

Abstract
Severe hypocapnia increases the risk of DKA-related cerebral injury in children, but the reason for this association is unclear. To determine whether the effects of hypocapnia on the brain are altered during hyperglycemia or ketosis, we induced hypocapnia (pCO2 20 ± 3 mmHg) via mechanical ventilation in three groups of juvenile rats: 25 controls, 22 hyperglycemic rats (serum glucose 451± 78 mg/dL) and 15 ketotic rats (beta-hydroxy butyrate 3.0 ± 1.0 mmol/L). We used magnetic resonance imaging to measure cerebral blood flow (CBF) and apparent diffusion coefficient (ADC) values in these groups and in 17 ventilated rats with normal pCO2 (40±3 mmHg). In a subset (n=35), after 2 hrs of hypocapnia, pCO2 levels were normalized (40±3 mmHg) and ADC and CBF measurements repeated. Declines in CBF with hypocapnia occurred in all groups. Normalization of pCO2 after hypocapnia resulted in striatal hyperemia. These effects were not substantially altered by hyperglycemia or ketosis, however, declines in ADC during hypocapnia were greater during both hyperglycemia and ketosis. We conclude that brain cell swelling associated with hypocapnia is increased by both hyperglycemia and ketosis, suggesting that these metabolic conditions may make the brain more vulnerable to injury during hypocapnia.
Introduction
Sub-clinical cerebral edema occurs commonly in children during treatment of diabetic ketoacidosis (DKA).(1; 2) Studies have shown that this edema is vasogenic with elevated apparent diffusion coefficient (ADC) values on magnetic resonance (MR) diffusion weighted imaging and values consistent with increased cerebral perfusion on MR perfusion weighted imaging.(2) Recent studies using near infrared spectroscopy also demonstrate increased cerebral regional oxygen saturation values during DKA treatment, consistent with cerebral hyperperfusion.(3; 4)
The cause of vasogenic edema during DKA treatment in children is not understood. Past hypotheses suggesting that DKA-related cerebral edema may be caused by osmotic fluctuations would not explain these findings because edema caused by osmotic fluid shifts would be expected to result in cell swelling (cytotoxic edema), rather than vasogenic edema.(5-7) Animal studies suggest that cerebral blood flow is low in untreated DKA and that metabolic alterations in the brain in untreated DKA are similar to those observed in hypoxic-ischemic brain injury.(8; 9) We have therefore proposed that cerebral hypoperfusion during untreated DKA and the effects of reperfusion during DKA treatment may be responsible for DKA-related cerebral injury.
Children with DKA typically present with hypocapnia as respiratory compensation for metabolic acidosis. Studies in patients with other medical conditions have demonstrated that hypocapnia can substantially reduce cerebral blood flow and that this reduction maybe of sufficient severity to cause cerebral ischemia.(10-12) Furthermore, return of CO2 levels to normal after a period of hypocapnia may be associated with elevations in cerebral blood flow above control values.(10; 13) Previous studies in our laboratory have demonstrated that hyperglycemia and ketosis also reduce cerebral blood flow and may independently cause mild brain cell swelling.(14) We undertook the current studies to investigate how the interactions of hypocapnia, hyperglycemia and ketosis affect cerebral blood flow and cerebral edema. We hypothesized that either hyperglycemia or ketosis or both might magnify the effects of pCO2 in altering cerebral blood flow and/or that brain cell swelling in the setting of hypocapnia might be increased by either hyperglycemia or ketosis.
Methods
Juvenile Sprague Dawley rats (Charles River Laboratories, Wilmington, MA) 4-6 weeks of age were used for all experiments. Cerebral blood flow (CBF) and apparent diffusion coefficient (ADC) values were compared among rats in four experimental groups; normal control (n=17), hypocapnia (n=25), hypocapnia plus hyperglycemia (n=22) and hypocapnia plus ketosis (n=15). Hyperglycemia and ketosis were induced in the rats according to the procedures described below. All studies were conducted in accordance with the Animal Use and Care Guidelines issued by the National Institute of Health using protocols approved by the Animal Use and Care Committee at the University of California Davis.
Hyperglycemia model
Rats were treated with an intraperitoneal injection of Streptozotocin (STZ) in 0.05mol/L citric acid, pH 4.3; 115 mg/kg for rats <140 grams; 130 mg/kg for rats >140 grams. Rats were given unlimited access to D10W (water with 10% dextrose, Fisher Scientific, Santa Clara, CA) in the first 24 hrs after STZ injection to prevent hypoglycemia and were subsequently allowed unlimited access to tap water and standard rat chow. Twenty-four hours after STZ injection, rats were treated with subcutaneous insulin (Novolin 70/30 insulin 3 units daily for rats <100 grams; 4 units daily for rats 100-200 grams, and 5 units daily for rats >200 grams) for a period of 5 days to allow for resolution of any non-pancreatic toxicities of STZ. Urine glucose and ketoacids (acetoacetate) were measured daily using Multistix urinalysis strips (BAYER, Fisher Scientific, Santa Clara, CA) up to and including the day of imaging. Animals with elevated urine acetoacetate concentrations (above “trace” levels; 0.5 mmol/L) at any time prior to imaging were not used for the hyperglycemia model. One day prior to imaging studies, rats received no insulin treatment.
Ketosis model
Five days before imaging studies, rats were fed a diet consisting of 50% standard rat chow (LabDiet 5001, Commercial Chow, Richmond, IN) and 50% high fat diet (60% fat, Research Diets, Inc., OpenSource Diets #D12492) for 1 day. For an additional 3 days, rats consumed only the high fat diet. Twenty-four hours prior to imaging, rats were fasted and allowed access only to tap water. Using this protocol, we were able to reliably generate ketosis (β-hydroxybutyrate concentrations ≥2.0 mM, measured using Precision Xtra blood ketone test, Abbott Laboratories, Abbott Park, IL). Rats that failed to develop the previously specified level of ketosis were not used in the experiments.
