

Abstract
Background
Disrupted in Schizophrenia 1 (DISC1) is a protein implicated in schizophrenia, bipolar disorder, major depressive disorder, and autism. To date, most of research examining DISC1 function has focused on its role in neurodevelopment, despite its presence throughout life. DISC1 also regulate cAMP signaling by increasing PDE4 catabolism of cAMP when cAMP concentrations are high. In this study, we tested the hypothesis that DISC1, through its regulation of cAMP, modulates I-SK and I-TRPC channel-mediated ionic currents that we have shown previously to regulate the activity of mature prefrontal cortical pyramidal neurons.
Methods
We used patch-clamp recordings in prefrontal cortical slices from adult rats in which DISC1 function was reduced in vivo by shRNA viral knockdown or in vitro by dialysis of DISC1 antibodies.
Results
We found that DISC1 disruption resulted in an increase of: mGluR-induced intracellular Ca2+ waves, SK-mediated hyperpolarization and a decrease of TRPC-mediated sustained depolarization. Consistent with a role for DISC1 in regulation of cAMP signaling, forskolin-induced cAMP production also increased intracellular Ca2+ waves, I-SK and decreased I-TRPC. Lastly, inhibiting cAMP generation with guanfacine, an α2A-noradrenergic agonist, normalized the function of SK and TRPC channels.
Conclusions
Based on our findings, we propose that diminished DISC1 function, such as occurs in some mental disorders, can lead to the disruption of normal patterns of PFC activity through the loss of cAMP regulation of mGluR-mediated intracellular Ca2+ waves, SK and TRPC channel activity.
Keywords: mGluR5, IP3, Ca2 waves, DISC1, persistent activity, prefrontal cortex
INTRODUCTION
The etiology of mental disorders, such as schizophrenia, is extremely complex due in part to polygenic-mediated aberrations in multiple biochemical signaling pathways that contribute to cognitive dysfunction. Mental illness often involves cognitive dysfunction of the prefrontal cortex (PFC), whose neuronal circuits and patterns of neuronal activity are needed for working memory (1). Various genetic risk factors for schizophrenia are associated with altered PFC circuits and working memory deficits (2–6). In this study, we examine how manipulating one such gene, Disrupted in Schizophrenia 1 (DISC1), alters PFC pyramidal excitability through its regulation of intrinsic ionic conductances that are activated by Group 1 metabotropic glutamate receptors (mGluRs).
Recent studies of primate dorsolateral PFC (dlPFC) have shown extensive DISC1 labeling in dendritic spines in layer III and in layer V, the microcircuits greatly afflicted in schizophrenia (7–9). These data revealed DISC1 co-localization with PDE4A and HCN channels near the synapse and in the spine neck, as well as near the spine apparatus that stores internal Ca2+ (10, 11). Thus, electron microscopy data suggest that DISC1 is positioned to regulate synaptic efficacy and excitability (12–14), e.g. regulating cAMP-induced loss of firing during stress exposure (10). Despite the possibility that DISC1 might contribute to a myriad of cAMP-dependent biochemical and ionic mechanisms that regulate the activity of PFC neurons, little is known about how DISC1 influences the functional properties of mature neurons.
As a first step in determining this role, we focused on whether DISC1 regulates two channels: small-conductance K+ (SK) channels and Transient Receptor Potential C (TRPC) channels, both of which have been proposed to contribute to patterns of activity that encode working memory and that are modulated by changes in [cAMP] (15–17). Both channels are also Ca2+-dependent and activated by mGluR5. To test this hypothesis, we performed patch-clamp recordings and high-speed Ca2+ fluorescence imaging on layer V pyramidal neurons in mPFC slices from rats (12–20 weeks old) infused with an shRNA viral construct targeted to DISC1 mRNA and from control rats. We show that disruption of DISC1 leads to enhancement of IP3-mediated intracellular Ca2+ waves, of SK-mediated hyperpolarization and to suppression of TRPC-mediated depolarization elicited by activation of Group 1 mGluRs. Consistent with the hypothesis that DISC1 is capable of regulating these channels through its ability to regulate cAMP, we found that raising cAMP concentrations with forskolin in control neurons also enhanced IP3-mediated intracellular Ca2+ waves, SK-mediated hyperpolarization and suppressed TRPC-mediated depolarization, while inhibition of cAMP signaling normalized SK and TRPC currents (Isk and Itrpc, respectively) following loss of DISC1 function. Based on these findings, we propose that loss of DISC1 leads to disinhibition of intracellular cAMP signaling, which in turn leads to dysregulation of IP3, SK and TRPC channels, and ultimately, disruption of the appropriate patterns of PFC activity for encoding working memory function.
METHODS and MATERIALS
All procedures described have been published elsewhere (15, 18) and followed NIH guidelines outlined in “Preparation and Maintenance of Higher Animals During Neuroscience Experiments” (publication 91-3207) and were approved by the Institutional Animal Care and Use Committee at the Yale University School of Medicine. For details see text in Supplement.
AAV-shRNA targeting DISC1
shRNA constructs
Five plasmid DNA constructs were generated expressing short hairpin RNAs directed against DISC1 (shRNA 1–5) from an H1 promoter and coexpressing lacZ from a CMV promoter, flanked by ITR sites. These shRNA constructs were co-transfected with a plasmid coding for full length DISC1 with a C-terminal hemagglutanin epitope tag into HEK-293 cells. As controls, cells were also transfected with an shRNA construct with a hairpin sequence that does not match any known gene (scrambled) or with an empty expression plasmid (negative) (Figure 1A). Additional analysis of the efficacy of the shRNA constructs was performed by overexpressing rat DISC1 in a 1:4 ratio with each shRNA plasmid or with a plasmid expressing a scrambled shRNA sequence, using Effectene transfection reagent in HEK-293 cells (Figure 1B).
Figure 1.

DISC1 shRNA knockdown efficacy. A. Five plasmid DNA constructs were generated coding for short hairpin RNAs directed against DISC1 (shRNA 1–5). As controls, cells were also transfected with an shRNA construct with a hairpin sequence that does not match any known gene (“scr”) and with an empty expression plasmid (“neg”). The active shRNA constructs (1–5) knocked down expression of DISC1 to varying extents with constructs 1–3 showing highest efficacy. The scrambled control showed no effects on DISC1 expression. Transfection with an empty plasmid demonstrated that the antibody was specific for overexpressed DISC1. B. Overexpression of DISC1 in HEK-293 cells was knocked down by DISC1-kd. C. Diagram schematic, modified with permission from the rat atlas of Paxinos and Watson (72), showing the infusion location in rat PFC. D. β-Gal histochemical staining shows transduced pyramidal neurons in PFC. E. Infusion of active shRNA constructs (1 or 2) into rat PFC effectively eliminate DISC1 expression. In this case, unilateral infusion of active shRNA eliminated DISC1 expression in the infused PFC without altering expression in the SHAM hemisphere. F. Infusion of the active shRNA construct into PFC has no effect on nearby area M2 (right panel), but completely eliminated DISC1 expression in PFC (left panel).
