

1. Introduction
Endocannabinoid signaling begins with the on-demand synthesis and release of endogenous ligands, the two most abundant being anandamide (AEA) and 2-arachadonoylglycerol (2-AG). These retrograde lipid messengers traverse the synapse to interact with cannabinoid receptors on the pre-synaptic terminal [1]. The burden of signal termination falls upon two serine hydrolases, fatty acid amide hydrolase (FAAH; metabolizes AEA) and monoacylglycerol lipase (MAGL; metabolizes 2-AG). Dysregulation within the cannabinoid system has been implicated in numerous neurological conditions (pain, addiction, schizophrenia, anxiety) but pharmacological agonism or antagonism of cannabinoid receptors without adverse side effects has been elusive [2]. In rodent studies, genetic or pharmacological blockade of FAAH or MAGL has demonstrated analgesic, anti-nociceptive and anti-anxiety phenotypes lacking the negative effects associated with administration of Δ9–tetrahydrocannabinol (THC; the psychoactive ingredient of cannabis sativa) [3]. Thus, as gatekeepers of endocannabinoid tone, FAAH and MAGL have garnered much interest as pharmacological targets [4].
Selective and potent inhibitors of FAAH have been developed (for review, see [5,6]) with some reaching clinical evaluation [7]. Although far fewer, the number of MAGL inhibitors reported is steadily growing [8–11]. To assess the effectiveness of FAAH inhibitors in the clinic, blood/plasma sampling was conducted to determine alterations to endocannabinoid concentrations [7]. Unfortunately, given the short biological half-life and locally acting nature of endocannabinoids within the living brain, these measurements are likely an insufficient assessment of central inhibitory activity. A non-invasive biomarker would be an asset for investigating the role FAAH or MAGL may play in central nervous system (CNS) diseases or for assessing inhibitor efficacy during clinical trials.
Molecular imaging by positron emission tomography (PET) [12,13] is capable of quantifying FAAH or MAGL activity provided suitable radiotracers are developed. To date, a number of promising FAAH radiotracers for PET have been reported [14–20]. To our knowledge, two of those radiotracers have been evaluated in humans, the irreversible radioligand [11C]CURB, which has been validated in modeling studies [18], and [11C]MK-3168, a reversible radioligand prepared at Merck [20]. While clinical translation is planned for two additional FAAH radiotracers [17,19], it is noteworthy that there have been no reported attempts to prepare MAGL radiotracer for imaging studies.
To date, most MAGL inhibitors are carbamate- and urea-based suicide inhibitors forming covalent bonds between the carbamoyl moiety and Ser132 of the enzyme’s catalytic triad with the expulsion of a leaving group [21]. Logically then, the carbamoyl group provides an ideal site to radiolabel these inhibitors, ensuring retention of the radioisotope by the enzyme. To attain this outcome, an automated, one pot carbon-11 labeled (11C; β+; t½ = 20.4 min) CO2 fixation method can be employed [22]. Using this technique, [11C]carbamates [16,23,24], unsymmetrical [11C]ureas [19,25,26], and [11C]oxazolidines [27] have previously been prepared with sufficient yield, radiochemical purity and specific activity to complete preclinical and clinical evaluations. We herein report the preparation of carbamate- and urea-based MAGL inhibitors amenable to radiolabeling via [11C]CO2 fixation (seven known, five novel; Table 2). From those, five were selected for radiolabeling as the first candidate carbon-11 labeled radiotracers targeting MAGL (Figure 1). Additionally, ex vivo regional distribution and specificity of the above-mentioned radiotracers for MAGL in rats and mice are described.
Table 2.
One pot, two step triphosgene synthesis of MAGL inhibitors 7–18.
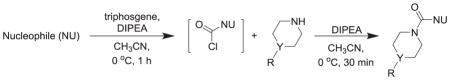
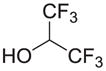
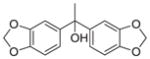
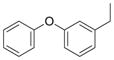
| |||||
|---|---|---|---|---|---|
| Nucleophile (NU) | R | Y | Product | hMAGL IC50 | hFAAH IC50 |
|
|
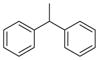
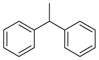
CH | 7a (KML29) | 5.9 nM | >50 μM |
|
N | 8a (JW642) | 3.7 nM | 21 μM | |
|
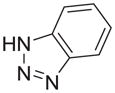
N | 9 | nd | nd | |
|
| |||||
|
|
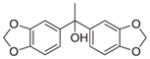
CH | 10 | nd | nd |
|
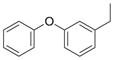
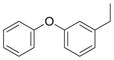
N | 11 | nd | nd | |
|
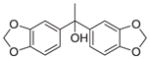
N | 12b (ML30) | 0.54 nM | 562 nM | |
|
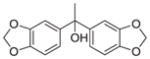
CH | 13 | nd | nd | |
|
| |||||

|
|
CH | 14 | nd | nd |
|
N | 15c (JJKK-006) | 0.076 nMd | nd | |
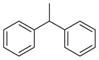
|
N | 16c (AKU-005) | 0.59 nMd | 63 nMd | |

|
CH | 17c (JJKK-0046) | 0.56 nM | 1 μM | |
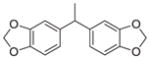
|
CH | 18 c (JJKK-0048) | 0.36 nM | 4.8 μM | |
Figure 1.