Magnetic Resonance (MR) Diffusion Weighted Imaging and Perfusion Weighted Imaging procedures
Brain apparent diffusion coefficient (ADC) values and cerebral blood flow (CBF) were measured using MR diffusion weighted imaging and perfusion weighted imaging. Four groups of rats were compared: normal control rats, rats with hypocapnia alone, rats with hyperglycemia and hypocapnia, and rats with ketosis and hypocapnia. Rats in all groups were anesthetized using Na pentobarbital (IP 55 mg/kg); then the left femoral vein and artery were cannulated with PE-50 polyethylene tubing as described previously.(9) Sodium pentobarbital was then administered intravenously at a dosage of 10-15 mg/kg every 30 minutes for the duration of the experiment, with the dosage adjusted to maintain constant anesthesia and immobility. The femoral artery cannula was used for blood sampling. A heating pad with circulating water (Gaymar Inc., Orchard Park, NY) was used to maintain body temperature at 36.8-37.0°C throughout the studies.
Hypocapnia
Rats were intubated and ventilated (Harvard Small Animal ventilator, Holliston, MA) either to generate hypocapnia or to maintain normal pCO2 according to the assigned group. Blood samples were taken for analysis of pCO2 and pH immediately after intubation, and the respiratory rate and tidal volume adjusted to maintain the pCO2 level within the desired range. Normal control rats were maintained at a pCO2 of 35-45 mmHg and hypocapnic rats at a pCO2 of 15-25 mmHg.
In a subset of experiments, after ADC and CBF data were collected, pCO2 levels in the hypocapnic rats were gradually increased to 35-45 mmHg over a period of approximately one hour by making incremental adjustments in respiratory rate and tidal volume (n=14 hypocapnic control rats, n=12 hyperglycemic rats, n=9 ketotic rats). ADC and CBF measurements were then repeated to determine the change in these measures when CO2 levels returned to normal after a period of hypocapnia.
Magnetic resonance diffusion-weighted spin echo images (DWI) were acquired using a 7-Tesla Bruker Biospec MRI system as described previously.(9) ADC values (10-6 cm2/sec) were determined from 6 × 4 pixel regions of interest (ROI) for 8 brain regions (6 cortex and 2 striatum) using Paravision 4.0 software with 4 gradient strengths of 5-95 mT/m.(15) In each rat, CBF (ml/100gm/sec) was also determined using perfusion weighted imaging (PWI) analysis with continuous arterial spin labeling (ASL) (9) and a standard Bruker PERFPACK2 protocol (Bruker, Billerica, MA). ASL data were acquired using the same field of view and slice thickness as for DWI, and the ASL ROIs were chosen from the 128 × 32 matrix so as to measure CBF and ADC on the same voxels. PWI images were acquired in 11.02 minutes using TE/TR 12.77 msec/1023 msec with 1-second Adiabatic-Fast-Passage labeling pulse in the presence of a 10 mT/m gradient to obtain inversion +/- 8515 Hz (+/- 2cm) from the isocenter (also slice center) for control and labeled images, respectively. T1 maps for the same voxels were also acquired from selected rats in each treatment group in order to correct CBF measurements for possible location- and treatment-dependent variations in T1.
Statistical analyses
Boxplots of ADC and CBF values were used to describe the distribution of these measures in the four groups of rats and to assess the presence of potential outliers(16) that may affect the robustness of conclusions from classical analysis of variance and covariance (ANOVA/ANCOVA) methods for comparing means.(17) In ANOVA/ANCOVA models, model F-tests, R-squares and root mean square errors are reported to aid interpretation of the pairwise contrasts that are our primary concern. Pairwise comparisons of means were tested for statistical significance using the Tukey-Kramer multiple comparisons procedure to control the familywise Type 1 error rate for the set of up to 6 pairwise comparisons per model. In ANOVA models, we also used Daytons pairwise comparisons on the Akaike information criterion procedure for exploratory selection of the best fitting model of group means. (18) This procedure is a valuable exploratory tool for selecting a parsimonious ordering of group means with desirable statistical properties. (19) ANCOVA models were used to assess whether hyperglycemia and ketosis had direct effects on ADC independent of the effects on CBF. Separate models were specified for the cortex and for the striatum. In each, the region-specific ADC measurement was specified as the response variable and the region-specific CBF measurement was included as a covariate in a model that also included the four-level group mean factor.
ADC and CBF values during hypocapnia were compared to values measured after return to normal pCO2 using paired t-tests. CBF values after recovery from hypocapnia were compared to normal control CBF values using classical and robust ANOVA methods. P values ≤ 0.05 were considered to represent statistical significance and p-values ≤ 0.10 were considered to represent a trend. Statistical analyses were conducted using Stata/SE version 12 (StataCorp, College Station, TX) and Version 9.3 of the SAS System for Windows (SAS Institute, Cary, NC). To assess the sensitivity of inferences to the presence of outliers, robust regression models were fit using the default settings in SAS PROC ROBUSTREG for M-estimation (for ANOVA models) and for S-estimation (for ANCOVA models).(20) In case inferences are sensitive to the presence of outliers, pairwise contrasts from regression models are reported with 98.33% confidence intervals to correspond to a Bonferroni correction for 3 pairwise comparisons per family of interest.
Results
Biochemical data for rats in each of the four experimental groups are presented in Table 1 and ADC and CBF values in Figures 1 and 2. In comparison to normal control rats, rats in all three hypocapnic groups (hypocapnia alone, hypocapnia plus hyperglycemia and hypocapnia plus ketosis) had reduced cortical CBF (Figure 1). In the striatum, mean reductions in CBF were statistically significant in the hyperglycemic and ketotic groups. In the group with hypocapnia alone, there was a trend toward reduced striatal CBF (p=0.06) that became statistically significant when outlier-resistant statistical analyses were used (p=0.016). Reductions in CBF were similar in all hypocapnic groups, suggesting that neither hyperglycemia nor ketosis substantially altered the effect of hypocapnia on CBF.