AAV-shRNA
shRNAs were co-transfected with adeno-associated virus 2 (AAV2). Virus was titered by transduction of camptothecin pre-treated HEK-293 cells followed by beta-galactosidase staining. Viral constructs were infused into the mPFC of anesthetized rats (1–2 μL; 0.25 μL/min). The active shRNA constructs (1–5) knocked down expression of DISC1 to varying extents with constructs 1–3 showing highest efficacy (Figure 1A). The scrambled control showed no effects on DISC1 expression.
Histochemistry and Immunolabeling
β-galactosidase. Standard histochemistry techniques were used to stain for β-galactosidase. Tissue was mounted onto glass slides and incubated in a humidified chamber at 37°C for 24 hours in a solution prepared by combining two premixed solutions. Solution A: 4% X-gal dissolved in N, N dimethylformamide (DMF); stored at −20°C in the dark. Solution B: potassium ferricyanide crystalline (5mM) potassium ferricyanide trihydrate (5mM) and magnesium chloride (2mM); dissolved in 0.1M PBS and stored at 4°C in the dark. Solution B was warmed to 37°C and solution A was added to it at a concentration of 1:40.
DISC1
DISC1 staining was performed by following standard immunohistochemical procedures. DISC1 labeling of rat tissue was achieved using a mixture of 2 different antibodies (both from Novus Biologicals, Littleton, CO). Rabbit polyclonal anti-DISC1 NB110-40773 which targets an internal region within residues 400–500 of the rat DISC1 protein and rabbit polyclonal anti-DISC1 NB110-40775 which targets an internal region within residues 700–800 of the mouse DISC1 protein. These primary antibodies were mixed because in our hands the x-775 antibody gave predominantly dendritic and punctuate staining while the x-773 predominantly stained the soma. Hence, a combination of both was expected to target different cellular compartments expressing DISC1 and give a more complete and accurate staining pattern.
Slice Preparation for Electrophysiological Recordings and Ca2+ Imaging
mPFC slices were extracted from 12–20 week-old male Sprague-Dawley rats (15) or 10–60 days following shRNA constructs infusion (see Text S1). Ca2+ waves were induced by a puff of DHPG on the primary apical dendrites. A low-affinity Ca2+ indicator dye was included in the recording pipette (fura-2FF) as well as an inert fluorescent dye (Alexa 488 or Alexa 564) for better visualization of neuronal processes. Dye fluorescence was imaged with high-speed cooled CCD camera. Images were collected at 25Hz with 5×5 pixel binning.
RESULTS
AAV2-shRNA Knockdown of DISC1 In Mature Rat mPFC
To examine the role of DISC1 protein in the cellular and ionic function of mature mPFC pyramidal neurons, we developed an AAV2 construct expressing shRNA (DISC1-shRNA) targeting the disc1 gene, as well as a control, scrambled shRNA viral construct (see Methods; Figure 1A,B). DISC1 expression was determined 10–14 days after the infusions were performed. Initially, viral transfections were confirmed by staining for β-galactosidase expression (Figure 1D; n=7). To assess the efficacy of the DISC1-shRNA on knocking down DISC1 protein (DISC1-kd) in vivo, we performed an immunohistochemical analysis of DISC1 protein expression on tissue in which animals (12–20 weeks old) were infused in mPFC with DISC1-shRNA (bilateral, n=6; unilateral, n=1) or saline (bilateral, n=2; unilateral, n=2); non-infused control animals were also assessed (n=3). In some cases, DISC1-shRNA was infused into one hemisphere and saline was infused into the other hemisphere (n=2). Subsequently, DISC1 immunolabeling analysis showed that DISC1 protein was almost entirely absent in mPFC infused with DISC1-shRNA (Figure 1E). The DISC1 protein was not effected in nearby cortical areas, M1 and M2 (Figure 1F), indicating that viral infusions were contained within the mPFC. Immunolabeling in disc1 knockout rats confirmed that the NB110-40775 antibody used to assess DISC1 expression was valid (see Figure S1), while a NB110-40773 antibody had mixed results (19)
Based on the efficacy of the DISC1-shRNA in eliminating DISC1 protein, we used the viral construct as a tool to probe the role of DISC1 in the functional properties of mature mPFC pyramidal neurons. In the following experiments, whole-cell patch-clamp recordings were made from layer V pyramidal neurons in acute mPFC slices extracted 10–60 days after bilateral infusion of DISC1-shRNA or scrambled-shRNA. As an additional control, we examined neurons in which an active AAV-shRNA targeting Wig1 (Wig1-kd), an irrelevant protein, was infused into the mPFC. Finally, we compared all results to neurons from control slices in which no manipulations were performed.
Cellular Characterization of Mature mPFC Pyramidal Neurons In DISC1-kd Rats
We first examined whether DISC1-kd alters normal basic membrane properties of layer 5 mPFC pyramidal neurons that control their ability to fire action potentials. More specifically, we assessed the following membrane properties in DISC1-kd cells, control cells, and scrambled-shRNA cells: resting membrane potential (RMP), input resistance (Rin), threshold for firing action potentials (rheobase), action potential kinetics, and the sag in membrane potential during hyperpolarizing current pulses (an estimate of hyperpolarization-activated cyclic nucleotide (HCN) gated channels activity) (see Table 1). We found that DISC1-kd neurons had a significantly higher Rin than control cells and scrambled-shRNA cells. Consistent with a higher Rin, DISC-kd cells required less current injection to fire an action potential compared to the other cells (Figure 2). Because there was no effect on action potential amplitude or kinetics, we conclude that DISC1 does not affect Na+ channels or fast K+ channels involved in spiking and repolarization. Although there did not appear to be an effect on the sag in membrane potential during hyperpolarizing current pulses, the interpretation of this measurement is confounded by the increase in Rin and by the fact that our patch-clamp recordings in the soma are not likely to capture HCN channel function occurring in distal dendrites and spines (20, 21).
Table 1.
