular events, such as alterations of peptidoglycan integrity or membrane-linked changes with DNA-processing mediated by the SWI2/SNF2 ATPase and the protein with the conserved glutamate and histidine residues. Thus, this system could, in principle, directly respond to either chemical agents or disruption of peptidoglycan by phages.
Neighborhood associations of genes encoding TerB-domain with a distinct-DNA processing module
A subfamily of TerB domain proteins are encoded in gene neighborhoods where they are combined with a further two-gene module with a potential role in DNA processing (Fig. 2C and D; called AAA+ two gene module). We further analyzed this subfamily of TerB proteins, the products of this two gene module and other associated genes to better apprehend their potential biochemistry and functions. The TerB proteins in this neighborhood are characterized by an N-terminal membrane-spanning helix followed by a predicted cytoplasmic region, which contains TerB domains sandwiched between two distinct globular domains with a predominantly α-helical structure (gi: 160900575, Fig. 3B). Of these, the N-terminal domain contains an absolutely conserved glutamate and partly overlaps with the imprecisely defined DUF4016 from the Pfam database (TerBN, Table 1). The C-terminal domain has not been reported before and displays multiple conserved acidic residues (TerBC). The presence of conserved acidic residues in both these domains suggests that they, like the TerB domain, might also chelate metals. These two domains might also occur together in the same polypeptide independently of the TerB domain in certain cases (Fig. 2D; see below).
The two gene module associated with these neighborhoods is characterized by the following genes:
-
(i)
The first gene encodes a protein with an AAA+ NTPase domain (gi:152987173; PSPA7_4430 of Pseudomonas aeruginosa; DUF2791 of Pfam). This version of the AAA+ domain is distinct from all other members of this superfamily and is characterized by the presence of an α-helical insert after the second strand-helix unit of the core AAA+ domain, comparable to that seen in the HslU/ClpX clade92. This insert is predicted to form a second torroidal structure that surrounds the central aperture of the AAA+ multimeric torroid93.
-
(ii)
The second gene encodes a superfamily-II helicase (SF-II) related to the DNA helicases involved in DNA repair typified by the RecQ helicase94. The SF-II helicases encoded by the two-gene module are distinguished by their domain architecture characterized by three additional C-terminal domains beyond the core helicase region (PSPA7_4429 of Pseudomonas aeruginosa) (Fig. 3C and Supplementary material). The first of these is a wHTH domain that is characteristic of all RecQ-like DNA helicases and binds DNA at the site of unwinding95–98. The second domain is a β-barrel domain with distantly related the PRC barrel (HHpred p=10−5, probability 90% hit to the PRC barrel domain from RimM, PDB: 3h9n) while the third is a multi-helical domain. Based on the structure of the RecQ-related helicases (e.g. Hel30895, 98) we propose that these addition domains help form a clasp around the unwound DNA. In some cases (e.g. Hoch_0604; GI: 262193925 from Haliangium ochraceum) there is a further 3′-5′ exonuclease domain fused at the C-terminus (Supplementary material).
In some of the neighborhoods, where the gene for the TerB domain protein is combined with the AAA+ two gene module, there are two further genes which also encode DNA-processing proteins. The first of these encodes a protein with an inactive N-terminal SF-I helicase domain and a C-terminal RecB-like REase fold domain related to the nuclease domain found in the Cas4 proteins from the CRISPR system35, 69. The second gene encodes a large SF-I helicase protein related to UvrD (Fig. 2C). These two genes might additionally occur by themselves in several bacteria (Supplementary material)
Beyond the association with genes for TerB domain proteins, the AAA+ two gene module might also occur as a standalone unit (Fig. 2D). In certain firmicutes it might be further combined with a gene coding for a protein with a YjbR domain (PDB: 2FKI;99) combined with the two α-helical domains flanking the TerB domain in the above-described proteins (Fig. 2D and Supplementary material).
Furthermore, in several cases the AAA+ two gene module is combined with the previously characterized mobile Phage growth limitation (Pgl) operons which have been implicated in restriction of phages34. In the Pgl-type operons these two genes occur adjacent to a gene for a protein with a REase fold nuclease fused to two C-terminal STY-kinase domains and a RNA polymerase α-subunit C-terminal domain (a module comprised of two HhH repeats), and the “core” Pgl genes, which code for a N-6 adenine DNA-methylase, a further AAA+ NTPase named PglY, and the PglZ-type phosphoesterase (Fig. 3C). A subset of these operons might also encode potential toxin proteins, such as a Mut7-C RNase (DUF82 in Pfam) domain protein, which is related to the PIN domain RNases, and a DOC domain protein which modifies proteins by transfer NMP moieties to target serines or threonines100 (Table 1).