Table 1. Biochemical Values; Mean (SD).
| Normal Control N (n=17) |
Hypocapnia H (n=25) |
Hyperglycemia + Hypocapnia G (n= 19) |
Ketosis + Hypocapnia K (n=15) |
Tukey-Kramer Words* | |
|---|---|---|---|---|---|
| glucose (mg/dL) | 143 (14) | 146 (35) | 451 (78) | 122 (24) | G, HNK |
| β hydroxy butryrate (mmol/L) | --------- | ---------- | -------- | 3.0 (1.0) | -------- |
| pH | 7.43 (0.05) | 7.71 (0.08) | 7.61 (0.09) | 7.69 (0.09) | HK, G, N |
| pCO2 (mmHg) | 40 (3) | 20 (2) | 20 (3) | 19 (4) | N, GHK |
| serum sodium (mmol/L) | 140 (3) | 140 (3) | 141 (4) | 141 (3) | KGHN |
| serum chloride (mmol/L) | 105 (3) | 107 (4) | 108 (6) | 108 (2) | KGHN |
| serum bicarbonate (mmol/L) | 26 (2) | 26 (3) | 20 (4) | 22 (2) | HN, KG |
| serum potassium (mmol/L) | 4.3 (0.4) | 4.0 (0.5) | 3.7 (0.4) | 3.7 (0.4) | NH, HGK |
| Serum urea nitrogen (mg/dL) | 12 (4) | 13 (5) | 19 (7) | 7 (4) | G, HN, K |
Tukey-Kramer multiple comparisons procedure used for pairwise comparisons of group means in oneway AN OVA for unbalanced groups. Words are formed by group letters (as indicated in italics in column headings), in descending order of group means. For each outcome, a pairwise contrast is statistically significant (p<0.05) if and only if the corresponding pair of letters do not appear together in any of the words for that outcome. For example, for the glucose outcome only those pairwise contrasts involving Group G are significant, while for the pH outcome the H vs. K pairwise contrast is not significant but all other pairwise contrasts are.
Figure 1. Declines in CBF in the cerebral cortex and striatum during hypocapnia are similar under control conditions, hyperglycemia or ketosis.
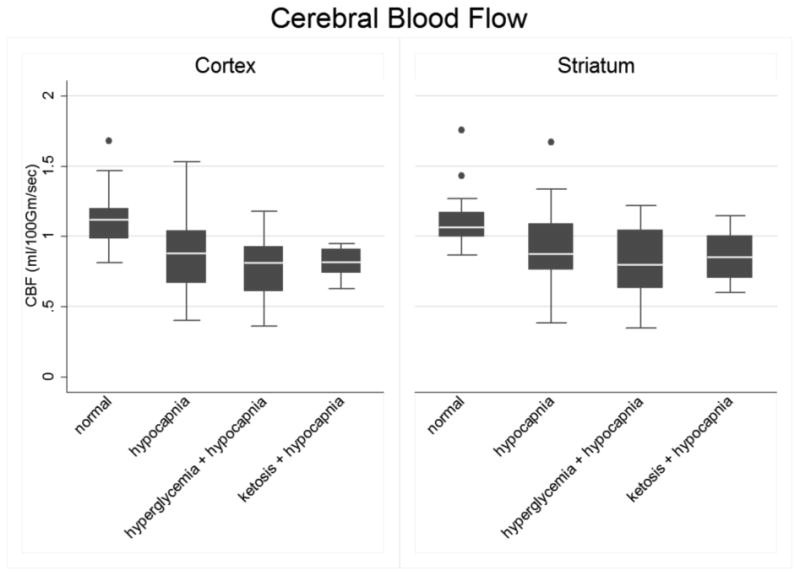
Normal controls (N, n=17), hypocapnia (H, n=25), hyperglycemia plus hypocapnia (G, n=22) and ketosis plus hypocapnia (K, n=15). The line in the middle of each box indicates the median (50% percentile), the top and bottom indicate the 75th / 25th percentiles. The whiskers (1.5 interquartile ranges) describe the maximum and minimum values. Individual values beyond the whiskers are possible outliers. Pairwise contrasts in means (95% confidence intervals) and p-values were estimated using Tukey-Kramer multiple comparisons procedure to control the familywise Type-1 error rate, with statistical significance declared for simultaneous p < 0.05. Cortex: Oneway ANOVA omnibus F(3,75)=9.49, p<0.001. Model R-square=0.28; root mean square error=0.22. Pairwise contrasts involving normal controls: N vs. H = 0.27 ( 0.08, 0.45 ), p<0.001; N vs. K = 0.31 ( 0.10, 0.52 ), p=0.001 and N vs. G = 0.37 ( 0.18, 0.56 ), p=0.002. Pairwise contrasts involving hypocapnic rats: H vs. K = 0.04 ( -0.15, 0.23 ), p=0.94; H vs. G = 0.11 ( -0.07, 0.28 ), p=0.38; K vs. G = 0.06 ( -0.13, 0.26), p=0.83. Best fitting model (Daytons PCAIC procedure) group means ordered as N > H = K > G . Striatum: Omnibus F(3,75)=4.82, p=0.004. Model R-square=0.16; root mean square error=0.25. Pairwise contrast in means (95% CI) involving normal controls: N vs. H: 0.20 ( -0.004, 0.41 ), p=0.06. N vs. K 0.25 ( 0.02, 0.48 ), p=0.03; N vs. G 0.29 ( 0.08, 0.51 ), p=0.003. Pairwise contrast in means involving hypocapnic groups: H vs. K = 0.05 ( -0.17, 0.26), p=0.93 ; H vs. G = 0.09 ( -0.10, 0.28 ), p=0.61; K vs. G = 0.04 ( -0.18, 0.26 ), p=0.96. Outlier-resistant robust ANOVA, N vs. H (98.33% CI) = 0.19 ( 0.002, 0.38 ), unadjusted p=0.016. Best fitting model for group means (Daytons PCAIC procedure): N > H = K = G .
Figure 2. Declines in ADC values in the cerebral cortex and striatum during hypocapnia are augmented by hyperglycemia or ketosis.