Summary of membrane potential changes in mPFC DISC1-kd pyramidal neurons.
| condition | resting membrane potential (mV) | input resistance (mΩ) | rheobase (pA) | 1st action potential latency (ms) | mean spike width (ms) | mean sag amplitude (mV) |
|---|---|---|---|---|---|---|
| control | −61±0.5 (23) | 75.6±4.6 (23) | 207±17.4 (25) | 13±1.4 (25) | 3.5±0.4 (25) | 2.1±0.1 (25) |
|
| ||||||
| DISC1-kd | −61.4±0.4 (16) | 127.5±12.4 (21)a** | 121±10 (21)a**/c* | 11±1.8 (21)c | 2.5±0.3 (21) | 2.5±0.5 (21) |
| scrambled | −60±0.9 (11) | 82.5±6 (11)b* | 183±16.6 (11) | 17±1.2 (11) | 2.3±0.3 (11) | 1.9±0.2 (11) |
rheobase: threshold current for evoking an action potential
sag amplitude = depolarizing membrane potential during hyperpolarizing pulse (−250 pA)
Values were determined at the beginning of recordings (<5 min) and at the end (~30–45 min). ANOVA comparisons.
p<0.001 (compared to control)
p<0.01 (compared to DISC1-kd)
p<0.5;
p<0.01 (compared to scrambled)
Figure 2.

Basic membrane properties of layer V pyramidal neurons in DISC1-kd rats mPFC slices. A. Membrane voltage responses (upper panel) to current steps (100 pA steps; lower panel) recorded in cells from control, DISC-kd and scrambled animals. Red traces show the minimal current injection for triggering action potentials. The current for initiating action potentials in DISC1-kd cells was lower (121 ± 10 pA, n=21) compared to control (207 ± 10 pA, n=27) and scrambled cells (183 ± 16 pA, n=9). B. Summary data showing the minimal current injection necessary for triggering an action potential (middle panel). DISC1-kd cells had a higher input resistance (Rin = 127.5 ± 12, n=20) compared to control cells (Rin = 75.63 ± 4.6, n=23, p<0.001) and scrambled cells (Rin = 82.54 ± 6, n=11, p<0.05) (left panel). The membrane potential threshold for initiating an action potential did not differ across treatments (right panel).
Disruption of DISC1 Enhances Isk-mediated Hyperpolarization and Diminishes Itrpc-mediated Depolarization
Our general hypothesis is that disruption of DISC1 protein contributes to cognitive dysfunction through disruption of patterns of activity that encode salient information. Because it is not plausible to accurately replicate mPFC activity in a reduced preparation, we examined how regulation of ionic currents that control spiking activity might be altered in DISC1-kd neurons. Based on our previous findings showing that mGluR-mediated, IP3R-dependent intracellular Ca2+ waves can activate Isk and Itrpc, ionic conductances that can potently regulate excitability of mPFC neurons (15, 17), we tested whether DISC1 contributes to their regulation.
Application of the Group 1 mGluR agonist DHPG (400 μM) to mPFC pyramidal neurons (RMP~−63 mV) elicited a transient SK channel-mediated hyperpolarization and a CAN channel-like sustained depolarization (Figure 3), which we have shown in hippocampal pyramidal cells to be due to activation of TRPC channels ((17); see Figure S2). A similar biphasic response was elicited in scrambled and Wig1-kd treated animals that did not differ significantly from control cells. In contrast, neurons from DISC1-kd animals exhibited a robust hyperpolarization, but only a small depolarization. Analysis showed that the duration of the Isk-mediated hyperpolarization increased significantly in DISC-kd cells compared to control and scrambled/Wig1-kd cells (DISC1-kd, 2454.5 ± 110 ms, n=9; control, 1952 ± 116 ms, n=9; scrambled/Wig1-kd, 2005 ± 124 ms, n=7; p<0.05), which was reflected in a significant increase in the integrated area of the hyperpolarization in DISC1-kd neurons (DISC1-kd, 2.3 ± 0.2 × 105 mV·ms, n=9; control, 1.6 × 105 ± 0.1 × 105 mV·ms, n=12; scrambled/Wig1-kd, 1.56 ± 0.2 × 105 mV·ms, n=7; p<0.05). We also found that the Itrpc-mediated depolarization was significantly smaller in DISC1-kd cells compared to control and scrambled or Wig1-kd treated cells (DISC1-kd, 0.27±0.05 mV, n=9; control, 2.06±0.12 mV, n=12; scrambled or Wig1-kd, 2.15±0.2 mV, n=7; p<0.0001) (Figure 3).
Figure 3.

DISC1 knockdown enhances mGluR-mediated SK hyperpolarization and suppresses TRPC depolarization. A. mGluR activation of IP3-dependent intracellular Ca2+ waves triggers a transient hyperpolarization followed by sustained depolarization in layer V neurons (holding potential ~ −65 mV). In DISC1-kd neurons (middle panel), the hyperpolarization was enhanced and the sustained depolarization was suppressed compared to control (left panel) and scrambled (right panel). Images of the recorded fura-2FF filled neurons are to the left of electrical and fluorescence responses. B–E. Summary graphs show that DISC1-kd neurons were hyperpolarized for longer and had larger hyperpolarization area (B, C), and a suppressed depolarization amplitude (E), compared to controls. There was no change in hyperpolarization amplitude (D). (mean ± SEM; *p<0.05, **p<0.01, ***p< 0.0001).
We have shown previously that Isk correlates with the rise in [Ca2+]i (15, 17). Analysis of mGluR-mediated, IP3R-dependent rises in [Ca2+]i in the soma showed a significant increase in the area of [Ca2+]i in DISC1-kd (780 ± 95 ΔF/F.ms, n= 9) as compared to controls (580 ± 80 ΔF/F.ms, n= 12, p<0.01, T-test); and to scrambled/Wig1-kd (584 ± 64 ΔF/F.ms, n=7, p=0.01, T-test). This suggests that the increase in Isk in DISC1-kd cells is correlated with an enhancement of intracellular Ca2+ release in the somatic region.
To validate whether the changes in Isk and Itrpc, were due to disruption of DISC1 protein and not to an off-target effect of the shRNA on other mRNA transcripts, we suppressed DISC1 function acutely by dialyzing recorded neurons with a combination of antibodies targeting DISC1 (anti-DISC1; NB110-40773 and NB110-40775; 1:200 dilution). Anti-DISC1 was included in the recording pipette and the mGluR-elicited hyperpolarization and depolarization were evaluated during dialysis of the antibody. Approximately 10 min after going into whole-cell mode there was a significant increase in hyperpolarization duration (172±12.67%, n=16, p<0.0001) and a significant decrease in depolarization amplitude (25±4.42%, n=16; p<0.0001) when compared to responses recorded within the first 5 min (Figure 4). The hyperpolarization amplitude was not significantly affected by anti-DISC1 (113±8.6%, n=16, p=0.11).
Figure 4.

Intracellular immunoblockade with DISC1 antibodies enhanced SK hyperpolarization and suppressed TRPC depolarization. A. Electrical and fluorescence responses recorded over time from a neuron dialyzed with two different DISC1 antibodies (1:200 dilution). 10–15 min after going into whole cell mode, there was an enhancement of the hyperpolarization and a suppression of the depolarization. B. There were no obvious differences in neurons dialyzed with heat-inactivated anti-DISC1. C. Summary data showing that anti-DISC1 alters mGluR-mediated membrane potential changes compared to controls. (***p<0.0001). Addition of heat-inactivated anti-DISC1 or anti-IgG did not affect the hyperpolarization or depolarization.