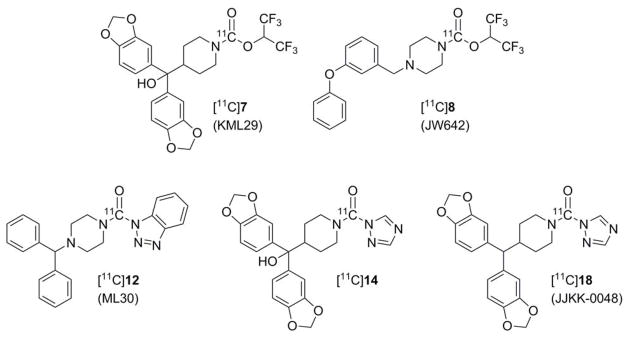
Five candidate carbon-11 labeled MAGL radiotracers prepared herein.
2. Materials and Methods
2.1. General materials and methods
All reagents were used as purchased from commercial sources unless otherwise stated. A Scanditronix MC 17 cyclotron was used for radionuclide production. Following a 5 min, 40 μA bombardment of a 14N target (14N(p,α)11C nuclear reaction), [11C]CO2 was concentrated from the gas target in a stainless steel coil cooled to −178 °C. Upon warming, the [11C]CO2 in a stream of N2 gas was passed through a NOx trapping column and a drying column of P2O5 prior to use [28]. Purifications and analyses of radioactive mixtures were performed by high performance liquid chromatography (HPLC) with an in-line UV detector (254 nm) in series with a NaI crystal radioactivity detector. Isolated radiochemical yields were determined with a dose calibrator (Capintec CRC-712M). Automated radiosyntheses were controlled by Labview™ software. Unless otherwise stated, all radioactivity measurements were corrected for radioactive decay. POCl3 was distilled under reduced pressure prior to use. Flash chromatography was completed using a Biotage Isolera instrument with a gradient elution (solvents: A = ethyl acetate; B = hexane; C = methanol + 5% triethylamine; D = dichloromethane + 1% triethylamine). Gradient was measured in column volumes (CV). Proton NMR spectra were recorded at 25 °C on a Bruker Advance 400 mHz spectrometer. High-resolution mass spectra were obtained via electrospray ionization mass spectrometry conducted with a JEOL AccuTOF or Agilent 6538 Q-TOF instrument. All animal experiments were carried out under humane conditions, with approval from the Animal Care Committee at the Centre for Addiction and Mental Health and in accordance with the guidelines set forth by the Canadian Council on Animal Care. Mice (male, C57BL/6) and rats (male, Sprague Dawley) were kept on a reversed 12 h light/12 h dark cycle and allowed food and water ad libitum.
2.2. Synthesis of piperidine and piperazine precursors
Piperidines tert-butyl (4-bis(benzo[d][1,3]dioxol-5-yl)(hydroxyl)methyl)piperidine-1-carboxylate (1), bis(benzo[d][1,3]dioxol-5-yl)piperidin-4-yl)methanol (2) and 4-(bis(benzo[d][1,3]dioxol-5-yl)methylene)piperidine (5) were prepared following procedures referenced in [9,11]. Piperazines 1-(3-phenoxybenzyl)piperazine (3) and 1-benzhydrylpiperzine (4) were prepared following procedures referenced in [9,10,29].
2.2.1 Synthesis of 4-(bis(benzo[d][1,3]dioxol-5-yl)methyl)piperidine (6)
Catalytic transfer hydrogenation was effected by heating a solution of 5 (200 mg, 0.54 mmol), 10% palladium on carbon (171 mg, 0.017 mmol), and ammonium formate (169 mg, 2.7 mmol) in methanol (3 mL) to 100 °C in a Biotage Initiator microwave for 35 min. The solution was filtered through a Celite plug, the solvent removed under reduced pressure, and the residue was partitioned between CH2Cl2 and saturated aqueous NaHCO3 (20 mL each). After a second wash of the organic layer with brine, the organic layer was dried (Na2SO4), filtered and removed under reduced pressure. Crude oil was purified via flash chromatography with a gradient elution (Start 2% C in D for 1 CV, increase to 20% C over 10 CV). Combination of pure fractions and removal of solvent yielded a brown oil (151 ± 15 mg, 83 ± 4%, n = 3): 1H NMR (400 MHz, CDCl3) matched reported values [11]. HRMS C20H22NO4 [M+H+] Observed 340.1544; Calculated 340.1549.
2.3. General synthesis of piperidinyl and piperazinyl carbamates and ureas (7–18)
Hexafluoroisopropanol (HFIP), benzotriazole, or 1,2,4-triazole (0.36 mmol) was added to a chilled solution (0 °C, ice water bath) of triphosgene (30 mg, 0.1 mmol) and diisopropylethyl amine (DIPEA; 105 μL, 0.6 mmol) in anhydrous acetonitrile (CH3CN; 1 mL) under N2. After 1 h, a solution of piperidine or piperazine (0.3 mmol) and DIPEA (52 μL, 0.3 mmol) in anhydrous CH3CN (1 mL) was added dropwise and after 30 min the solvent was removed under vacuum and the residue partitioned between CH2Cl2 and water (20 mL each). The organic layer was washed with brine (20 mL), dried (Na2SO4), filtered, and the solvent was removed under vacuum. Crude products were purified by gradient flash chromatography. Pure fractions were combined, the solvent removed and, when possible, solids were precipitated from ethyl acetate/hexane solutions. Purity of all compounds was >95% as measured by HPLC. Proton NMR spectroscopic and HRMS data for reported compounds (7, 8, 12, 15–18) matched literature values [9–11].