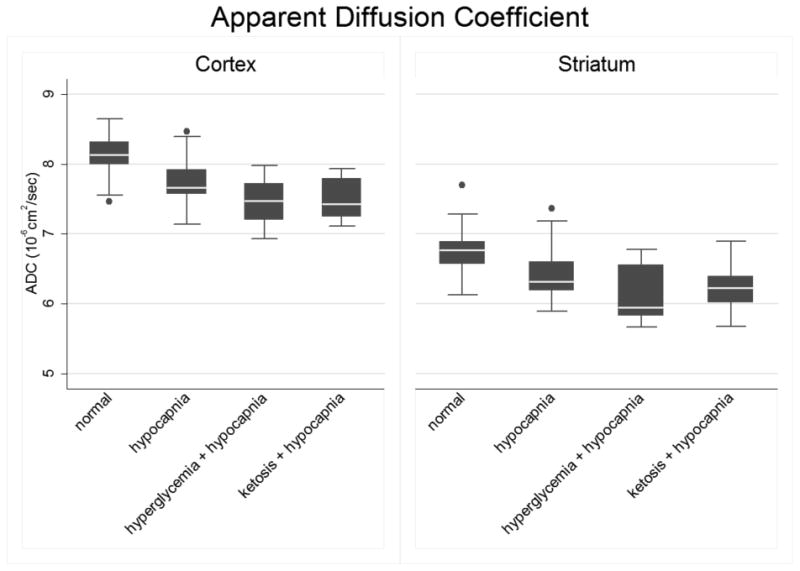
Normal controls (N, n=17), hypocapnia (H, n=25), hyperglycemia plus hypocapnia (G, n=22) and ketosis plus hypocapnia (K, n=15). The line in the middle of the box indicates the median (50% percentile), top and bottom borders indicate the 75th / 25th percentiles. The whiskers (1.5 interquartile ranges) describe maximum and minimum values. Individual values beyond the whiskers are possible outliers. Pairwise contrasts in means, (95% CI) and p-values were estimated using Tukey-Kramer multiple comparisons procedure to control the familywise Type-1 error rate, with statistical significance declared for simultaneous p < 0.05. Cortex: Oneway ANOVA omnibus F(3,75)=15.47, p<0.001. Model R-square=0.38; root mean square error=0.32. Pairwise contrasts involving normal controls: N vs. H = 0.35 (0.08, 0.61), p=0.006; N vs. K = 0.61 (0.31, 0.91), p<0.001; and N vs. G = 0.65 (0.37, 0.92), p<0.001. Among hypocapnic groups: H vs. K = 0.26 ( -0.01, 0.54 ), p=0.07; H vs. G = 0.30 (0.06, 0.55), p=0.01; and K vs. G = 0.04 ( -0.25, 0.32), p=0.98. Best fitting model for group means (Daytons PCAIC procedure): N > H > K = G. Striatum: Omnibus F(3,75)=9.76, p<0.001. Model R-square=0.38; root mean square error=0.38. N vs. H pairwise contrast in means (95% CI): 0.29 ( -0.03 – 0.60 ) p=0.09. N vs. G: 0.63 ( 0.30, 0.95 ), p<0.001, N vs. K: 0.51 ( 0.16, 0.87 ), p=0.002. Among hypocapnic groups: H vs. K = 0.23 ( -0.10, 0.56), p=0.27; H vs. G = 0.34 ( 0.05, 0.64), p=0.02; K vs. G = 0.12 ( -0.22, 0.45 ), p=0.81. In outlier-resistant robust ANOVA, the N vs. H contrast (98.33% CI) = 0.32 ( 0.02, 0.63 ), unadjusted p=0.012. Best fitting model for group means (Dayton's PCAIC procedure): N > H > K = G.
Cortical ADC values were significantly reduced during hypocapnia in all groups compared with normal controls (Figure 2). Reductions in cortical ADC were significantly greater in the hyperglycemic group compared to the group with hypocapnia alone. There was also a trend toward greater reduction in cortical ADC in the ketotic group compared to the group with hypocapnia alone (p=0.07). In the striatum, statistically significant reductions in ADC values during hypocapnia were observed in the hyperglycemic and ketotic groups in comparison with normal controls. In the group with hypocapnia alone, there was a trend toward reduced CBF in the striatum (p=0.09) that became statistically significant when outlier-resistant statistical analyses were used (p=0.012). In both the cortex and the striatum, the best fitting model for ordering of group means suggested that ADC values are reduced in the group with hypocapnia compared to normal control values, and that further reductions in ADC result when hypocapnia occurs in the setting of either hyperglycemia or ketosis.
ADC and CBF values were positively correlated (in striatum, r =0.34, 95% CI: 0.13, 0.53, p=0.002; in cortex, r=0.50, 95% CI: 0.31, 0.65, p<0.001, Figure 3), raising the question of whether the ADC reductions in the experimental groups might mainly result from differences in CBF. In ANCOVA models adjusted for region-specific CBF, the effect of hyperglycemia on ADC continued to be significant in both the cortex and striatum (Figure 3 legend). There was a trend toward a significant effect of ketosis in the cortex (p=0.08) but not in the striatum.
Figure 3. Correlation between CBF and ADC values under different physiologic conditions.