The products of the two-gene module, namely the AAA+ NTPase and the SF-II DNA helicase related to RecQ, could be potentially involved in manipulating DNA in relation to recombination or cleavage. In particular the AAA+ domain might function analogous to the clamp-loaders in the assembly of a DNA-associated complex. The TM segments in the TerB domain proteins associated with them suggest that, as in the case of the other systems discussed in this article, the DNA-manipulating activity might respond to a signal, such as the presence of a metal ion, being sensed close to the membrane (Fig. 6). The combination of the two-gene module with the Pgl system and with the proteins with a Cas4-like REase domain also points to a potential role for its activity in anti-phage defense. In this context, its DNA manipulating activity might be directed at restriction of invading phages by unwinding their DNA, facilitating attack by associated nuclease domains or alternatively by acting suicidally on self DNA. The toxin modules found in some of these operons could also augment suicidal action by cleaving particular RNAs or modifying proteins.
Other architectures and gene neighborhoods of the core ter components and transfers to eukaryotes
The TerB, TerC and TerD domains occur outside of the above systems in certain notable domain architectural and gene-neighborhood contexts. For example, in several proteobacteria a version of the TerB domain, which has lost its metal-binding sites occurs fused to a distinctive α/β-hydrolase domain (e.g. gi: 158421812; termed domain of unknown function DUF3141 in proteobacteria; Fig 2B). In a phylogenetically diverse group of bacteria, the TerD domain occurs fused to the C-terminus of certain proteins with a divergent version of the α-helical ROT/TROVE (Fig. 3A). Some of these proteins additionally contain the recently described bacterial-type RING finger domain101. As ROT/TROVE domains have been implicated in binding RNA and regulating RNA stability and translation102, it is possible that these proteins couple sensing of a stress signal with RNA interaction. TerD domains have also been transferred to eukaryotes on multiple occasions. In eukaryotes two notable associations are discernible: (i) There are multiple fusions with calcium and lipid-binding C2 domains103 in several microbial eukaryotes, like ciliates, diatoms, Trichomonas and Naegleria, which are suggestive of its involvement in membrane-associated Ca2+-recognition (Fig. 3A). (ii) In several animals and Naegleria the TerD domain is fused to an ubiquitin-like domain (Fig. 3A). The animal versions have a further N-terminal RING finger domain suggesting that they function as E3 Ub-ligases. In certain fungi and chlorophyte algae, the TerD domain is combined in a large protein with an E2 Ub-ligase domain and a F-box domain, which is a component of SCF-type E3 Ub-ligases (Fig. 3A). Thus, this protein as a whole is predicted to function as part of a SCF-type Ub ligase. It is conceivable that the eukaryotic versions of the TerD domain also have a role in stress response by recruiting Ca2+-dependent or Ub-conjugation-based signaling pathways. The prior evidence for the TerD domains binding cAMP in Dictyostelium38 suggests that in eukaryotes they might have also been recruited to the cyclic nucleotide signaling system.
Architectures and gene neighborhoods of TelA-containing systems
Other than the ter gene clusters with multiple TerD paralogs, there is one conserved gene neighborhood, seen in several proteobacteria and Gram-positive bacteria, where a TelA gene is linked with single TerD genes along with genes for TerC and one encoding a protein with a standalone vWA domain related to that found in TerF (Fig. 2B). The strong gene-neighborhood association of TelA with the TerF-like vWA domain suggests that it might interact specifically with this vWA domain in the membrane-associated ter complex. The TelA domain is a highly polar domain predicted to adopt an all α-helical fold, with at least 10 conserved helices, that typically occurs as a standalone protein (Supplementary material). TelA genes co-occur, mostly mutually exclusively, with two genes respectively of the YceG and XpaC family, both in the context of the core ter genes and independently of them (Fig. 2A and B). This indicates that the TelA protein specifically interacts either with the YceG or XpaC protein. The YceG proteins contain two copies of an α+β domain, with a nearly absolutely conserved aspartate embedded in an otherwise strongly hydrophobic segment, which could potentially interact with the membrane. The XpaC family proteins are characterized by a tetrahelical domain combined with one or more N-terminal membrane-spanning helices (Supplementary material). While the XpaC proteins are annotated as proteins mediating the hydrolysis of 5-bromo-4-chloroindolyl phosphate bonds, there is no published evidence in support of such activity. Nevertheless, consistent with a role in stress response, the Bacillus subtilis TelA and XpaC genes have been shown to be induced by high salt stress104. Studies on stress response in Several of the XpaC-containing operons, also encode two secreted proteins, one with just a periplasmic-binding protein-II (PBP-II) domain (e.g. gi: 160896213, Fig. 2B)105 and another with a PBP-II domain fused to a vWA domain (e.g. gi: 160896215, Fig. 2B). Further support for a membrane-linked function for TelA comes from a group of conserved gene neighborhoods, in which TelA is encoded alongside a predicted membrane protein, which is an inactive version of the cysteine-containing TM domain found in the disulfide-bond reductase DsbD (gi: 237654700; Fig. 2B)106. These neighborhoods often contain additional tightly linked genes encoding multiple secreted proteins with PBP-II or vWA domains (Fig. 2B and 5). These might occur either by themselves or combined together in the same polypeptide, or in polypeptides combining the OmpA and PBP-II domains. Some of these predicted operons (Fig. 2B) might also include genes for an ABC transporter ATPase and its associated membrane-spanning permease protein or a protein with one or two CoQ4 domains (see above for this domain). These associations strongly suggest that TelA could form multiple membrane-associated complexes along with XpaC, YceG and other proteins either by itself or in conjunction with the core ter components (Fig. 6). In some of these complexes, the multiple extracellular binding proteins with PBP-II, OmpA and vWA domains could potentially form an array of extracellular sensors that bind different soluble compounds or peptidoglycan. These signals could then be relayed on the cytoplasmic surface of the membrane by the TelA complex. The ABC transporter ATPase encoded by some of these operons could potentially aid in the efflux of the deleterious compounds, whereas the CoQ4 could bind isoprenoids or alter membrane composition.