2.3.1. 1,1,1,3,3,3-hexafluoropropan-2-yl 4-benzhydrlpiperazine-1-carboxylate (9)
Gradient: Started at 5% A in B for 1 CV, increased to 40% A over 10 CV, held at 40% A for 2 CV. White solid, 58 mg, 44%: mp 209–210 °C. 1H NMR (400 MHz, CDCl3): δ ppm 7.40 (d, J = 7.2 Hz, 4H), 7.28 (t, J = 7.8 Hz, 4H), 7.19 (t, J = 7.3 Hz, 2H), 5.72 (septet, J = 6.2 Hz, 1H), 4.26 (s, 1H), 3.55 (quartet, J = 4.8 Hz, 4H), 2.44–2.38 (m, 4H). 13C NMR (100 MHz, CDCl3): δ pppm 149.2, 135.3, 129.4, 128.4, 120.7 (quart, 2JC-F = 284 Hz), 74.2, 67.7 (septet, 3JC-F = 33.6 Hz), 66.3. HRMS C21H21N2O2F6 [M+H+] Observed 447.1509; Calculated 447.1507.
2.3.2. (1H-benzo[d][1,2,3]triazol-1-yl)(4-(bis(benzo[d][1,3]dioxol-5-yl)(hydroxy)methyl) piperidin-1-yl)methanone (10)
Gradient: Started at 8% A in B for 1 CV, increased to 80% A over 10 CV, held at 80% A for 2 CV. Clear oil, 50 mg, 33%: 1H NMR (400 MHz, CDCl3): δ ppm 8.07 (d, J = 8.32 Hz, 1H), 7.95 (d, J = 8.3 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1 H) 7.43 (t, J = 7.2, 1H), 6.95–6.92 (m, 4H), 6.75 (d, J = 8.7 Hz, 2H), 5.92 (s, 4H), 4.59 (d, J = 12.9 Hz, 2H), 3.16 (br s, 2H), 2.60 (br s, 1H), 1.79–1.59 (m, 4 H). 13C NMR (100 MHz, CDCl3): δ ppm 149.5, 148.1, 146.6, 145.6, 139.8, 129.6, 125.4, 120.1, 119.1, 113.8, 108.2, 107.0, 101.4, 79.5, 44.7, 31.5, 27.0. HRMS C27H23N4O5 [M+-H2O] Observed 483.1669; Calculated 483.1668.
2.3.3. (1H-benzo[d][1,2,3]triazol-1-yl)(4-(3-phenoxybenzyl)piperazin-1-yl)methanone (11)
Gradient: Started at 8% A in B for 1 CV, increased to 75% A over 10 CV, held at 75% A for 2 CV. Clear oil, 94 mg, 76%: 1H NMR (400 MHz, CD3OD): δ ppm 8.09 (d, J = 8.2 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1 H) 7.47 (t, J = 7.2, 1H), 7.36–7.27 (m, 3H), 7.13–6.99 (m, 5H), 3.92 (br s, 4H), 3.57 (s, 2H), 2.65–2.62 (m, 4H). 13C NMR (100 MHz, CD3OD): δ ppm 158.4, 156.4, 149.0, 145.2, 133.0, 130.6, 130.2, 129.8, 129.6, 126.0, 125.5, 123.8, 120.6, 119.8, 119.3, 114.2, 113.5, 59.9, 51.1, 47.8. HRMS C24H24N5O2 [M+H+] Observed 414.1922; Calculated 414.1925.
2.3.4. (1H-benzo[d][1,2,3]triazol-1-yl)(4-(bis(benzo[d][1,3]dioxol-5-yl)methylene)piperidin-1-yl)methanone (13)
Gradient: Started at 8% A in B for 1 CV, increased to 80% A over 10 CV, held at 80% A for 2 CV. Clear oil, 47 mg, 32%. 1H NMR (400 MHz, CDCl3): δ ppm 8.08 (d, J = 8.32 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.60 (t, J = 7.2 Hz, 1 H) 7.45 (t, J = 7.2, 1H), 6.92–6.87 (m, 1H), 6.76 (t, J = 7.6 Hz, 2H), 6.63–6.57 (m, 4H), 5.94 (s, 4H), 4.37 (br s, 1H), 3.88 (br s, 4H), 2.60 (t, J = 5.8 Hz, 4H). 13C NMR (100 MHz, CDCl3): δ ppm 147.6, 146.6, 135.8, 132.5, 125.3, 123.0,120.0, 113.7, 110.1, 108.2, 106.8, 101.2, 79.3, 44.4, 32.1. HRMS C27H23N4O5 [M+H+] Observed 483.1658; Calculated 483.1668.
2.3.5. (4-(bis(benzo[d][1,3]dioxol-5-yl)(hydroxy)methyl)piperidin-1-yl)(1H-1,2,4-triazol-1-yl) methanone (14)
Gradient: Started at 18% A in B for 1 CV, increased to 100% A over 6 CV, held at 80% A for 6 CV. White solid, 67 mg, 49%: mp 134–135 °C. 1H NMR (400 MHz, CDCl3): δ ppm 8.74 (s, 1H), 7.96 (s, 1H), 6.93–6.90 (m, 4H), 6.76–6.74 (m, 2H), 5.93 (s, 4H), 4.60 (br s, 1H), 3.04–3.02 (m, 2H), 2.57–5.54 (m, 1H), 1.68–1.66 (m, 2H), 1.59–1.54 (m, 4H). 13C NMR (100 MHz, CDCl3): δ ppm 151.9, 148.5, 147.8, 146.6, 146.3, 139.5, 118.8, 107.9, 106.7, 101.1, 79.3, 48.4, 44.4, 26.7. HRMS C22H20N2O5 [M+-H2O] Observed 433.1510; Calculated 433.1506.