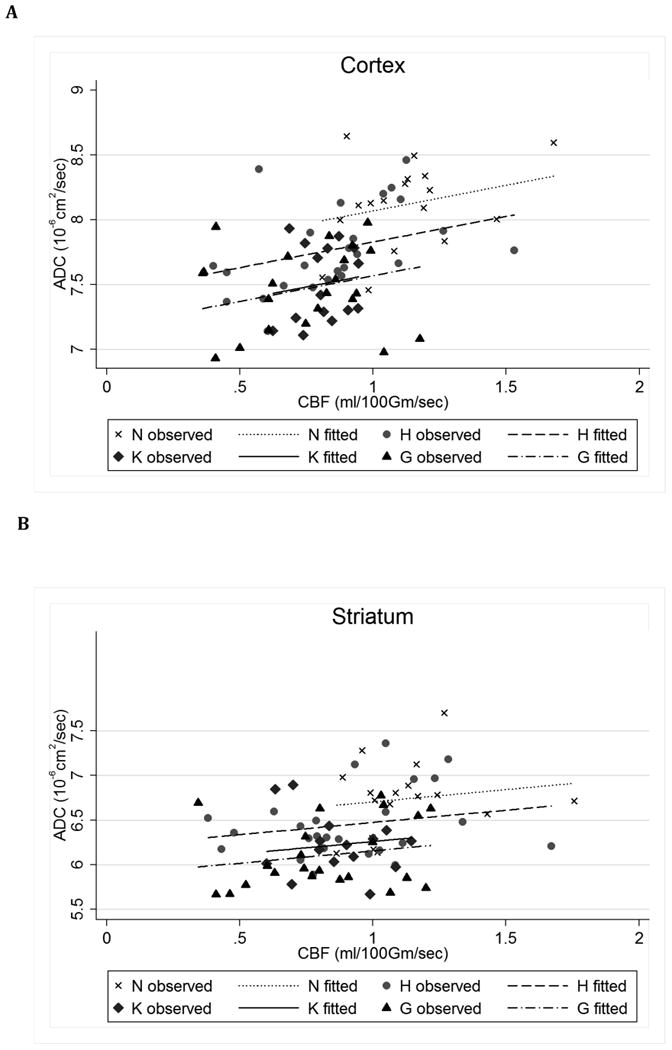
ANCOVA models with region-specific ADC (dependent variable), adjusting for region-specific CBF, to estimate adjusted mean differences (AMD) and (simultaneous 95% CI) for each of the three groups vs the reference group of hypocapnia. For AMD, the Tukey-Kramer multiple comparisons procedure was used to estimate 95% CI (shown in parentheses) and p-values, with statistical significance declared for simultaneous p < 0.05 to control the familywise Type-1 error rate. Cortex: ANCOVA model F(4,74)=13.92, p<0.001. Model R-square=0.43, root mean square error=0.31. AMD for N vs. H = 0.24 ( -0.04, 0.52 ), p=0.12; G vs. H = -0.26 ( -0.50, -0.02 ), p=0.03; K vs. H = -0.25 ( -0.51, 0.02 ), p=0.08 . Striatum: ANCOVA model F(4,74)=8.08, p<0.001. Model R-square=0.30; root mean square error=0.38. AMD (95% CI): N vs. H = 0.23 ( -0.10, 0.56 ), p=0.26; G vs. H = -0.32 ( -0.61, -0.02 ), p=0.03; K vs. H = -0.21 ( -0.54, 0.11 ), p=0.32.
When we adjusted pCO2 levels to normal after 2 hours of hypocapnia, significant increases in both cortical and striatal ADC and CBF were observed in all groups (Figures 4 & 5, paired t-test p ≤ 0.02 for each group's mean change during recovery). Recovery from hypocapnia was associated with elevations in striatal CBF above normal control values (p=0.04), in the group exposed to hypocapnia alone, consistent with cerebral hyperemia. In the cortex, elevations in CBF after recovery from hypocapnia were less robust and these values did not rise to levels significantly above normal. ADC values after recovery from hypocapnia were not significantly elevated above those of normal controls in any group. In general, ADC values in the hyperglycemic and ketotic groups after recovery from hypocapnia returned to levels below those of rats in the group exposed to hypocapnia alone. These differences were statistically significant in both cortex and striatum in the group exposed to hypocapnia alone vs. hypocapnia plus hyperglycemia, and in the striatum only the group exposed to hypocapnia alone vs. hypocapnia plus ketosis (Figure 5 Legend).
Figure 4. Changes in CBF with normalization of CO2 after hypocapnia are similar under control conditions, hyperglycemia or ketosis.
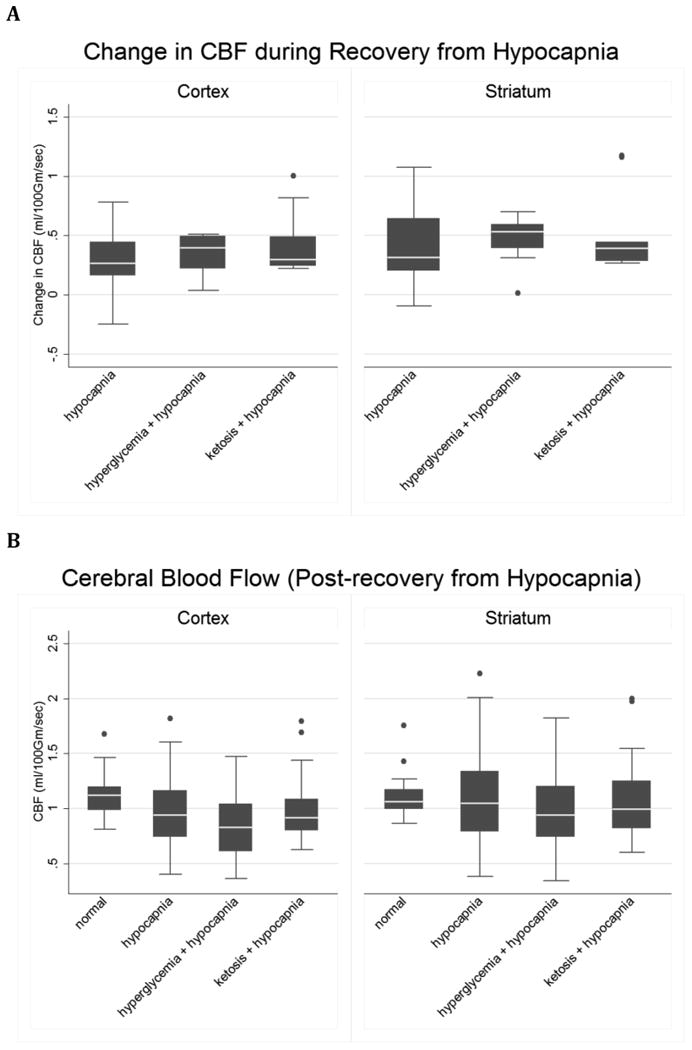
A. Control (n=14), hyperglycemia (n=12) and ketosis (n=9). The line in the middle of each box indicates the median (50% percentile), the top and bottom indicate the 75th / 25th percentiles. The whiskers (1.5 interquartile ranges) describe the maximum and minimum values. Individual values beyond the whiskers are possible outliers. Cortex: Oneway ANOVA omnibus F-test: F(2,32)=1.37, p= 0.27. Striatum: Omnibus F(2,32)=0.35, p= 0.70. B. CBF in the cortex and striatum in control (n=17), and in hypocapnic rats after CO2 normalization: hypocapnia only (H), hyperglycemia plus hypocapnia (G) and ketosis plus hypocapnia (K). Pairwise contrasts in means (95% CI) and p-values were estimated using Tukey-Kramer multiple comparisons procedure to control the familywise Type-1 error rate, with statistical significance declared for simultaneous p < 0.05. Cortex: Omnibus F(3, 48) = 1.61, p = 0.20. Model R-square=0.09; root mean square error=0.30. K vs. N = 0.16 ( -0.17, 0.48 ), p=0.58; H vs. N = 0.09 ( -0.20, 0.37 ), p=0.84; G vs. N = -0.10 ( -0.40, 0.19 ), p=0.79 ; K vs. H = 0.07 ( -0.27, 0.40 ), p=0.96 ; K vs. G = 0.26 ( -0.09, 0.61 ), p=0.20 ; H vs. G = 0.19 ( -0.12, 0.50 ), p=0.36 . Striatum: Omnibus F(3, 48) = 3.15, p=0.03. Model R-square=0.16; root mean square error=0.34. H vs. N=0.34 ( 0.01, 0.67 ), p=0.04; K vs. N=0.31 ( -0.07, 0.69 ), p=0.14; G vs. N=0.12 ( -0.23, 0.46 ), p=0.80; H vs. K=0.03 ( -0.36, 0.43 ), p=0.995; H vs. G = 0.23 ( -0.13, 0.59 ), p=0.35; K vs. G = 0.19 ( -0.21, 0.59), p=0.59.