A SYNTHETIC OVERVIEW AND GENERAL CONCLUSIONS
While a number of studies in bacteria have pointed to the importance of the ter components and TelA in natural tolerance to different stresses, little is known about how they achieve their functions. Recent structural studies on TerD and TerB have helped in understanding certain structural features and biochemical properties of the ter system23, 24. However, their implications for the ability of the system to counter a mechanistically diverse array of stresses remained unclear. Our analysis based on comparative genomics and sequence-structure comparisons detailed in this study suggests that the ter components and TelA are at the center of a rather extensive system of interacting proteins and small-molecules whose “tentacles” extend to a wide range of biological processes. Thus, in a sense the ter components and TelA appear to resemble the complexes that have been uncovered via chemogenomics studies in eukaryotes that provide robustness to the system against a diverse array of mechanistically distinct chemical stresses2.
Specifically, our studies clarify a number of distinct points regarding the action of ter- and TelA- centered systems. First, different lines of evidence suggest that key ter components such as those with TerD, TerB, TelA, XpaC and vWA domains are likely to form multi-subunit complexes proximal to the inner surface of the membrane (Fig. 6). This achieved either with a membrane-spanning element like TerC or other TM segments or by means of different lipid-binding domains. Second, several of these domains like TerD, TerB and vWA possess predicted metal-binding sites, suggesting they might have the ability to directly sense metals or use cations to regulate their interactions (Fig. 5 and 6). These inferences of a membrane associated complex-metal binding are consistent with early observations on the ter system, which pointed to the membrane-proximal reduction of tellurium with its crystallization on the outer surface of the membrane and resistance to pore-forming colicins12, 26, 27. In some cases the complexes in the inner membrane are also complemented by an array of secreted proteins with PBP-II, vWA and OmpA domains that could form an extracellular system to sense a variety of compounds, and also possibly the state of the peptidoglycan. This work also clarifies the role of the TerY-P triad, revealing it to be at the center of a signaling switch, which could relay the membrane-proximal events sensed by the core ter components/TelA within the cell through phosphorylation and dephosphorylation of proteins. Furthermore, analysis of the domain architectures and operons of TerB, TerD and TerY-like vWA domains reveals that on multiple occasions they have been combined with manifold DNA-processing enzyme complexes. This suggests that the sensing of xenobiotics proximal to the membrane by these ter components could directly trigger a repair response to withstand their potential mutagenic effect. Importantly, this connection to the DNA-processing components also explains a key aspect of the ter system, i.e. natural resistance against several unrelated phages. We posit that these DNA-processing components, with one or more DNase domains, by themselves or in conjunction with certain previously characterized systems such as the Pgl systems33, 34, could restrict phages by targeting their DNA. It is also possible that some of the nucleases in these systems limit phage infection through “apoptotic” action by cleaving the cellular genome or inhibit translation by cleaving or binding different RNA molecules.
Perhaps one of the most striking aspects of our investigation is the recovery of biosynthetic systems that function in conjunction with the ter components. The evidence from the presence of the PRTases and phosphatases in this system strongly imply a nucleotide-derived metabolite; however, the question arises as to what might be the role of this nucleotide-like metabolite? The previous report on the poorly studied eukaryotic TerD homologs binding cAMP38 is consistent with TerD possibly binding a nucleotide generated by this system. Likewise, the indole-3-carbaldehyde in the structure of TerB is also consistent with it binding a nucleotide-derived ligand (Fig. 5). Thus, at least some of the TerD and TerB domains could possibly bind the ligands produced by the biosynthetic system and influence the sensing of stress signals. Further, the PRTase fused to the pelota domain could bind mRNA and act as a ligand-influenced translation regulator (Fig. 6). Alternatively, but not mutually exclusively, the solute produced by this biosynthetic system could play a more active role as an extremolyte. Extremolytes such as the ectoine and hydroxyectoine, which are pyrimidine-derived compounds, have been shown to protect bacteria from high osmolality107. In a similar vein, the product of the biosynthetic operons could also have a stress-protective role. Finally, it should be noted that PRTases, such as ComF, have also been implicated in competence for DNA uptake by transformation in cyanobacteria68. The PRTases encoded by the shorter two-gene biosynthetic modules embedded in ter system are closely related to ComF PRTases. Other than occurring in the ter system, these PRTases occur in competence-related operons along with a gene for the competence protein Smf/DprA and are fused to N-terminal SF-II DNA helicase modules (e.g. E.coli c4223, gi: 26250047) or to Zn-ribbon domains (e.g. Pseudomonas aeruginosa PA0489, GI: 15595686). The DprA/Smf proteins act downstream of the DNA-uptake machinery and have been shown to bind ssDNA108, 109. While the exact role of the PRTases in DNA uptake remains unresolved, we propose that this functional connection is of importance and might be related to the ability of the PRTases to bind single stranded nucleic acids55–58. Thus, a direct interaction between components of the biosynthetic module and invading DNA, such as those of phages cannot be ruled out. In any case, further biochemical analysis of the biosynthetic system reported here is likely to be a promising area for uncovering an entirely unexpected aspect of bacterial stress response.