2.3. General Radiosynthesis of [11C-carbonyl]carbamates and ureas ([11C]7, [11C]8, [11C]12, [11C]14, and [11C]18)
A conical vial (1 mL) sealed with a Teflon-lined silicone septum was purged with N2 (10 mL/min for 5 min) 10 min prior to end-of-bombardment and then charged with a solution of appropriate precursor amine and phosphazene base BEMP (2-tert-butylimino-2-diethylamino-1,3-dimethylperhydro-1,3,2-diazaphosphorine) in anhydrous CH3CN (100 μL). Carbon-11 labeled CO2 was dispensed in a stream of nitrogen (7 mL/min) into the conical vial until radioactivity peaked. One min later, a solution of POCl3 in CH3CN (100 μL) was added followed 1 min later by a CH3CN solution (100 μL) of the appropriate nucleophile (HFIP, benzotriazole, or 1,2,4-triazole). After 1 min, the reaction was quenched with a 1:1 mixture of mobile phase in water (700 μL) and then purified on a Phenomenex Luna C18 10 μm, 10 x 250 mm HPLC column. Specific reagent concentrations along with HPLC mobile phase and flow rates can be found in Table 1. The desired product was collected, evaporated, and formulated in 5% v/v Tween80® in saline (10 mL) and passed through a Millipore 0.22 μm filter into a sterile vial containing 1N NaHCO3 solution (0.5 mL). The identity and radiochemical purity of the radiotracer were verified by co-injection with the respective authentic compound using several reverse-phase HPLC conditions (different solvents, pH, wavelengths, columns). Lipophilicities of the radiotracers (log P7.4) were measured by a literature shake flask procedure (n = 8/radiotracer) [30].
Table 1.
Reagent concentrations used for radiolabeling experiments along with semipreparative HPLC mobile phases, flow rates, and retention time.
| Precursor (mg/mL) | [BEMP] (% v/v) | Nucleophile | [POCl3] (% v/v) | Mobile Phase (CH3CN/H2O) | Flow Rate (mL/min) | tR (min) | |
|---|---|---|---|---|---|---|---|
| [11C]7 | 2 (10) | 6.4 | 1.5% (v/v) HFIP | 0.2 | 55/45a | 9 | 8.5 |
| [11C]8 | 3 (10) | 7.5 | 1.5% (v/v) HFIP | 0.3 | 65/35a,b | 10 | 6.2 |
| [11C]12 | 4 (2) | 4 | 10 mg/mL Benzotriazole | 0.2 | 70/30c | 10 | 7.7 |
| [11C]14 | 2 (2) | 4 | 6 mg/mL 1,2,4-Triazole | 0.2 | 50/50b | 8 | 5.5 |
| [11C]18 | 6 (4) | 4 | 4 mg/mL 1,2,4-Triazole | 0.4 | 60/40b | 8 | 4.7 |
Mobile phase contains:
1% formic acid;
0.1N ammonium formate;
0.05N ammonium formate
2.4. Ex vivo biodistribution studies in rodents
2.4.1. Brain uptake and pharmacological blocking of [11C]7 in rats
In two separate experiments, conscious male Sprague-Dawley rats (330–370 g; n = 4/group), held in a restraining box, received 28–34 MBq of high specific activity [11C]7 (208 GBq/μmol) in 0.3 mL of 5% v/v Tween80® buffered saline via warm water bath vasodilated tail-veins. Rats were sacrificed by decapitation at 2 and 40 min after radiotracer administration and blood was collected from the trunk. A third group, having been pretreated 1 h prior to radiotracer administration with 7 (2 mg/kg in 5% DMSO and 5% Tween80® saline; ip), were also sacrificed at 40 min post radiotracer injection. The brain was quickly removed, stored on ice and dissected. Brain regions (whole cortex, cerebellum, hypothalamus and remainder of brain) were excised, blotted, and weighed. Radioactivity in the tissues was assayed in an automated gamma counter, back corrected to the time of injection using diluted aliquots of the initial injected dose as standards. Tails were counted in a dose calibrator and the injected dose was corrected for residual activity.
2.4.2. Brain uptake and pharmacological blocking of all radiotracers in mice
For each radiotracer, two groups of conscious male C57BL/6 mice (20–30 g; n = 4/group) received 3.0–11.5 MBq of high specific activity (70–147 GBq/μmol) radiotracer in 0.2 mL of 5% v/v Tween80® buffered saline via heating lamp vasodilated tail-veins. Mice were sacrificed by cervical dislocation and decapitated at 2 and 40 min after radiotracer administration. A third group of mice, having received an intraperitoneal (ip) injection of the appropriate unlabeled inhibitors (2.0 mg/kg in 5% DMSO and 5% Tween80® buffered saline) 30 min prior to radiotracer administration, were sacrificed at 40 min post radiotracer injection. Blood was collected from the trunk of all mice, whole brains were quickly removed, blotted, and weighed. Radioactivity in the tissues was assayed as described above. Statistical analysis was completed using the Student t-test and any p < 0.05 was considered significant.
3. Results
3.1. Chemistry
Piperidine and piperazine compounds 1–6 were synthesized by adapted literature procedures [9–11,29] in comparable yields and purities. It should be noted that attempts to remove the Boc group from 1 using HCl or trifluoroacetic acid solely produced 5. Iodotrimethylsilane preferentially removed the Boc group versus the hydroxyl group to give the desired product, 2, if excess N-methylmorpholine was added prior to the silane [9]. For the reduction of the piperidine 5, a catalytic transfer hydrogenation was employed in conjunction with microwave heating to decrease the reaction time [11]. Following the triphosgene procedure outlined in Table 2, carbamates 7–9 and ureas 10–18 were prepared through reacting piperidine and piperazine precursors (2–6) with carbanochloridate or carbamylchloride generated in situ at 0 °C. Consumption of precursors was instantaneous and the desired carbamates 7–9 and the ureas 10–18 were isolated in moderate to good yields with high purity (>95% measured by HPLC).