Figure 5. Changes in ADC with normalization of CO2 after hypocapnia are similar under control conditions, hyperglycemia or ketosis.
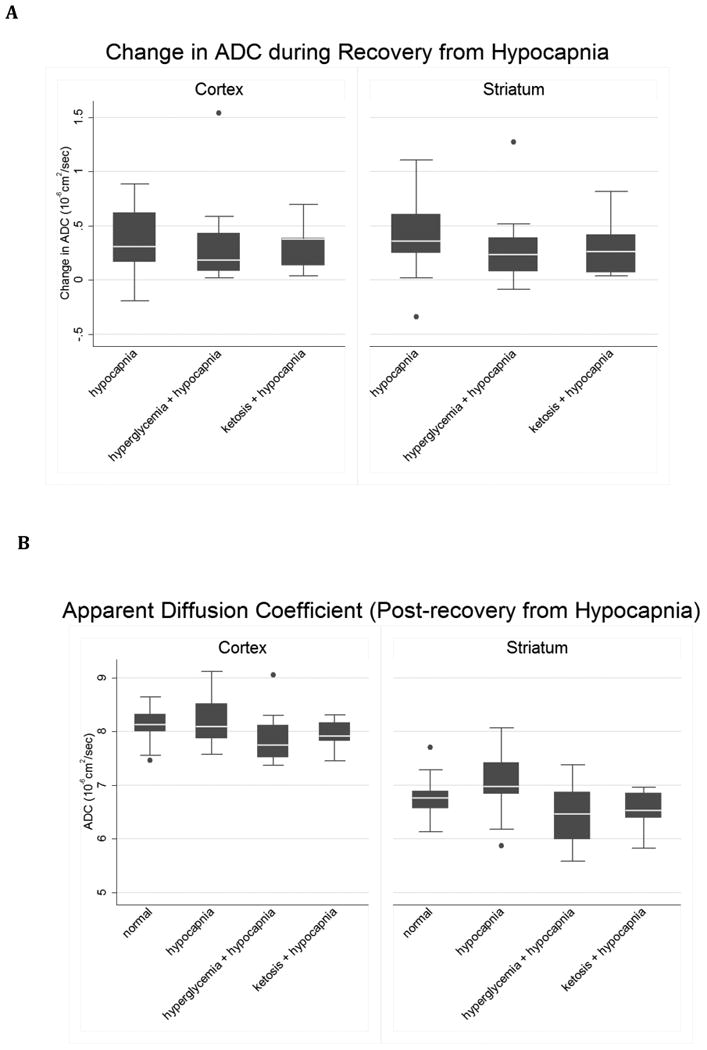
A. control (n=14), hyperglycemia (n=12) and ketosis (n=9). Cortex: Oneway ANOVA omnibus F-test: F(2,32)=0.07, p=0.94. Striatum: omnibus F(2,32)=0.40, p=0.67. B. ADC in normal control (n=17) and in hypocapnic rats after CO2 normalization: hypocapnia only (H), hyperglycemia plus hypocapnia (G) and ketosis plus hypocapnia (K).). Pairwise contrasts in means, (95% CI) and p-values were estimated using Tukey-Kramer multiple comparisons procedure to control the familywise Type-1 error rate, with statistical significance declared for simultaneous p < 0.05. Cortex: Oneway ANOVA omnibus F(3,48) = 2.18, p=0.10. Model R-square=0.12; root mean square error=0.42. H vs. N= 0.13 ( -0.27, 0.53 ), p=0.82; K vs. N= -0.19 ( -0.65, 0.27 ), p=0.69; G vs. N = -0.25 ( -0.67, 0.17 ), p=0.41; H vs. K = 0.32 ( -0.15, 0.80 ), p=0.29; H vs. G = 0.38 ( -0.06, 0.82 ), p=0.11; K vs. G = 0.06 ( -0.43, 0.55 ), p=0.99. Outlier-resistant robust ANOVA, H vs. G contrast (unadjusted 98.33% CI) = 0.40 ( 0.01, 0.79 ), unadjusted p=0.014. Best fitting model of group means (Dayton's PCAIC procedure): H = N > K = G. Striatum: Omnibus F(3,48) = 3.16, p=0.03. Model R-square=0.17; root mean square error=0.49. Pairwise contrasts involving normal controls: H vs. N=0.27 ( -0.20, 0.74 ), p=0.43; K vs. N = -0.22 ( -0.76, 0.32 ), p=0.70; G vs. N = -0.27 ( -0.76, 0.22 ), p=0.48. Pairwise contrasts among the hypocapnia groups: H vs. K = 0.49 ( -0.07,1.05 ), p=0.10; H vs. G = 0.54 ( 0.03, 1.06 ), p=0.04; K vs. G = 0.05 ( -0.53, 0.63 ), p=0.996. Outlier-resistant robust ANOVA, H vs. K contrast (98.33% CI) = 0.54 ( 0.07, 1.02 ), unadjusted p=0.006. Best fitting model of group means (Dayton's PCAIC procedure): H > N > K = G .