Correlating with the increased hyperpolarization duration in anti-DISC1 cells, there was an increase in the duration of intracellular Ca2+ waves ~10 min after breaking into cells relative to initial break-in (soma, 121±7.84%, n=16, p<0.05; proximal apical dendrite, 124.06±8.4 %, n=16, p<0.01; apical dendrite, 168±19%, n=16, p<0.01). A similar pattern was observed with just the NB110-40775 antibody, but not the NB110-40773 antibody, indicating that the NB110-40775 antibody was responsible for the enhancement of IP3-mediated intracellular Ca2+ waves, for the increased of the SK-mediated hyperpolarization and for the decreased of TRPC-mediated depolarization (Figure S3).
Control experiments in which inactivated anti-DISC1 (heated to 90° for 10 min; 1:200 dilution) or IgG (1:100 dilution) were loaded into neurons did not significantly affect the intracellular Ca2+ waves, the hyperpolarization or the depolarization: hyperpolarization duration (inactivated anti-DISC1, 102±2%, n=7; IgG, 106±12%, n=6; p>0.5), hyperpolarization amplitude (inactivated anti-DISC1, 101±5%, n=7; IgG, 110±4%, n=6; p>0.5), or depolarization amplitude (inactivated anti-DISC1, 95±7%, n=7; IgG, 98±5.5%, n=6; p>0.5). We further examined whether the DISC1 affect on TRPC-mediated depolarization occurred independently from the affect on SK-mediated hyperpolarization by blocking SK channels. We found that application of the SK channel blocker with apamin (100 nM) did not affect the ability of anti-DISC1 to disrupt the TRPC-mediated depolarization (Figure S4). This results suggest that DISC1 regulates distinct channels. The results of these experiments—using disparate strategies for disrupting DISC1 protein—show that DISC1 interacts with mGluR-mediated, IP3R-dependent intracellular Ca2+ waves regulation of mature mPFC pyramidal neuron function by enhancing an Isk-mediated hyperpolarization and suppressing an Itrpc depolarization, leading to reduced PFC activity.
Raising cAMP Enhances SK-mediated Hyperpolarization and Suppresses TRPC-mediated Depolarization
Based on evidence that DISC1 regulates cAMP through its ability to tether and release PDE4B when [cAMP] is high (22–24), and based on evidence that cAMP can regulate Isk and Itrpc/can (17, 25, 26), we tested whether raising [cAMP] mimics the effects of disrupted DISC1 on mGluR-mediated intracellular Ca2+ waves, Isk and Itrpc. Consistent with this prediction, raising cAMP with bath application of forskolin (10 μM) resulted in a significant increase in the duration of mGluR-mediated hyperpolarization and a significant decrease in depolarization amplitude compared to baseline (hyperpolarization duration, 115±3%; depolarization amplitude, 30±3%, n=7; p<0.001) (Figure 5A). Forskolin treatment also significantly enhanced the area of [Ca2+]i in all cell compartments (soma, 150 ± 5%, n=7, p<0.05; soma-dendrite border, 179±8%, n=7, p<0.05; and dendrites, 123.5±4%, n=7, p<0.05). Thus, the loss of DISC1 function or pharmacologically increasing cAMP signaling with forskolin produced a common effect by enhancing [Ca2+]i release, Isk-mediated hyperpolarization and suppressing Itrpc-mediated depolarization in layer V mPFC pyramidal neurons. Similar effects were seen with PDE4 inhibitor, Rolipram (see Figure S4.
Figure 5.

Role of cAMP in DISC1 regulation of SK-mediated hyperpolarization and TRPC-mediated depolarization. A. Raising cAMP increased SK-mediated hyperpolarization and suppressed TRPC-mediated depolarization. A representative neuron showing that bath application of forskolin (10 μM) mimicked the effects of DISC1-kd, by enhancing mGluR-mediated hyperpolarization and suppressing mGluR-mediated depolarization (left panels). Summary data showing the effects of forskolin-induced rises in cAMP on SK hyperpolarization and TRPC depolarization (right panel). (*p<0.05, **p< 0.001.) B. Suppressing cAMP rescued DISK1-kd effects on mGluR-mediated SK- and TRPC-mediated membrane potential changes. An example of a neuron dialyzed with anti-DISC1 before and after bath application of guanfacine (10–20 μM) (top panel). Addition of anti-DISC1 increased SK-mediated hyperpolarization and suppressed TRPC-mediated depolarization 10 min after the start of dialysis (top, middle trace). Bath application of guanfacine normalized both SK-mediated hyperpolarization duration and TRPC-mediated depolarization amplitude to control levels (top, right trace). Summary data showing that guanfacine rescued SK and TRPC channel function that was altered by immunoblockade with anti-DISC1 (bottom panels). (**p< 0.0001)
Using an alternative approach to test whether DISC1 might be acting through cAMP to regulate IP3-mediated [Ca2+]i release, Isk and Itrpc, we suppressed DISC1 function with anti-DISC1 antibodies, and then attempted to normalize excitability by pharmacologically decreasing cAMP production with bath application of the α2A-noradrenergic receptor agonist, guanfacine, which inhibits adenylate cyclase activity (Figure 5B). Dialysis of recorded neurons with anti-DISC1 for 10–15 minutes resulted in a significant increase in hyperpolarization duration (anti-DISC1 5 min, 2251±186 ms; anti-DISC1 10min, 3980±223 ms, n=8, p<0.0001) and a significant reduction in the TRPC-mediated depolarization (anti-DISC1 5 min, 2.4±0.2 mV; anti-DISC1 10min, 0.6±0.1 mV, n=8, p<0.0001). Following dialysis of anti-DISC1, bath application of guanfacine (10–20 μM) resulted in the rescue of the hyperpolarization duration by reducing its duration (anti-DISC1+guanfacine 20 min, 2433±129 ms, n=8) to baseline duration (p=0.5 when compared to anti-DISC1 5 min; p<0.0001 when compared to anti-DISC1 10 min). Similarly, guanfacine restored depolarization amplitude (anti-DISC1+guanfacine 20 min, 1.9±0.3, n=8) to baseline amplitude (p=0.2 when compared to anti-DISC1 5 min and p<0.0001 when compared to anti-DISC1 10 min). Guanfacine, however did not appear to change intracellular Ca2+ waves under these conditions.