In conclusion our computational analysis of the ter system and associated genes has uncovered multifaceted stress response and anti-viral systems that are widely distributed across bacteria. The phyletic patterns of these systems point to an extensive dispersion mainly through lateral transfer, pointing to the potential selective advantage conferred by these systems across a wide phylogenetic range of bacteria. To some degree these systems have also been acquired by eukaryotes, although in them the individual components appear to have been coupled with alternative signaling mechanisms in conjunction with the ubiquitin and calcium-based signaling system. Our analysis also provides the first indications as to how the ter system could simultaneously provide natural resistance against mechanistically disparate stresses by mobilizing different types of repair, anti-viral defense and signaling systems both on the cell-surface and within the cell. We hope this analysis would help further biochemical explorations on this unprecedented class of systems and potentially aid in development of novel counter-bacterial therapeutics.
MATERIAL AND METHODS
Iterative sequence profile searches were performed using the PSI-BLAST110 and JACKHMMER programs run against the non-redundant (NR) protein database of National Center for Biotechnology Information (NCBI). Similarity-based clustering for both classification and culling of nearly identical sequences was performed using the BLASTCLUST program (ftp://ftp.ncbi.nih.gov/blast/documents/blastclust.html). The HHpred program was used for profile-profile comparisons111. Structure similarity searches were performed using the DaliLite program112. Multiple sequence alignments were built by the Kalign and PCMA programs113, 114, followed by manual adjustments on the basis of profile-profile and structural alignments. Secondary structures were predicted using the JPred program115. For previously known domains, the Pfam database was used as a guide116, though the profiles were augmented by addition of newly detected divergent members that were not detected by the original Pfam models116. Clustering with BLASTCLUST followed by multiple sequence alignment and further sequence profile searches were used to identify other domains that were not present in the Pfam database. Signal peptides and transmembrane segments were detected using the TMHMM and Phobius programs117, 118. Contextual information from prokaryotic gene neighborhoods was retrieved using custom Perl scripts that extract the upstream and downstream genes of the query gene, along with their orientation, from GenBank files. A combination BLASTCLUST and sequence profile searches were then used to cluster the proteins to identify conserved gene-neighborhoods. Phylogenetic analysis was conducted using an approximately-maximum-likelihood method implemented in the FastTree 2.1 program under default parameters119. Structural visualization and manipulations were performed using the PyMol (http://www.pymol.org) program. The in-house TASS package, which comprises a collection of Perl scripts, was used to automate aspects of large-scale analysis of sequences, structures and genome context. The principal component analysis of the phyletic patterns of ter and associated genes was performed using the R statistical language (http://www.r-project.org/). Supplementary material can also be accessed at: ftp://ftp.ncbi.nih.gov/pub/aravind/Ter/Ter.html
Supplementary Material
Acknowledgments
The authors are supported by the intra-mural funds of the National Library of Medicine, National Institutes of Health, USA.
References
- 1.Venancio TM, Bellieny-Rabelo D, Aravind L. Front Genet. 2012;3:47. doi: 10.3389/fgene.2012.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Venancio TM, Balaji S, Geetha S, Aravind L. Mol Biosyst. 2010;6:1475–1491. doi: 10.1039/c002567b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venancio TM, Balaji S, Aravind L. Mol Biosyst. 2010;6:175–181. doi: 10.1039/b911821g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Venancio TM, Aravind L. Bioinformatics. 2010;26:149–152. doi: 10.1093/bioinformatics/btp647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roemer T, Davies J, Giaever G, Nislow C. Nat Chem Biol. 2012;8:46–56. doi: 10.1038/nchembio.744. [DOI] [PubMed] [Google Scholar]