Discussion
The cause of cerebral edema and cerebral injury in children with DKA remains poorly understood. Multiple biochemical and physiological derangements occur during DKA, including hyperglycemia, ketosis, acidosis, circulatory volume depletion, hypocapnia and electrolyte depletion. The extent to which any or all of these derangements contribute to cerebral edema and/or cerebral injury is unclear. The current study represents an initial attempt to address this question by examining the effects of hypocapnia, alone or in combination with hyperglycemia or ketosis, on cerebral water distribution and cerebral blood flow. In previous studies, we have demonstrated that both hyperglycemia and ketosis reduce CBF. Our current data, however, demonstrate that the effect of pCO2 on CBF appears to dominate those of either hyperglycemia or ketosis. Hyperemia may occur when pCO2 levels are returned to normal after a period of hypocapnia but this effect similarly does not appear to be significantly modified by hyperglycemia or ketosis. Brain cell swelling, however, was significantly increased when hypocapnia occurred in combination with either hyperglycemia or ketosis. These data imply that hyperglycemia and/or ketosis may make brain cells more vulnerable to injury during the reductions in CBF that occur with hypocapnia.
Hypocapnia occurs in children with DKA, in response to metabolic acidosis. Hypocapnia maybe profound, with pCO2 levels as low as 5-10 mmHg.(21) In spite of systemic acidosis in children with DKA, cerebrospinal fluid pH is relatively normal as a result of the relative impermeability of the blood brain barrier to hydrogen and bicarbonate ions, but relatively unrestricted permeability to CO2.(22; 23) During DKA, CO2 levels continue to modulate CBF via changes in CSF and extracellular fluid pH.(24) Previous studies in our laboratory have demonstrated a significant positive correlation between pCO2 and CBF in a rat model of DKA.(9)
Multiple studies have documented that prolonged hypocapnia can reduce CBF sufficiently to cause cerebral ischemia and may worsen various types of brain injuries.(10; 12; 25; 26) Prophylactic hyperventilation, administered as a means of avoiding increased intracranial pressure, has been shown to worsen outcomes of traumatic brain injury, stroke and other types of brain injury, and prolonged hypocapnia during anesthesia has been shown to be associated with neurocognitive dysfunction.(10; 11; 27-29) Hypocapnia, rather than hypoxia, has also been thought to be responsible for cognitive impairment and brain injuries caused by high altitude exposure.(30) Most importantly, hypocapnia has been found to be a risk factor for cerebral edema/cerebral injury in children with DKA.(31; 32)
We and others hypothesized that hypocapnia may play a role in causing brain injury in children with DKA via decreases in CBF.(31; 32) Although it is possible that prolonged hypocapnia alone might be responsible for cerebral injury, we undertook the current studies to determine whether either hyperglycemia and/or ketosis might make the brain more vulnerable to injury during DKA. In previous studies in our laboratory, we found that both hyperglycemia and ketosis independently decrease CBF, as well as causing declines in cerebral high-energy phosphate levels and mild brain cell swelling.(14) In the current studies, we found that brain cell swelling was significantly greater when hypocapnia occurred in the setting of hyperglycemia or ketosis. These data suggest that hyperglycemia and/or ketosis may result in adverse conditions under which the reduction in CBF resulting from hypocapnia might more readily result in brain injury.
During prolonged hypocapnia, buffering occurs with a decrease in bicarbonate levels in the CSF. This buffering then results in a rise in CBF toward normal.(11; 24) Previous investigators have suggested that a rise in CO2 levels after a period of prolonged hypocapnia may result in cerebral hyperperfusion.(13; 25; 33) Data collected in children using perfusion weighted MR imaging during DKA treatment with insulin and saline suggest elevated CBF(2) and studies using near infrared spectroscopy during DKA treatment in children similarly suggest cerebral hyperperfusion. (3; 4) In the current studies, we observed an increase in striatal CBF, with values exceeding those of normal controls, when pCO2 was returned to normal after a period of hypocapnia. This response, however, was not increased by hyperglycemia or ketosis. Furthermore, changes in CBF were not accompanied by significant elevations in ADC, suggesting that vasogenic cerebral edema did not occur. These findings suggest that the conditions of the current experiments are not sufficient to create vasogenic edema and additional factors present during DKA must contribute.
The current study has some limitations. There was substantial variability in CBF among individual animals in each experimental group and therefore differences in CBF of smaller magnitude were difficult to detect. It is possible that some of the observed differences among groups in CBF would have been statistically significant if the number of animals in each group had been larger. Furthermore, due to the technical limitations of maintaining pCO2 levels within a narrow range in very small animals, the duration of hypocapnia in the current studies was shorter than that which would be typical for many children with DKA. Although allometric arguments for differences in rat and human time would diminish this concern, it is possible that longer duration of hypocapnia might have resulted in more substantial changes in ADC and/or detection of greater differences between groups.
In summary, the current study demonstrates that reductions in cerebral blood flow resulting from hypocapnia are not substantially modified by either hyperglycemia or ketosis. Although cerebral hyperemia may occur when pCO2 levels return to normal after a period of hypocapnia, this effect is not increased by hyperglycemia or ketosis. Hypocapnia, however, results in brain cell swelling (reduced ADC values) and this swelling is significantly increased in the setting of either hyperglycemia or ketosis. These findings suggest that hyperglycemia and/or ketosis may make the brain more vulnerable to injury when hypocapnia occurs in children with DKA.
Acknowledgments
We are grateful to Abbott Laboratories for the generous donation of Precision Xtra test strips for blood glucose and ketone testing in these experiments. These studies were funded by American Diabetes Association research award #1-09-RA-52 (to NG). Funding for nuclear magnetic resonance equipment was provided in part by NSF OSTI 97-24412. The investigation was conducted in part in a facility constructed with support from Research Facilities Improvement Program Grant Number C06 RR17348-01 from the National Center for Research Resources, National Institutes of Health. Dr. Nicole Glaser serves as guarantor for this article and takes full responsibility for the article's originality and all other aspects of the study.