To further confirm that loss of DISC1 induced an alteration of layer V PFC pyramidal neurons excitability via unregulated rises in cAMP, we performed another set of experiments to test whether the forskolin effect was occluded by dialysis of anti-DISC1 antibodies. Similar to our previous results, we found an increase of the hyperpolarization duration 10 min after going into whole-cell mode (5 min, 1891 ± 458 ms; 10 min, 3646 ± 409 ms; p<0.05) and a suppression of the depolarization (5 min, 1.79 ± 0.37 mV; 10 min, 0.39 ± 0.05 mV; p<0.01). Adding forskolin did not significantly increase the hyperpolarization (3857 ± 375 ms), nor did it have an effect on the depolarization (0.4 ± 0.05 mV). The presence of the anti-DISC1 antibodies also occluded forskolin’s effecst on the hyperpolarization amplitude (Figure 6).
Figure 6.
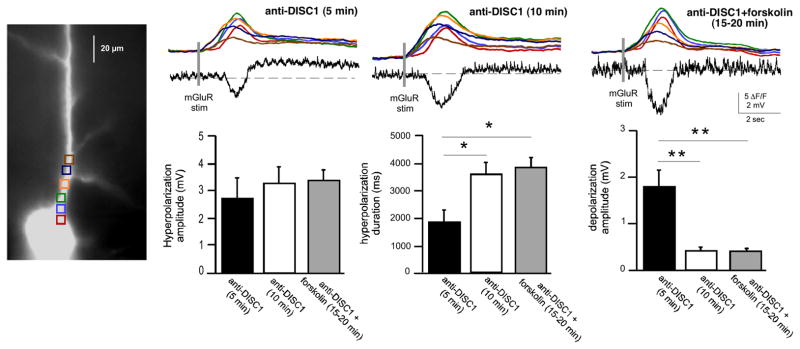
Forskolin effects are occluded in the presence of anti-DISC1 antibodies. Another example of a neuron dialyzed with a combination of antibodies targeting DISC1 (anti-DISC1; NB110-40773 and NB110-40775; 1:200 dilution) before and after bath application of forskolin (10 μM). Note that, consistent with the previous results, loss of DISC1 function resulted into an increase in the SK-mediated hyperpolarization and a suppression in the TRPC-mediated depolarization (middle panel). Bath application of forskolin did not induce further changes of the SK and TRPC channels function (right panel). (Below) Summary data showing that effects of forskolin are occluded in disrupted DISC1 conditions (*p<0.05; **p< 0.01).
DISCUSSION
The goal of this study was to determine the function of DISC1 protein in mature mPFC pyramidal neurons. We show that suppression of DISC1 protein with shRNA knockdown or by immunoblockade with antibodies targeting DISC1 leads to enhancement of SK channel-mediated hyperpolarization and suppression of TRPC channel-mediated sustained depolarization. Both of these ionic currents are activated by mGluR and IP3R-mediated intracellular Ca2+ waves. We also found an increase of IP3-mediated [Ca2+]i release in neurons loaded with anti-DISC1 antibody and in neurons treated with cAMP activators. Consistent with reports that DISC1 clamps cAMP levels in neurons, we found that pharmacologically raising cAMP mimicked the effect of disabling DISC1 function. Lastly we show that pharmacologically reducing cAMP in neurons with suppressed DISC1 function rescued normal SK and TRPC channel function. Taken together, these findings show that in the absence of DISC1 protein, there is an alteration of PFC pyramidal neuron excitability due to unregulated rises in cAMP.
DISC1 was initially identified when a loss-of-function translocation in the gene was discovered in Scottish families who suffered from mental disease (27–31). Subsequently, DISC1 has been implicated in bipolar disorder (32), MDD (33), and autism (34). DISC1 is expressed predominantly within pyramidal cell dendrites and spines of PFC and hippocampus—from early development through adulthood (28, 35–37). Analysis of its subcellular distribution in neurons shows that it associates with microtubules, mitochondria, the postsynaptic density, and the nucleus (28). Studies of DISC1 have shown that it plays a role in several developmental processes, including neurite outgrowth (38), axon targeting (39), and neuronal migration (36). It has also been shown that DISC1 is an AKAP scaffold-like protein that regulates cAMP signaling by releasing bound PDE4 when cAMP concentrations are high, leading to catabolism of cAMP (22, 24, 40). Immunoelectron microscopy of primate dlPFC supports this view, and indicates that DISC1 anchors PDE4 in dendrites and near the calcium-containing spine apparatus(10, 11). Although it is not known if these relationships are also found in rodent PFC neurons, many physiological properties of PFC neurons hold across species (41). Despite the possibility that DISC1 might contribute to a myriad of cAMP-dependent biochemical and ionic mechanisms that regulate the activity of PFC neurons, little is known about what role these complexes might play in the functional properties of mature neurons. In this study we tested the hypothesis that DISC1 might control the excitability of mature PFC pyramidal cells through its ability to regulate cAMP, and as a consequence, its ability to regulate Isk and Itrpc.
We and others have shown that activation of Isk and Itrpc by Group 1 mGluRs can potently regulate the firing of cortical and hippocampal pyramidal neurons (15, 17, 42) Glutamatergic activation of Gq-coupled Group 1 mGluRs triggers the IP3 signaling pathway and consequent activation of intracellular Ca2+ waves. Ca2+ wave propagation to the proximal apical dendrite/soma activates SK channels and a hyperpolarization time-locked to the rise in [Ca2+]i. Concomitant mGluR activation and a transient rise in [Ca2+]i, either through IP3R-mediated internal Ca2+ release or influx through VGCCs, can elicit a sustained depolarizing TRPC current that can boost firing in pyramidal neurons (15, 17). We have proposed previously that Isk might contribute to the termination of persistent activity, and that Itrpc is important for stabilizing persistent activity (15), consistent with computational models suggesting that a depolarizing boost is essential for maintaining sustained firing of neurons during working memory tasks (43, 44). Although there are not yet strong links between SK and TRPC channelopathies and psychiatric disease, both channels have been linked to schizophrenia and/or bipolar disease (45–49). Interestingly, antidepressants and phenothiazine antipsychotics have a high affinity for SK channels (50) and chronic lithium treatment alters TRPC channel expression (51).
cAMP is an ubiquitous second messenger that can regulate numerous ionic channels through activation of PKA. Because increasing cAMP with forskolin or cAMP analogs reduce a slow SK-like current (25, 26, 52, 53), it is possible that the increased in SK-mediated hyperpolarization we observed in DISC1 disrupted conditions is an indirect effect of enhanced intracellular Ca2+ waves due to PKA-dependent phosphorylation of IP3R and/or increased IP3Rs channel activity (54, 55). cAMP also may increase IP3Rs clustering at dendritic branch points (56). Little is known about cAMP regulation of TRPC channels. One study of snail burster cells found that forskolin suppressed a TRPC-like CAN current (Ca2+-dependent non-specific cation current; Ican) (16, 17), whereas another study found no effect of forskolin on mGluR-mediated Ican in hippocampal neurons (57). This negative result may be due to an insufficient rise in [cAMP] from repetitive forskolin application. Increased cAMP may also reduce firing and impair PFC cognitive function by increasing the open state of HCN (41) or KCNQ channels (58). However, as HCN channels are localized on distal dendrites and spines (20, 21, 41), they were not likely captured in the current experiments.