- 6.Ho CH, Piotrowski J, Dixon SJ, Baryshnikova A, Costanzo M, Boone C. Curr Opin Chem Biol. 2011;15:66–78. doi: 10.1016/j.cbpa.2010.10.023. [DOI] [PubMed] [Google Scholar]
- 7.Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, Brown GW, Kane PM, Hughes TR, Boone C. Nat Biotechnol. 2004;22:62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- 8.Shoemaker DD, Lashkari DA, Morris D, Mittmann M, Davis RW. Nat Genet. 1996;14:450–456. doi: 10.1038/ng1296-450. [DOI] [PubMed] [Google Scholar]
- 9.Masel J, Siegal ML. Trends Genet. 2009;25:395–403. doi: 10.1016/j.tig.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chasteen TG, Fuentes DE, Tantalean JC, Vasquez CC. FEMS Microbiol Rev. 2009;33:820–832. doi: 10.1111/j.1574-6976.2009.00177.x. [DOI] [PubMed] [Google Scholar]
- 11.Turner RJ, Weiner JH, Taylor DE. Microbiology. 1999;145(Pt 9):2549–2557. doi: 10.1099/00221287-145-9-2549. [DOI] [PubMed] [Google Scholar]
- 12.Taylor DE. Trends Microbiol. 1999;7:111–115. doi: 10.1016/s0966-842x(99)01454-7. [DOI] [PubMed] [Google Scholar]
- 13.Azeddoug H, Reysset G. Curr Microbiol. 1994;29:229–235. doi: 10.1007/BF01570159. [DOI] [PubMed] [Google Scholar]
- 14.Choudhury HG, Cameron AD, Iwata S, Beis K. Biochem J. 2011;435:85–91. doi: 10.1042/BJ20102014. [DOI] [PubMed] [Google Scholar]
- 15.Liu M, Turner RJ, Winstone TL, Saetre A, Dyllick-Brenzinger M, Jickling G, Tari LW, Weiner JH, Taylor DE. J Bacteriol. 2000;182:6509–6513. doi: 10.1128/jb.182.22.6509-6513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collins B, Joyce S, Hill C, Cotter PD, Ross RP. Antimicrob Agents Chemother. 2010;54:4658–4663. doi: 10.1128/AAC.00290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whelan KF, Colleran E, Taylor DE. J Bacteriol. 1995;177:5016–5027. doi: 10.1128/jb.177.17.5016-5027.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponnusamy D, Hartson SD, Clinkenbeard KD. Vet Microbiol. 2011;150:146–151. doi: 10.1016/j.vetmic.2010.12.025. [DOI] [PubMed] [Google Scholar]
- 19.Lauzier A, Simao-Beaunoir AM, Bourassa S, Poirier GG, Talbot B, Beaulieu C. Mol Plant Pathol. 2008;9:753–762. doi: 10.1111/j.1364-3703.2008.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sanssouci E, Lerat S, Grondin G, Shareck F, Beaulieu C. Antonie Van Leeuwenhoek. 2011;100:385–398. doi: 10.1007/s10482-011-9593-y. [DOI] [PubMed] [Google Scholar]
- 21.Taylor DE, Rooker M, Keelan M, Ng LK, Martin I, Perna NT, Burland NT, Blattner FR. J Bacteriol. 2002;184:4690–4698. doi: 10.1128/JB.184.17.4690-4698.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nechooshtan G, Elgrably-Weiss M, Sheaffer A, Westhof E, Altuvia S. Genes Dev. 2009;23:2650–2662. doi: 10.1101/gad.552209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan YR, Lou YC, Seven AB, Rizo J, Chen C. J Mol Biol. 2011;405:1188–1201. doi: 10.1016/j.jmb.2010.11.041. [DOI] [PubMed] [Google Scholar]
- 24.Chiang SK, Lou YC, Chen C. Protein Sci. 2008;17:785–789. doi: 10.1110/ps.073389408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Burian J, Tu N, Kl’ucar L, Guller L, Lloyd-Jones G, Stuchlik S, Fejdi P, Siekel P, Turna J. Folia Microbiol (Praha) 1998;43:589–599. doi: 10.1007/BF02816374. [DOI] [PubMed] [Google Scholar]
- 26.Turner RJ, Weiner JH, Taylor DE. Can J Microbiol. 1995;41:92–98. doi: 10.1139/m95-012. [DOI] [PubMed] [Google Scholar]
- 27.Suzina NE, Duda VI, Anisimova LA, Dmitriev VV, Boronin AM. Arch Microbiol. 1995;163:282–285. doi: 10.1007/BF00393381. [DOI] [PubMed] [Google Scholar]
- 28.Osterman A, Overbeek R. Curr Opin Chem Biol. 2003;7:238–251. doi: 10.1016/s1367-5931(03)00027-9. [DOI] [PubMed] [Google Scholar]
- 29.Koonin EV, Wolf YI, Aravind L. Genome Res. 2001;11:240–252. doi: 10.1101/gr.162001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bork P, Dandekar T, Diaz-Lazcoz Y, Eisenhaber F, Huynen M, Yuan Y. J Mol Biol. 1998;283:707–725. doi: 10.1006/jmbi.1998.2144. [DOI] [PubMed] [Google Scholar]
- 31.Vavrova S, Valkova D, Drahovska H, Kokavec J, Mravec J, Turna J. Biometals. 2006;19:453–460. doi: 10.1007/s10534-005-4862-8. [DOI] [PubMed] [Google Scholar]
- 32.Makarova KS, Aravind L, Wolf YI, Tatusov RL, Minton KW, Koonin EV, Daly MJ. Microbiol Mol Biol Rev. 2001;65:44–79. doi: 10.1128/MMBR.65.1.44-79.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sumby P, Smith MC. Mol Microbiol. 2002;44:489–500. doi: 10.1046/j.1365-2958.2002.02896.x. [DOI] [PubMed] [Google Scholar]