3.2. Radiochemistry
We selected four MAGL inhibitors (7, 8, 12, and 18) with the highest reported affinity and selectivity [9–11] along with one novel compound (14) for radiolabeling. Carbonyl group of the carbamate or urea moiety were radiolabeled via a one-pot [11C]CO2 fixation method (Scheme 1) used previously to radiolabel comparable compounds [22]. Bubbling cyclotron produced [11C]CO2 into a vial with appropriate piperidine or piperazine in the presence of a phosphazene base (BEMP, an efficient CO2 capturing agent [24]) followed by the addition of POCl3 resulted in the formation of a putative mixed anhydride [19]. Lastly, these intermediates were reacted with the appropriate nucleophile (HFIP, benzotriazole, or 1,2,4-triazole) prior to quenching the reactions with 1:1 HPLC mobile phase in water. Optimized reaction conditions are presented in Table 1.
Scheme 1.
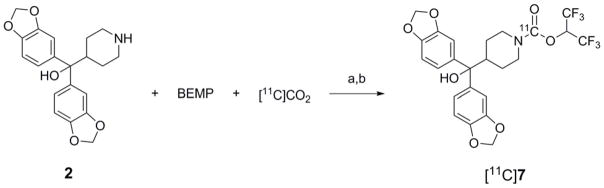
Representative [11C]CO2 fixation reaction forming candidate MAGL radiotracers [11C]7. a = POCl3, 1 min, rt; b = HFIP, 1 min, rt, then quenched with 1:1 water/HPLC mobile phase and purified by HPLC. All MAGL radiotracers (Figure 1) were prepared in a similar manner to the example shown.
All radiochemistry was carried out with an automated synthesis module requiring no heating or cooling, as previously described [23–27], and purified by preparative HPLC. Including formulation, the radiolabeled MAGL inhibitors were prepared in 19–30 min after end of bombardment with adequate radiochemical yields (5–13%, uncorrected for decay and based on starting [11C]CO2) and high radiochemical purity (>97%). Individual results for each radiotracer are summarized in Table 3. The formulated final products were prepared in clinically useful quantities (0.91–2.1 GBq) and of suitable specific activity (66–126 GBq/μmol at end of synthesis). The lipophilicities of the radiolabeled MAGL inhibitors were measured via a shake-flask method [30] as the partition coefficient between 1-octanol and 0.02 M phosphate buffered at pH = 7.4 (Log P7.4) and ranged from 2.7–5.2 (Table 3).
Table 3.
Radiochemical results for five candidate MAGL radiotracers. Data are expressed as means ± standard deviation.
| RCY (GBq; %) | Radiochemical Purity (%) | Specific Activity (GBq/umol) | Log P7.4 | Time (min) | n | |
|---|---|---|---|---|---|---|
| [11C]7 | 1.1 ± 0.2; 6.5 ± 0.9 | 99.3 ± 0.7 | 124.0 ± 28.6 | 4.67 ± 0.06 | 28 ± 3 | 7 |
| [11C]8 | 2.1 ± 0.5; 11.8 ± 2.8 | 99.9 ± 0.1 | 74.7 ± 16.0 | 5.0 ± 0.2 | 25 ± 4 | 4 |
| [11C]12 | 0.93 ± 0.27; 5.3 ± 1.5 | 99.8 ± 0.2 | 84.0 ± 8.5 | 3.61 ± 0.08 | 23 ± 4 | 3 |
| [11C]14 | 1.0 ± 0.3; 5.8 ± 1.9 | 99.0 ± 1.1 | 93.9 ± 47.1 | 2.76 ± 0.02 | 24 ± 1 | 6 |
| [11C]18 | 1.5 ± 0.3; 8.5 ± 1.5 | 98.9 ± 1.7 | 65.6 ± 7.1 | 3.32 ± 0.04 | 26 ± 6 | 3 |
RCY = radiochemical yield based upon starting [11C]CO2 and uncorrected for decay.
3.3. Ex vivo radiotracer distribution in rodents
Carbon-11 labeled MAGL inhibitors were then explored for potential as PET imaging agents in conscious rats ([11C]7 only) and mice. Animals were administered the radioactive compounds via vasodilated tail-vein injection and the whole brain uptake was measured by counting excised brains (for mice) and brain regions (for rats) at an early and late time point (2 and 40 min post-injection, respectively). For observed uptake, specificity was probed by pretreating another group of rodents with the matching unlabeled inhibitor (2 mg/kg, ip) 30 min prior to radiotracer administration and excising the brain at 40 min post-injection. Uptake of the radiotracers into whole brain and blood of mice is summarized in Figure 2.
Figure 2.

MAGL radiotracer binding to whole brain and blood of mice. Blocked groups were pretreated 30 min prior to radiotracer injection with appropriate unlabeled inhibitor (2 mg/kg, ip) and sacrificed 40 min post radiotracer injection. Data is expressed as mean SUV with standard deviation bars (n = 4). * = p < 0.05.
Initial experiments in rat with [11C]7 resulted in moderate radioactivity uptake (0.44–0.71 SUV at 2 and 40 min) that was homogeneously distributed throughout the brain and not blocked by pretreatment with unlabeled 7. In mice, both of the [11C]HFIP-carbamate radiotracers ([11C]7 and [11C]8) had moderate brain uptake (0.34–0.67 SUV at 2 and 40 min) which was not significantly attenuated by pretreatments with unlabeled compounds. Similar levels of non-specific brain uptake was observed for the triazole ureas [11C]12 and [11C]14. Unlike the [11C]HFIP-carbamates, the [11C]ureas exhibited high retention in mouse blood (1.9–11.1 SUV) which was blocked (85 and 90%) by pretreatment with unlabeled compound. The final triazole urea, [11C]18, had the highest retention in mouse brain (0.65–0.82 SUV) that was significantly reduced in the blocked group (42%, p < 0.05). Of the [11C]ureas, [11C]18 had the lowest retention in blood (1.1–1.5 SUV) and pretreatment with unlabeled 18 significantly reduced this retention (71%, p < 0.05).