High levels of cAMP in PFC, e.g. due to increased catecholaminie release during stress, impairs working memory performance (41, 59–61). Indeed, the same knockdown of DISC1 in rat PFC used in the present study has now been shown to lower the threshold for stress-induced working memory deficits (19), consistent with the altered PFC physiology observed in the current study. Moreover, our results showing that reducing cAMP with guanfacine rescues pyramidal cell function in DISC1 compromised neurons are consistent with numerous reports showing that guanfacine improves working memory in rats (60, 62) monkeys (41, 60, 63–66) and humans (67–70) through inhibition of cAMP signaling in the PFC (41, 60). Taken together, these findings provide a plausible mechanism for working memory deficits observed in mice and humans in which DISC1 is impaired (2, 71). More generally, impaired PFC function is associated with many mental disorders that have been linked to disruption of the disc1 gene. Understanding the functional role of DISC1 in mature neurons will help identify the physiological consequences of DISC1 loss, and possible therapeutic interventions for the treatment of these mental disorders.
Supplementary Material
Acknowledgments
Funding: NIAAA 1RL1AA017536 (AFTA and MFY); Kavli Foundation, NIMH RO1-MH067830 and P50-MH068789 (AFTA and MFY); NARSAD Distinguished Investigator Award (AFTA); NIH grant DC 01919 (LKK).
Footnotes
FINANCIAL DISCLOSURES
Yale University and AFTA receive royalties from the sales of extended release guanfacine (Intuniv™) from Shire Pharamceuticals. They do not receive royalties from immediate release guanfacine, the formulation used in this study. All other authors report no biomedical financial interests or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Bibliography
- 1.Goldman-Rakic PS. Cellular basis of working memory. Neuron. 1995;14:477–485. doi: 10.1016/0896-6273(95)90304-6. [DOI] [PubMed] [Google Scholar]
- 2.Cannon TD, Hennah W, van Erp TG, Thompson PM, Lonnqvist J, Huttunen M, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- 3.Gasperoni TL, Ekelund J, Huttunen M, Palmer CG, Tuulio-Henriksson A, Lonnqvist J, et al. Genetic linkage and association between chromosome 1q and working memory function in schizophrenia. Am J Med Genet B Neuropsychiatr Genet. 2003;116B:8–16. doi: 10.1002/ajmg.b.10757. [DOI] [PubMed] [Google Scholar]
- 4.Tan HY, Chen Q, Sust S, Buckholtz JW, Meyers JD, Egan MF, et al. Epistasis between catechol-O-methyltransferase and type II metabotropic glutamate receptor 3 genes on working memory brain function. Proc Natl Acad Sci U S A. 2007;104:12536–12541. doi: 10.1073/pnas.0610125104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wedenoja J, Tuulio-Henriksson A, Suvisaari J, Loukola A, Paunio T, Partonen T, et al. Replication of association between working memory and Reelin, a potential modifier gene in schizophrenia. Biol Psychiatry. 67:983–991. doi: 10.1016/j.biopsych.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sigurdsson T, Stark KL, Karayiorgou M, Gogos JA, Gordon JA. Impaired hippocampal-prefrontal synchrony in a genetic mouse model of schizophrenia. Nature. 464:763–767. doi: 10.1038/nature08855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 8.Broadbelt K, Byne W, Jones LB. Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr Res. 2002;58:75–81. doi: 10.1016/s0920-9964(02)00201-3. [DOI] [PubMed] [Google Scholar]
- 9.Bleich-Cohen M, Kupchik M, Gruberger M, Kotler M, Hendler T. Never resting region--mPFC in schizophrenia. Schizophr Res. 140:155–158. doi: 10.1016/j.schres.2012.06.030. [DOI] [PubMed] [Google Scholar]
- 10.Arnsten AF, Wang MJ, Paspalas CD. Neuromodulation of thought: flexibilities and vulnerabilities in prefrontal cortical network synapses. Neuron. 76:223–239. doi: 10.1016/j.neuron.2012.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Paspalas CD, Wang M, Arnsten AF. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: potential substrate for working memory deficits in schizophrenia. Cereb Cortex. 23:1643–1654. doi: 10.1093/cercor/bhs152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi-Takagi A, Takaki M, Graziane N, Seshadri S, Murdoch H, Dunlop AJ, et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat Neurosci. 13:327–332. doi: 10.1038/nn.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang Q, Charych EI, Pulito VL, Lee JB, Graziane NM, Crozier RA, et al. The psychiatric disease risk factors DISC1 and TNIK interact to regulate synapse composition and function. Mol Psychiatry. 16:1006–1023. doi: 10.1038/mp.2010.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei J, Graziane NM, Wang H, Zhong P, Wang Q, Liu W, et al. Regulation of N-Methyl-D-Aspartate Receptors by Disrupted-in-Schizophrenia-1. Biol Psychiatry. doi: 10.1016/j.biopsych.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hagenston AM, Fitzpatrick JS, Yeckel MF. MGluR-mediated calcium waves that invade the soma regulate firing in layer V medial prefrontal cortical pyramidal neurons. Cereb Cortex. 2008;18:407–423. doi: 10.1093/cercor/bhm075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Partridge LD, Swandulla D, Muller TH. Modulation of calcium-activated non-specific cation currents by cyclic AMP-dependent phosphorylation in neurones of Helix. J Physiol. 1990;429:131–145. doi: 10.1113/jphysiol.1990.sp018248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.El-Hassar L, Hagenston AM, D’Angelo LB, Yeckel MF. Metabotropic glutamate receptors regulate hippocampal CA1 pyramidal neuron excitability via Ca(2)(+) wave-dependent activation of SK and TRPC channels. J Physiol. 589:3211–3229. doi: 10.1113/jphysiol.2011.209783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hagenston AM, Rudnick ND, Boone CE, Yeckel MF. 2-Aminoethoxydiphenyl-borate (2-APB) increases excitability in pyramidal neurons. Cell Calcium. 2009;45:310–317. doi: 10.1016/j.ceca.2008.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gamo NJ, Duque A, Paspalas CD, Kata A, Fine R, Boven L, et al. Role of disrupted in schizophrenia 1 (DISC1) in stress-induced prefrontal cognitive dysfunction. Transl Psychiatry. 