- 34.Makarova KS, Wolf YI, Snir S, Koonin EV. J Bacteriol. 2011;193:6039–6056. doi: 10.1128/JB.05535-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Makarova KS, Aravind L, Wolf YI, Koonin EV. Biol Direct. 2011;6:38. doi: 10.1186/1745-6150-6-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang D, Iyer LM, Aravind L. Nucleic Acids Res. 2011;39:4532–4552. doi: 10.1093/nar/gkr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fodje MN, Hansson A, Hansson M, Olsen JG, Gough S, Willows RD, Al-Karadaghi S. J Mol Biol. 2001;311:111–122. doi: 10.1006/jmbi.2001.4834. [DOI] [PubMed] [Google Scholar]
- 38.Kay CA, Noce T, Tsang AS. Proc Natl Acad Sci U S A. 1987;84:2322–2326. doi: 10.1073/pnas.84.8.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bateman A. Trends Biochem Sci. 1997;22:12–13. doi: 10.1016/s0968-0004(96)30046-7. [DOI] [PubMed] [Google Scholar]
- 40.Ok SH, Yoo KS, Shin JS. Plant Signal Behav. 2012;7 doi: 10.4161/psb.19945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lucas M, Encinar JA, Arribas EA, Oyenarte I, Garcia IG, Kortazar D, Fernandez JA, Mato JM, Martinez-Chantar ML, Martinez-Cruz LA. J Mol Biol. 2010;396:800–820. doi: 10.1016/j.jmb.2009.12.012. [DOI] [PubMed] [Google Scholar]
- 42.De Angeli A, Moran O, Wege S, Filleur S, Ephritikhine G, Thomine S, Barbier-Brygoo H, Gambale F. J Biol Chem. 2009;284:26526–26532. doi: 10.1074/jbc.M109.005132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Walsh P, Bursac D, Law YC, Cyr D, Lithgow T. EMBO Rep. 2004;5:567–571. doi: 10.1038/sj.embor.7400172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Im YJ, Hurley JH. Dev Cell. 2008;14:902–913. doi: 10.1016/j.devcel.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Patino-Lopez G, Aravind L, Dong X, Kruhlak MJ, Ostap EM, Shaw S. J Biol Chem. 2010;285:8675–8686. doi: 10.1074/jbc.M109.086959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Martens S, Sabel K, Lange R, Uthaiah R, Wolf E, Howard JC. J Immunol. 2004;173:2594–2606. doi: 10.4049/jimmunol.173.4.2594. [DOI] [PubMed] [Google Scholar]
- 47.Chacinska A, van der Laan M, Mehnert CS, Guiard B, Mick DU, Hutu DP, Truscott KN, Wiedemann N, Meisinger C, Pfanner N, Rehling P. Mol Cell Biol. 2010;30:307–318. doi: 10.1128/MCB.00749-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dym O, Eisenberg D. Protein Sci. 2001;10:1712–1728. doi: 10.1110/ps.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marbois B, Gin P, Gulmezian M, Clarke CF. Biochim Biophys Acta. 2009;1791:69–75. doi: 10.1016/j.bbalip.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aravind L, Walker DR, Koonin EV. Nucleic Acids Res. 1999;27:1223–1242. doi: 10.1093/nar/27.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Craig SP, 3rd, Eakin AE. J Biol Chem. 2000;275:20231–20234. doi: 10.1074/jbc.R000002200. [DOI] [PubMed] [Google Scholar]
- 52.Argos P, Hanei M, Wilson JM, Kelley WN. J Biol Chem. 1983;258:6450–6457. [PubMed] [Google Scholar]
- 53.Heroux A, White EL, Ross LJ, Kuzin AP, Borhani DW. Structure. 2000;8:1309–1318. doi: 10.1016/s0969-2126(00)00546-3. [DOI] [PubMed] [Google Scholar]
- 54.Anantharaman V, Koonin EV, Aravind L. Nucleic Acids Res. 2002;30:1427–1464. doi: 10.1093/nar/30.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kantardjieff KA, Vasquez C, Castro P, Warfel NM, Rho BS, Lekin T, Kim CY, Segelke BW, Terwilliger TC, Rupp B. Acta Crystallogr D Biol Crystallogr. 2005;61:355–364. doi: 10.1107/S090744490403389X. [DOI] [PubMed] [Google Scholar]
- 56.Chander P, Halbig KM, Miller JK, Fields CJ, Bonner HK, Grabner GK, Switzer RL, Smith JL. J Bacteriol. 2005;187:1773–1782. doi: 10.1128/JB.187.5.1773-1782.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Savacool HK, Switzer RL. J Bacteriol. 2002;184:2521–2528. doi: 10.1128/JB.184.9.2521-2528.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonner ER, D’Elia JN, Billips BK, Switzer RL. Nucleic Acids Res. 2001;29:4851–4865. doi: 10.1093/nar/29.23.4851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caban K, Kinzy SA, Copeland PR. Mol Cell Biol. 2007;27:6350–6360. doi: 10.1128/MCB.00632-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Iyer LM, Abhiman S, Maxwell Burroughs A, Aravind L. Mol Biosyst. 2009;5:1636–1660. doi: 10.1039/b917682a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berg JM, Tymoczko JL, Stryer L. Biochemistry. W.H. Freeman; New York: 2012. [Google Scholar]