4. Discussion
To expand the arsenal of PET probes for endocannabinoid signaling, we have prepared five MAGL radiotracer candidates. With far fewer inhibitors reported for MAGL compared to FAAH, the options for radiolabeled analogues were limited and the best available, based on potency and selectivity, were selected [9–11]. In addition to preparing known MAGL inhibitors, several novel compounds sharing similar scaffolds (9–11, 13, and 14; Table 2) were also synthesized to expand the available compounds for radiolabeling. Those previously reported carbamates and ureas prepared herein are reported to interact with MAGL via covalent bond formation between Ser132 and the carbonyl moiety [31], therefore, we elected to radiolabel selected inhibitors at the carbonyl position via [11C]CO2 fixation [22]. The one-pot automated synthesis (Scheme 1) gave the [11C]carbamates and [11C]ureas (Figure 1) in sufficient quantities and qualities (Table 3) to conduct biodistribution experiments in rats and mice.
In our laboratory, we typically evaluate new neurological PET radiotracers by ex vivo biodistribution studies in conscious rats. Based upon less than suitable in vivo properties of [11C]7 in rats and given that both JZL184 and 7 demonstrated a greater affinity for the mouse MAGL than rat MAGL homologue [9], subsequent evaluation of BBB permeability were conducted with mice. Mouse whole brain uptake and blood radioactivity levels were evaluated at 2 and 40 min post radiotracer administration to determine if the candidate could penetrate the blood-brain barrier (BBB). Pharmacological blockade of radioactivity uptake in both tissues by unlabeled inhibitors provided a measure for specificity of any observed uptake.
The BBB penetration of [11C]7 in mice was indistinguishable from that observed in rats. Another [11C]HFIP-carbamate, [11C]8 (an isotopologue of JW642 [9]) also showed low brain penetration with no appreciable reduction following pretreatment. At the time of these experiments, KML29 (7) and JW642 (8) were the two most potent and selective MAGL inhibitors reported, showing marked improvement over their previously described para-nitrophenol analogues (JZL 184 and JZL195) [31]. We suspected that the modest brain uptake and apparent lack of specific binding of [11C]7 and [11C]8 were attributable to their very high lipophilicity (Table 3). More recently, two separate syntheses of more hydrophilic MAGL inhibitors containing benzotriazole and 1,2,4-triazole ureas [10,11] were reported. Based upon these studies, we radiosynthesized three [11C-carbonyl]ureas, [11C]12, [11C]14, and [11C]18, as potential MAGL radiotracers.
In their synthesis of 1,2,4-triazole-urea MAGL inhibitors (15–18), Aaltonen et al. reported difficulties preparing piperidine 2 from 1 and therefore could not synthesis 14 [11]. Although the affinity and selectivity are therefore unreported, structural comparability with known MAGL inhibitors [9,11,31] would suggest 14 shares similar values but possesses an added benefit of reduced lipophilicity versus 18 (Table 3). Whole brain uptake of the three [11C]ureas was moderate and non-specific, with the exception of [11C]18 which demonstrated significant reduction of radioactivity in the blocked group (Figure 2). However, this reduction was only 42% for an estimated 0.71 specific to non-specific binding ratio; less than ideal for imaging studies [32]. Without a reference region for MAGL, the non-specific binding in this instance was represented by uptake of radioactivity into the brain following pharmacological blockade. Overestimation of non-specific binding could have occurred if there was increased radiotracer input into the brain following blockade of peripheral binding sites [33–35]. Given the failure of the radiotracer evaluated herein to effectively penetrate the BBB, in vitro affinity and selectivity of the novel compounds for MAGL was not determined as we are no longer pursuing this chemical scaffold.
Decreasing the lipophilicity did not appear to increase BBB penetration of the urea-based radiotracers. Perhaps the high retention of radioactivity in mouse blood was the cause of the low uptake. Interestingly, we observed a significant reduction to the binding of [11C]12, [11C]14, and [11C]18 to mouse blood upon pharmacological blockade (90, 85 and 71%, respectively; Figure 2). Saturable binding in blood is not wholly surprising given the high expression of MAGL in plasma and on leukocytes [36]. Furthermore, very high doses of inhibitors were required to raise the levels of 2-AG in rodents brains [9,37], suggesting limited BBB penetration. It should be noted that rapid peripheral metabolism of a radiotracer does not, per se, invalidate its usefulness for CNS imaging. A case in point is the radiotracers successfully developed for acetylcholinesterase that are quickly metabolized by cholinesterases in blood [38,39]. Nevertheless even in this system, too rapid metabolism leads to poor brain uptake of the radiotracer, limiting its utility [40]. Taken together with the limited brain uptake results of this study, rapid metabolism driven by high levels of saturable blood binding may preclude MAGL from being a viable target for neuroimaging studies via PET.
5. Conclusion
Radiosynthesis and ex vivo whole mouse brain uptake for five MAGL radiotracer candidates has been outlined above. To our knowledge, this work represents the first reported attempts to image MAGL via PET. High radiotracer lipophilicity and high MAGL expression in blood likely contributed to the observed low brain penetration. Despite the promising in vitro pharmacological profile of the reported MAGL inhibitors radiolabeled herein, none of the five candidate radiotracers exhibited suitable in vivo behavior for PET neuroimaging. Further radiotracer optimization is required to improve brain penetration and specific binding.