2013;3:e328. doi: 10.1038/tp.2013.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- 22.Millar JK, Pickard BS, Mackie S, James R, Christie S, Buchanan SR, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- 23.Murdoch H, Mackie S, Collins DM, Hill EV, Bolger GB, Klussmann E, et al. Isoform-selective susceptibility of DISC1/phosphodiesterase-4 complexes to dissociation by elevated intracellular cAMP levels. J Neurosci. 2007;27:9513–9524. doi: 10.1523/JNEUROSCI.1493-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Soda T, Frank C, Ishizuka K, Baccarella A, Park YU, Flood Z, et al. DISC1-ATF4 transcriptional repression complex: dual regulation of the cAMP-PDE4 cascade by DISC1. Mol Psychiatry. 18:898–908. doi: 10.1038/mp.2013.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ren Y, Barnwell LF, Alexander JC, Lubin FD, Adelman JP, Pfaffinger PJ, et al. Regulation of surface localization of the small conductance Ca2+-activated potassium channel, Sk2, through direct phosphorylation by cAMP-dependent protein kinase. J Biol Chem. 2006;281:11769–11779. doi: 10.1074/jbc.M513125200. [DOI] [PubMed] [Google Scholar]
- 26.Pedarzani P, Storm JF. PKA mediates the effects of monoamine transmitters on the K+ current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron. 1993;11:1023–1035. doi: 10.1016/0896-6273(93)90216-e. [DOI] [PubMed] [Google Scholar]
- 27.Millar JK, Wilson-Annan JC, Anderson S, Christie S, Taylor MS, Semple CA, et al. Disruption of two novel genes by a translocation co-segregating with schizophrenia. Hum Mol Genet. 2000;9:1415–1423. doi: 10.1093/hmg/9.9.1415. [DOI] [PubMed] [Google Scholar]
- 28.Ishizuka K, Paek M, Kamiya A, Sawa A. A review of Disrupted-In-Schizophrenia-1 (DISC1): neurodevelopment, cognition, and mental conditions. Biol Psychiatry. 2006;59:1189–1197. doi: 10.1016/j.biopsych.2006.03.065. [DOI] [PubMed] [Google Scholar]
- 29.Cash-Padgett T, Jaaro-Peled H. DISC1 mouse models as a tool to decipher gene-environment interactions in psychiatric disorders. Front Behav Neurosci. 7:113. doi: 10.3389/fnbeh.2013.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duff BJ, Macritchie KA, Moorhead TW, Lawrie SM, Blackwood DH. Human brain imaging studies of DISC1 in schizophrenia, bipolar disorder and depression: a systematic review. Schizophr Res. 2013;147:1–13. doi: 10.1016/j.schres.2013.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Hikida T, Gamo NJ, Sawa A. DISC1 as a therapeutic target for mental illnesses. Expert Opin Ther Targets. 16:1151–1160. doi: 10.1517/14728222.2012.719879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chubb JE, Bradshaw NJ, Soares DC, Porteous DJ, Millar JK. The DISC locus in psychiatric illness. Mol Psychiatry. 2008;13:36–64. doi: 10.1038/sj.mp.4002106. [DOI] [PubMed] [Google Scholar]
- 33.Hashimoto R, Numakawa T, Ohnishi T, Kumamaru E, Yagasaki Y, Ishimoto T, et al. Impact of the DISC1 Ser704Cys polymorphism on risk for major depression, brain morphology and ERK signaling. Hum Mol Genet. 2006;15:3024–3033. doi: 10.1093/hmg/ddl244. [DOI] [PubMed] [Google Scholar]
- 34.Kilpinen H, Ylisaukko-Oja T, Hennah W, Palo OM, Varilo T, Vanhala R, et al. Association of DISC1 with autism and Asperger syndrome. Mol Psychiatry. 2008;13:187–196. doi: 10.1038/sj.mp.4002031. [DOI] [PubMed] [Google Scholar]
- 35.Austin CP, Ky B, Ma L, Morris JA, Shughrue PJ. Expression of Disrupted-In-Schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience. 2004;124:3–10. doi: 10.1016/j.neuroscience.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 36.Kamiya A, Kubo K, Tomoda T, Takaki M, Youn R, Ozeki Y, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 37.Kirkpatrick B, Xu L, Cascella N, Ozeki Y, Sawa A, Roberts RC. DISC1 immunoreactivity at the light and ultrastructural level in the human neocortex. J Comp Neurol. 2006;497:436–450. doi: 10.1002/cne.21007. [DOI] [PubMed] [Google Scholar]
- 38.Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T, et al. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry. 2003;8:685–694. doi: 10.1038/sj.mp.4001352. [DOI] [PubMed] [Google Scholar]
- 39.Faulkner RL, Jang MH, Liu XB, Duan X, Sailor KA, Kim JY, et al. Development of hippocampal mossy fiber synaptic outputs by new neurons in the adult brain. Proc Natl Acad Sci U S A. 2008;105:14157–14162. doi: 10.1073/pnas.0806658105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Millar JK, Mackie S, Clapcote SJ, Murdoch H, Pickard BS, Christie S, et al. Disrupted in schizophrenia 1 and phosphodiesterase 4B: towards an understanding of psychiatric illness. J Physiol. 2007;584:401–405. doi: 10.1113/jphysiol.2007.140210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, et al. Alpha2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 42.Yeckel MFSA, Fitzpatrick JS, Hertle D, Hagenston AM, Garner RT. Intracellular Calcium Waves Transmit Synaptic Information to the Nucleus in Hippocampal Pyramidal Neurons. In: Dudek SM, editor. Transcirptional Regulation By Neuronal Activity. Springer Science Publishing; 2008. pp. 73–89. [Google Scholar]
- 43.Tegner J, Compte A, Wang XJ. The dynamical stability of reverberatory neural circuits. Biol Cybern. 2002;87:471–481. doi: 10.1007/s00422-002-0363-9. [DOI] [PubMed] [Google Scholar]
- 44.Wang XJ. Synaptic reverberation underlying mnemonic persistent activity. Trends Neurosci. 2001;24:455–463. doi: 10.1016/s0166-2236(00)01868-3. [DOI] [PubMed] [Google Scholar]
- 45.Gargus JJ, Fantino E, Gutman GA. A piece in the puzzle: an ion channel candidate gene for schizophrenia. Mol Med Today. 1998;4:518–524. doi: 10.1016/s1357-4310(98)01358-6. [DOI] [PubMed] [Google Scholar]
- 46.Wittekindt O, Jauch A, Burgert E, Scharer L, Holtgreve-Grez H, Yvert G, et al. The human small conductance calcium-regulated potassium channel gene (hSKCa3) contains two CAG repeats in exon 1, is on chromosome 1q21.3, and shows a possible association with schizophrenia. Neurogenetics. 1998;1:259–265. doi: 10.1007/s100480050038. [DOI] [PubMed] [Google Scholar]