- 62.Ose T, Watanabe K, Mie T, Honma M, Watanabe H, Yao M, Oikawa H, Tanaka I. Nature. 2003;422:185–189. doi: 10.1038/nature01454. [DOI] [PubMed] [Google Scholar]
- 63.Smith CV, Huang CC, Miczak A, Russell DG, Sacchettini JC, Honer zu Bentrup K. J Biol Chem. 2003;278:1735–1743. doi: 10.1074/jbc.M209248200. [DOI] [PubMed] [Google Scholar]
- 64.Howard BR, Endrizzi JA, Remington SJ. Biochemistry. 2000;39:3156–3168. doi: 10.1021/bi992519h. [DOI] [PubMed] [Google Scholar]
- 65.Goulding CW, Bowers PM, Segelke B, Lekin T, Kim CY, Terwilliger TC, Eisenberg D. J Mol Biol. 2007;365:275–283. doi: 10.1016/j.jmb.2006.09.086. [DOI] [PubMed] [Google Scholar]
- 66.Appleby TC, Kinsland C, Begley TP, Ealick SE. Proc Natl Acad Sci U S A. 2000;97:2005–2010. doi: 10.1073/pnas.259441296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Burroughs AM, Allen KN, Dunaway-Mariano D, Aravind L. J Mol Biol. 2006;361:1003–1034. doi: 10.1016/j.jmb.2006.06.049. [DOI] [PubMed] [Google Scholar]
- 68.Nakasugi K, Svenson CJ, Neilan BA. Microbiology. 2006;152:3623–3631. doi: 10.1099/mic.0.29189-0. [DOI] [PubMed] [Google Scholar]
- 69.Aravind L, Makarova KS, Koonin EV. Nucleic Acids Res. 2000;28:3417–3432. doi: 10.1093/nar/28.18.3417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Iyer LM, Babu MM, Aravind L. Cell Cycle. 2006;5:775–782. doi: 10.4161/cc.5.7.2629. [DOI] [PubMed] [Google Scholar]
- 71.Leung CC, Glover JN. Cell Cycle. 2011;10:2461–2470. doi: 10.4161/cc.10.15.16312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Feng H, Parker JM, Lu J, Cao W. Biochemistry. 2004;43:12648–12659. doi: 10.1021/bi049451c. [DOI] [PubMed] [Google Scholar]
- 73.Snyder L. Mol Microbiol. 1995;15:415–420. doi: 10.1111/j.1365-2958.1995.tb02255.x. [DOI] [PubMed] [Google Scholar]
- 74.Aravind L, Anantharaman V, Balaji S, Babu MM, Iyer LM. FEMS Microbiol Rev. 2005;29:231–262. doi: 10.1016/j.femsre.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 75.Pieper U, Groll DH, Wunsch S, Gast FU, Speck C, Mucke N, Pingoud A. Biochemistry. 2002;41:5245–5254. doi: 10.1021/bi015687u. [DOI] [PubMed] [Google Scholar]
- 76.Panne D, Raleigh EA, Bickle TA. J Mol Biol. 1999;290:49–60. doi: 10.1006/jmbi.1999.2894. [DOI] [PubMed] [Google Scholar]
- 77.Pieper U, Schweitzer T, Groll DH, Pingoud A. Biol Chem. 1999;380:1225–1230. doi: 10.1515/BC.1999.155. [DOI] [PubMed] [Google Scholar]
- 78.Gast FU, Brinkmann T, Pieper U, Kruger T, Noyer-Weidner M, Pingoud A. Biol Chem. 1997;378:975–982. doi: 10.1515/bchm.1997.378.9.975. [DOI] [PubMed] [Google Scholar]
- 79.Pieper U, Brinkmann T, Kruger T, Noyer-Weidner M, Pingoud A. J Mol Biol. 1997;272:190–199. doi: 10.1006/jmbi.1997.1228. [DOI] [PubMed] [Google Scholar]
- 80.Anantharaman V, Abhiman S, de Souza RF, Aravind L. Gene. 2011;475:63–78. doi: 10.1016/j.gene.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lintner NG, Frankel KA, Tsutakawa SE, Alsbury DL, Copie V, Young MJ, Tainer JA, Lawrence CM. J Mol Biol. 2011;405:939–955. doi: 10.1016/j.jmb.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simeone R, Bottai D, Brosch R. Curr Opin Microbiol. 2009;12:4–10. doi: 10.1016/j.mib.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 83.Pallen MJ, Chaudhuri RR, Henderson IR. Curr Opin Microbiol. 2003;6:519–527. doi: 10.1016/j.mib.2003.09.005. [DOI] [PubMed] [Google Scholar]
- 84.Iyer LM, Zhang D, Rogozin IB, Aravind L. Nucleic Acids Res. 2011;39:9473–9497. doi: 10.1093/nar/gkr691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pallen MJ. Trends Microbiol. 2002;10:209–212. doi: 10.1016/s0966-842x(02)02345-4. [DOI] [PubMed] [Google Scholar]
- 86.Zhang D, de Souza RF, Anantharaman V, Iyer LM, Aravind L. Biol Direct. 2012;7:18. doi: 10.1186/1745-6150-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iyer LM, Makarova KS, Koonin EV, Aravind L. Nucleic Acids Res. 2004;32:5260–5279. doi: 10.1093/nar/gkh828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yusufzai T, Kadonaga JT. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Park JS, Lee WC, Yeo KJ, Ryu KS, Kumarasiri M, Hesek D, Lee M, Mobashery S, Song JH, Kim SI, Lee JC, Cheong C, Jeon YH, Kim HY. FASEB J. 2012;26:219–228. doi: 10.1096/fj.11-188425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parsons LM, Lin F, Orban J. Biochemistry. 2006;45:2122–2128. doi: 10.1021/bi052227i. [DOI] [PubMed] [Google Scholar]