Acknowledgments
This work was supported by NIH Grant # 1R21MH094424-01 to AAW. We would like to thank Armando Garcia, Winston Stableford and Min Wong as well as Frank Giuliano and Alvina Ng for their assistance with the radiochemistry and animal dissection experiments, respectively. Lastly, we thank Dr. Christopher Fowler (Umea University, Umea, Sweden) for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katona I, Freund TF. Multiple functions of endocannabinoid signaling in the brain. Annu Rev Neurosci. 2012;35:529–558. doi: 10.1146/annurev-neuro-062111-150420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pacher P, Kunos G. Modulating the endocannbinoid system in human health and disease - successes and failures. FEBS J. 2013;280:1918–1943. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blankman JL, Cravatt BF. Chemical probes of endocannabinoid metabolism. Pharmacol Rev. 2013;65:849–871. doi: 10.1124/pr.112.006387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stratton HJ, Allen DL, Wu J, Shafer MS. Modulation of the endogeneous cannabinoid system as a therapeutic target in the treatment of mental health disorders. Biochem Pharmacol. 2013;S1:008. doi: 10.4172/2167-0501.S1-008. [DOI] [Google Scholar]
- 5.Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- 6.Bisogno T, Maccarrone M. Latest advances in the discovery of fatty acid amide hydrolase inhibitors. Expert Opin Drug Discov. 2013;8:509–522. doi: 10.1517/17460441.2013.780021. [DOI] [PubMed] [Google Scholar]
- 7.Huggins JP, Smart TS, Langman S, Taylor L, Young T. An efficient randomised, placebo-controlled clinical trial with the irreversible fatty acid amide hydrolase-1 inhibitor PF-044577845, which modulates endocannabinoids but fails to induce effective analgesia in patients with pain due to osteoarthritis of the knee. Pain. 2012;153:1837–1846. doi: 10.1016/j.pain.2012.04.020. [DOI] [PubMed] [Google Scholar]
- 8.Bisogno T, Ortar G, Petrosino S, Morera E, Palazzo E, Nalli M, et al. Development of a potent inhibitor of 2-arachidonoylglycerol hydrolysis with antinociceptive activity in vivo. Biochim Biophys Acta - Mol Cell Biol Lipids. 2009;1791:53–60. doi: 10.1016/j.bbalip.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 9.Chang JW, Niphakis MJ, Lum KM, Cognetta ABI, Wang C, Matthews ML, et al. Highly selective inhibitors of monoacylglycerol lipase bearing a reactive group that is bioisosteric with endocannabinoid substrates. Chem Biol. 2013;19:579–588. doi: 10.1016/j.chembiol.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morera L, Labar G, Ortar G, Lambert DM. Development and characterization of endocannabinoid hydrolases FAAH and MAGL inhibitors bearing benzotriazol-1-yl carboxamide scaffold. Bioorg Med Chem. 2012;20:6260–6275. doi: 10.1016/j.bmc.2012.09.011. [DOI] [PubMed] [Google Scholar]
- 11.Aaltonen N, Savinainen JR, Riera Ribas C, Rökkö J, Kuusisto A, Korhone J, et al. Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem Biol. 2013;20:379–390. doi: 10.1016/j.chembiol.2013.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Cherry SR. Fundamentals of positron emission tomography and applications in preclinical drug development. J Clin Pharmacol. 2001;41:482–491. doi: 10.1177/00912700122010357. [DOI] [PubMed] [Google Scholar]
- 13.Patel S, Gibson R. In vivo site-directed radiotracers: a mini-review. Nucl Med Biol. 2008;35:805–815. doi: 10.1016/j.nucmedbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 14.Wilson AA, Garcia A, Parkes J, Houle S, Tong J, Vasdev N. [11C]CURB: Evaluation of a novel radiotracer for imaging fatty acid amide hydrolase by positron emission tomography. Nucl Med Biol. 2011;38:247–253. doi: 10.1016/j.nucmedbio.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 15.Skaddan MB, Zhang L, Johnson DS, Zhu A, Zasadny KR, Coelho RV, et al. The synthesis and in vivo evaluation of [18F]PF-9811: a novel PET ligand for imaging brain fatty acid amide hydrolase (FAAH) Nucl Med Biol. 2012;39:1058–1067. doi: 10.1016/j.nucmedbio.2012.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson AA, Hicks JW, Sadovski O, Parkes J, Tong J, Houle S, et al. Radiosynthesis and evaluation of [11C-carbonyl]-labeled carbamates as fatty acid amide hydrolase radiotracers for positron emission tomography. J Med Chem. 2013;56:201–209. doi: 10.1021/jm301492y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadovski O, Hicks JW, Parkes J, Raymond R, Nobrega J, Houle S, et al. Development and characterization of a promising fluorine-18 labelled radiopharmaceutical for in vivo imaging of fatty acid amide hydrolase. Bioorg Med Chem. 2013;21:4351–4357. doi: 10.1016/j.bmc.2013.04.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rusjan PM, Wilson AA, Mizrahi R, Boileau I, Chavez SE, Lobaugh NJ, et al. Mapping human brain fatty acid amide hydrolase activity with PET. J Cereb Blood Flow Metab. 2013;33:407–414. doi: 10.1038/jcbfm.2012.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hicks JW, Parkes J, Sadovski O, Tong J, Houle S, Vasdev N, et al. Synthesis and preclinical evaluation of [11C-carbonyl]PF-04457845 for neuroimaging of fatty acid amide hydrolase. Nucl Med Biol. 2013;40:740–746. doi: 10.1016/j.nucmedbio.2013.