- 47.Chandy KG, Fantino E, Wittekindt O, Kalman K, Tong LL, Ho TH, et al. Isolation of a novel potassium channel gene hSKCa3 containing a polymorphic CAG repeat: a candidate for schizophrenia and bipolar disorder? Mol Psychiatry. 1998;3:32–37. doi: 10.1038/sj.mp.4000353. [DOI] [PubMed] [Google Scholar]
- 48.Cardno AG, Bowen T, Guy CA, Jones LA, McCarthy G, Williams NM, et al. CAG repeat length in the hKCa3 gene and symptom dimensions in schizophrenia. Biol Psychiatry. 1999;45:1592–1596. doi: 10.1016/s0006-3223(99)00033-5. [DOI] [PubMed] [Google Scholar]
- 49.Yoon IS, Li PP, Siu KP, Kennedy JL, Macciardi F, Cooke RG, et al. Altered TRPC7 gene expression in bipolar-I disorder. Biol Psychiatry. 2001;50:620–626. doi: 10.1016/s0006-3223(01)01077-0. [DOI] [PubMed] [Google Scholar]
- 50.Stocker M, Hirzel K, D’Hoedt D, Pedarzani P. Matching molecules to function: neuronal Ca2+-activated K+ channels and afterhyperpolarizations. Toxicon. 2004;43:933–949. doi: 10.1016/j.toxicon.2003.12.009. [DOI] [PubMed] [Google Scholar]
- 51.Andreopoulos S, Wasserman M, Woo K, Li PP, Warsh JJ. Chronic lithium treatment of B lymphoblasts from bipolar disorder patients reduces transient receptor potential channel 3 levels. Pharmacogenomics J. 2004;4:365–373. doi: 10.1038/sj.tpj.6500266. [DOI] [PubMed] [Google Scholar]
- 52.Madison DV, Nicoll RA. Cyclic adenosine 3′,5′-monophosphate mediates beta-receptor actions of noradrenaline in rat hippocampal pyramidal cells. J Physiol. 1986;372:245–259. doi: 10.1113/jphysiol.1986.sp016007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Storm JFPP, Haug T, Winther T. Modulation of K+ Channels in Hippocampal Neurons: Transmitters Acting via Cyclic AMP Enhance the Excitability of Hippocampal Neurons Through Kinase-Dependent and -Independent Modulation of AHP- and h-Channels. In: Kuba K, et al., editors. Slow Synaptic Responses and Modulation ©. Vol. 2000 Springer; Japan: 2000. [Google Scholar]
- 54.DeSouza N, Reiken S, Ondrias K, Yang YM, Matkovich S, Marks AR. Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complex. J Biol Chem. 2002;277:39397–39400. doi: 10.1074/jbc.M207059200. [DOI] [PubMed] [Google Scholar]
- 55.Foskett JK, White C, Cheung KH, Mak DO. Inositol trisphosphate receptor Ca2+ release channels. Physiol Rev. 2007;87:593–658. doi: 10.1152/physrev.00035.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fitzpatrick JS, Hagenston AM, Hertle DN, Gipson KE, Bertetto-D’Angelo L, Yeckel MF. Inositol-1,4,5-trisphosphate receptor-mediated Ca2+ waves in pyramidal neuron dendrites propagate through hot spots and cold spots. J Physiol. 2009;587:1439–1459. doi: 10.1113/jphysiol.2009.168930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Congar P, Khazipov R, Ben-Ari Y. Direct demonstration of functional disconnection by anoxia of inhibitory interneurons from excitatory inputs in rat hippocampus. J Neurophysiol. 1995;73:421–426. doi: 10.1152/jn.1995.73.1.421. [DOI] [PubMed] [Google Scholar]
- 58.Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- 59.Taylor JR, Birnbaum S, Ubriani R, Arnsten AF. Activation of cAMP-dependent protein kinase A in prefrontal cortex impairs working memory performance. J Neurosci. 1999;19:RC23. doi: 10.1523/JNEUROSCI.19-18-j0001.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ramos BP, Stark D, Verduzco L, van Dyck CH, Arnsten AF. Alpha2A-adrenoceptor stimulation improves prefrontal cortical regulation of behavior through inhibition of cAMP signaling in aging animals. Learn Mem. 2006;13:770–776. doi: 10.1101/lm.298006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–422. doi: 10.1038/nrn2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Franowicz JS, Arnsten AF. Actions of alpha-2 noradrenergic agonists on spatial working memory and blood pressure in rhesus monkeys appear to be mediated by the same receptor subtype. Psychopharmacology (Berl) 2002;162:304–312. doi: 10.1007/s00213-002-1110-6. [DOI] [PubMed] [Google Scholar]
- 63.Franowicz JS, Arnsten AF. The alpha-2a noradrenergic agonist, guanfacine, improves delayed response performance in young adult rhesus monkeys. Psychopharmacology (Berl) 1998;136:8–14. doi: 10.1007/s002130050533. [DOI] [PubMed] [Google Scholar]
- 64.Franowicz JS, Arnsten AF. Treatment with the noradrenergic alpha-2 agonist clonidine, but not diazepam, improves spatial working memory in normal young rhesus monkeys. Neuropsychopharmacology. 1999;21:611–621. doi: 10.1016/S0893-133X(99)00060-3. [DOI] [PubMed] [Google Scholar]
- 65.Arnsten AF, Dudley AG. Methylphenidate improves prefrontal cortical cognitive function through alpha2 adrenoceptor and dopamine D1 receptor actions: Relevance to therapeutic effects in Attention Deficit Hyperactivity Disorder. Behav Brain Funct. 2005;1:2. doi: 10.1186/1744-9081-1-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arnsten AF, Cai JX, Goldman-Rakic PS. The alpha-2 adrenergic agonist guanfacine improves memory in aged monkeys without sedative or hypotensive side effects: evidence for alpha-2 receptor subtypes. J Neurosci. 1988;8:4287–4298. doi: 10.1523/JNEUROSCI.08-11-04287.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kolar D, Keller A, Golfinopoulos M, Cumyn L, Syer C, Hechtman L. Treatment of adults with attention-deficit/hyperactivity disorder. Neuropsychiatr Dis Treat. 2008;4:389–403. doi: 10.2147/ndt.s6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Jerie P. Clinical experience with guanfacine in long-term treatment of hypertension. Part II: adverse reactions to guanfacine. Br J Clin Pharmacol. 1980;10(Suppl 1):157S–164S. doi: 10.1111/j.1365-2125.1980.tb04924.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kaminer D, Seedat S, Stein DJ. Post-traumatic stress disorder in children. World Psychiatry. 2005;4:121–125. [PMC free article] [PubMed] [Google Scholar]
- 70.Kiechel JR. Pharmacokinetics and metabolism of guanfacine in man: a review. Br J Clin Pharmacol. 1980;10(Suppl 1):25S–32S. doi: 10.1111/j.1365-2125.1980.tb04901.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6. San Diego, CA: Academic Press; 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.