- 91.Catterall WA. Neuron. 2010;67:915–928. doi: 10.1016/j.neuron.2010.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Iyer LM, Leipe DD, Koonin EV, Aravind L. J Struct Biol. 2004;146:11–31. doi: 10.1016/j.jsb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 93.Diemand AV, Lupas AN. J Struct Biol. 2006;156:230–243. doi: 10.1016/j.jsb.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 94.Bernstein KA, Gangloff S, Rothstein R. Annu Rev Genet. 2010;44:393–417. doi: 10.1146/annurev-genet-102209-163602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kitano K, Kim SY, Hakoshima T. Structure. 2010;18:177–187. doi: 10.1016/j.str.2009.12.011. [DOI] [PubMed] [Google Scholar]
- 96.Oyama T, Oka H, Mayanagi K, Shirai T, Matoba K, Fujikane R, Ishino Y, Morikawa K. BMC Struct Biol. 2009;9:2. doi: 10.1186/1472-6807-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Richards JD, Johnson KA, Liu H, McRobbie AM, McMahon S, Oke M, Carter L, Naismith JH, White MF. J Biol Chem. 2008;283:5118–5126. doi: 10.1074/jbc.M707548200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Buttner K, Nehring S, Hopfner KP. Nat Struct Mol Biol. 2007;14:647–652. doi: 10.1038/nsmb1246. [DOI] [PubMed] [Google Scholar]
- 99.Singarapu KK, Liu G, Xiao R, Bertonati C, Honig B, Montelione GT, Szyperski T. Proteins. 2007;67:501–504. doi: 10.1002/prot.21297. [DOI] [PubMed] [Google Scholar]
- 100.Anantharaman V, Aravind L. Genome Biol. 2003;4:R81. doi: 10.1186/gb-2003-4-12-r81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Burroughs AM, Iyer LM, Aravind L. Mol Biosyst. 2011;7:2261–2277. doi: 10.1039/c1mb05061c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wurtmann EJ, Wolin SL. Proc Natl Acad Sci U S A. 2010;107:4022–4027. doi: 10.1073/pnas.1000307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang D, Aravind L. Gene. 2010;469:18–30. doi: 10.1016/j.gene.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Petersohn A, Brigulla M, Haas S, Hoheisel JD, Volker U, Hecker M. J Bacteriol. 2001;183:5617–5631. doi: 10.1128/JB.183.19.5617-5631.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tam R, Saier MH., Jr Microbiol Rev. 1993;57:320–346. doi: 10.1128/mr.57.2.320-346.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cho SH, Parsonage D, Thurston C, Dutton RJ, Poole LB, Collet JF, Beckwith J. MBio. 2012;3 doi: 10.1128/mBio.00291-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lentzen G, Schwarz T. Appl Microbiol Biotechnol. 2006;72:623–634. doi: 10.1007/s00253-006-0553-9. [DOI] [PubMed] [Google Scholar]
- 108.Tadesse S, Graumann PL. BMC Microbiol. 2007;7:105. doi: 10.1186/1471-2180-7-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Mortier-Barriere I, Velten M, Dupaigne P, Mirouze N, Pietrement O, McGovern S, Fichant G, Martin B, Noirot P, Le Cam E, Polard P, Claverys JP. Cell. 2007;130:824–836. doi: 10.1016/j.cell.2007.07.038. [DOI] [PubMed] [Google Scholar]
- 110.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Nucleic Acids Research. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Soding J, Biegert A, Lupas AN. Nucleic Acids Research. 2005;33:W244–248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Holm L, Kaariainen S, Rosenstrom P, Schenkel A. Bioinformatics. 2008;24:2780–2781. doi: 10.1093/bioinformatics/btn507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Pei J, Sadreyev R, Grishin NV. Bioinformatics. 2003;19:427–428. doi: 10.1093/bioinformatics/btg008. [DOI] [PubMed] [Google Scholar]
- 114.Lassmann T, Frings O, Sonnhammer EL. Nucleic Acids Research. 2009;37:858–865. doi: 10.1093/nar/gkn1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Cole C, Barber JD, Barton GJ. Nucleic Acids Research. 2008;36:W197–201. doi: 10.1093/nar/gkn238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A. Nucleic Acids Research. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. J Mol Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 118.Kall L, Krogh A, Sonnhammer EL. Nucleic Acids Research. 2007;35:W429–432. doi: 10.1093/nar/gkm256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Price MN, Dehal PS, Arkin AP. PLoS One. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.