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu P, Hamill TG. Discovery of MK-3168: a PET tracer for imaging brain fatty acid amide hydrolase. ACS Med Chem Lett. 2013;4:509–513. doi: 10.1021/ml4000996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Long JZ, Nomura DK, Cravatt BF. Characterization of monoacylglycerol lipase inhibition reveals differences in central and peripheral endocannabinoid metabolism. Chem Biol. 2009;16:744–753. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rotstein BH, Liang SH, Holland JP, Collier TL, Hooker JM, Wilson AA, et al. 11CO2 Fixation: A Renaissance in PET Radiochemistry. Chem Commun. 2013;49:5621–5630. doi: 10.1039/c3cc42236d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hooker JM, Reibel AT, Hill SM, Schueller MJ, Fowler JS. One-pot direct incorporation of [11C]CO2 into carbamates. Angew Chem Int Ed. 2009;48:3482–3485. doi: 10.1002/anie.200900112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson AA, Garcia A, Houle S, Vasdev N. Direct fixation of [11C]CO2 by amines: formation of [11C-carbonyl]-methylcarbamates. Org Biomol Chem. 2010;8:428–432. doi: 10.1039/b916419g. [DOI] [PubMed] [Google Scholar]
- 25.Wilson AA, Garcia A, Houle S, Sadovski O, Vasdev N. Synthesis and application of isocyanates radiolabeled with carbon-11. Chem-Eur J. 2011;17:259–264. doi: 10.1002/chem.201002345. [DOI] [PubMed] [Google Scholar]
- 26.Hicks JW, Wilson AA, Rubie EA, Woodgett JR, Houle S, Vasdev N. Towards the preparation of radiolabeled 1-aryl-3-benzyl ureas: Radiosynthesis of [11C-carbonyl] AR-A014418 by [11C]CO2 fixation. Bioorg Med Chem Lett. 2012;22:2099–2101. doi: 10.1016/j.bmcl.2011.12.139. [DOI] [PubMed] [Google Scholar]
- 27.Vasdev N, Sadovski O, Garcia A, Dolle F, Meyer JH, Houle S, et al. Radiosynthesis of [11C]SL25. 1188 via [11C]CO2 fixation for imaging monoamine oxidase. B J Label Compd Radiopharm. 2011;54:678–680. [Google Scholar]
- 28.Tewson TJ, Banks W, Franceschini M, Hoffpauir J. A trap for the removal of nitrogen oxides from carbon-11 carbon dioxide. Appl Radiat Isot Int J Radiat Appl Instrum Part A. 1989;40:765–768. [Google Scholar]
- 29.Moussa IA, Banister SD, Beinat C, Giboureau N, Reynolds AJ, Kassiou M. Design, Synthesis, and Structure Affinity Relationships of Regioisomeric N-Benzyl Alkyl Ether Piperazine Derivatives as σ-1 Receptor Ligands. J Med Chem. 2010;53:6228–6239. doi: 10.1021/jm100639f. [DOI] [PubMed] [Google Scholar]
- 30.Wilson AA, Jin L, Garcia A, DaSilva JN, Houle S. An admonition when measuring the lipophilicity of radiotracers using counting techniques. Appl Radiat Isot. 2001;54:203–208. doi: 10.1016/s0969-8043(00)00269-4. [DOI] [PubMed] [Google Scholar]
- 31.Long JZ, Jin X, Adibekian A, Li W, Cravatt BF. Characterization of tunable piperidine and piperazine carbamates as inhibitors of endocannabinoid hydrolases. J Med Chem. 2010;53:1830–1842. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hargreaves RJ, Rabiner EA. Translational PET imaging research. Neurobiol Dis. 2014;61:32–38. doi: 10.1016/j.nbd.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 33.Suhara T, Sudo Y, Yoshida K, Okubo Y, Fukuda H, Obata T, et al. Lung as reservoir for antidepressants in pharmacokinetic drug interactions. The Lancet. 1998;351:332–335. doi: 10.1016/S0140-6736(97)07336-4. [DOI] [PubMed] [Google Scholar]
- 34.Imaizumi M, Briard E, Zoghbi SS, Gourley JP, Hong J, Musachio JL, et al. Kinetic evaluation in nonhuman primates of two new PET ligands for peripheral benzodiazepine receptors in brain. Synapse. 2007;61:595–605. doi: 10.1002/syn.20394. [DOI] [PubMed] [Google Scholar]
- 35.Wilson AA, Garcia A, Parkes J, McCormick P, Stephenson KA, Houle S, et al. Radiosynthesis and initial evaluation of [18F]-FEPPA for PET imaging of peripheral benzodiazepine receptors. Nucl Med Biol. 2008;35:305–314. doi: 10.1016/j.nucmedbio.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 36.Fielding CJ. Monoglyceride hydrolase activitys in rat plasma and platelets. J Biol Chem. 1981;256:876–881. [PubMed] [Google Scholar]
- 37.Long JZ, Li W, Booker L, Burston JL, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kilbourn MR, Synder SE, Sherman PS, Kuhl DE. In vivo studies of acetylcholinesterase activity using a labeled substrate, N-[11C]methylpiperdin-4-yl propionate ([11C]PMP) Synapse. 1996;22:123–31. doi: 10.1002/(SICI)1098-2396(199602)22:2<123::AID-SYN5>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 39.Namba H, Fukushi K, Nagatsuka S, Iyo M, Shinotoh H, Tanada S, et al. Positron emission tomography: quantitative measurement of brain acetylcholinesterase activity using radiolabeled substrates. Methods. 2002;27:242–50. doi: 10.1016/s1046-2023(02)00081-6. [DOI] [PubMed] [Google Scholar]
- 40.Shao X, Lisi JM, Butch ER, Kilbourn MR, Snyder SE. N-methylpiperidinemethyl, N-methylpyrrolidyl and N-methylpyrrolidinemethyl esters as PET radiotracers for acetylcholinesterase activity. Nucl Med Biol. 2003;30:293–302. doi: 10.1016/s0969-8051(02)00438-9. [DOI] [PubMed] [Google Scholar]