Footnotes
Author Contributions: NG- planned/supervised studies, analyzed data, wrote manuscript, secured funding for studies, AB- conducted studies, assisted with data entry and analysis, reviewed manuscript, contributed to discussion, SA- supervised and assisted with technical aspects of studies related to MR imaging, assisted with data interpretation, reviewed/revised manuscript, contributed to discussion, DT – assisted in data interpretation and planning aspects of study design, conducted statistical analyses of data, reviewed/revised manuscript, WL- conducted studies, assisted with data entry and analysis, reviewed/revised manuscript, contributed to discussion, MO- conducted studies, assisted with data entry, reviewed manuscript, contributed to discussion, MO'D- planned/supervised studies, assisted with data analysis and interpretation, reviewed/revised manuscript.
References
- 1.Glaser N, Wooton-Gorges S, Buonocore M, Marcin J, Rewers A, Strain J, DiCarlo J, Neely E, Barnes P, Kuppermann N. Frequency of Sub-Clinical Cerebral Edema in Children with Diabetic Ketoacidosis. Pediatr Diab. 2006;7:75–80. doi: 10.1111/j.1399-543X.2006.00156.x. [DOI] [PubMed] [Google Scholar]
- 2.Glaser N, Gorges S, Marcin J, Buonocore M, DiCarlo J, Neely E, Barnes P, Bottomly J, Kuppermann N. Mechanism of Cerebral Edema in Children with Diabetic Ketoacidosis. J Pediatr. 2004;145:164–171. doi: 10.1016/j.jpeds.2004.03.045. [DOI] [PubMed] [Google Scholar]
- 3.Roberts J, Vavilala M, Schenkman K, Shaw D, Martin L, Lam A. Cerebral hyperemia and impaired cerebral autoregulation associated with diabetic ketoacidosis in critically ill children. Crit Care Med. 2006;34:2217–2223. doi: 10.1097/01.CCM.0000227182.51591.21. [DOI] [PubMed] [Google Scholar]
- 4.Glaser N, Tancredi D, Marcin J, Caltagirone R, Lee Y, Murphy C, Kuppermann N. Cerebral hyperemia measured with near infrared spectroscopy during treatment of diabetic ketoacidosis in children. J Pediatr. 2013 doi: 10.1016/j.jpeds.2013.06.008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sevick R, Kanda F, Mintorovitch J, Arieff A, Kucharczyk J, Tsuruda J, Norman D, Moseley M. Cytotoxic brain edema: Assessment with diffusion-weighted MR imaging. Radiology. 1992;185:687–690. doi: 10.1148/radiology.185.3.1438745. [DOI] [PubMed] [Google Scholar]
- 6.Righini A, Ramenghi L, Zirpoli S, Mosca F, Triulzi F. Brain apparent diffusion coefficient decrease during correction of severe hypernatremic dehydration. Am J Neuroradiol. 2005;26:1690–1694. [PMC free article] [PubMed] [Google Scholar]
- 7.Vajda Z, Pedersen M, Doczi T, Sulyok E, Stodkilde-Jorgensen H, Frokiaer J, Nielsen S. Effects of centrally administered arginine vasopressin and atrial natriuretic peptide on the development of brain edema in hyponatremic rats. Neurosurgery. 2001;49:697–705. doi: 10.1097/00006123-200109000-00031. [DOI] [PubMed] [Google Scholar]
- 8.Glaser N, Yuen N, Anderson S, Tancredi D, O'Donnell M. Cerebral Metabolic Alterations in Rats with Diabetic Ketoacidosis: Effects of Treatment with Insulin and Intravenous Fluids and Effects of Bumetanide. Diabetes. 2010;59:702–709. doi: 10.2337/db09-0635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yuen N, Anderson S, Glaser N, O'Donnell M. Cerebral Blood Flow and Cerebral Edema in Rats with Diabetic Ketoacidosis. Diabetes. 2008;57:2588–2594. doi: 10.2337/db07-1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Curley G, Kavanagh B, Laffey J. Hypocapnia and the injured brain: More harm than benefit. Crit Care Med. 2010;38:1348–1359. doi: 10.1097/CCM.0b013e3181d8cf2b. [DOI] [PubMed] [Google Scholar]
- 11.Laffey J, Kavanagh B. Hypocapnia. New Engl J Med. 2002;347:43–54. doi: 10.1056/NEJMra012457. [DOI] [PubMed] [Google Scholar]
- 12.Stocchetti N, Maas A, Chieregato A, van der Plas A. Hyperventilation in head injury: a review. Chest. 2005;127:1812–1827. doi: 10.1378/chest.127.5.1812. [DOI] [PubMed] [Google Scholar]
- 13.Muizelaar J, Marmarou A, Ward J, Kontos H, Choi S, Becker D, Gruemer H, Young H. Adverse effects of prolonged hyperventilation in patients with severe head injury: a randomized clinical trial. J Neurosurg. 1991;75:731–739. doi: 10.3171/jns.1991.75.5.0731. [DOI] [PubMed] [Google Scholar]
- 14.Glaser N, Ngo C, Anderson S, Yuen N, Trifu A, O'Donnell M. Effects of hyperglycemia and effects of ketosis on cerebral perfusion, cerebral water distribution and cerebral metabolism. Diabetes. 2012;61:1831–1837. doi: 10.2337/db11-1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tatlisumak T, Takano K, Carano R, Fisher M. Effect of basic fibroblast growth factor on experimental focal ischemia studied by diffusion-weighted and perfusion imaging. Stroke. 1996;27:2292–2298. doi: 10.1161/01.str.27.12.2292. [DOI] [PubMed] [Google Scholar]
- 16.Tukey J. Exploratory data analysis. Reading, MA: Addison-Wesley; 1977. [Google Scholar]
- 17.Neter J, Kutner M, Wasserman W, Nachtsheim C. Applied linear statistical models: regression, analysis of variance, and experimental designs. Homewood, IL: McGraw Hill; 1990. [Google Scholar]
- 18.Dayton C. Information criteria for pairwise comparisons. Psychological Methods. 2003;8:61–71. doi: 10.1037/1082-989x.8.1.61. [DOI] [PubMed] [Google Scholar]
- 19.Kuiper R, Hoijtink H. Comparisons of means using exploratory and confirmatory approaches. Psychological Methods. 2010;15:69–86. doi: 10.1037/a0018720. [DOI] [PubMed] [Google Scholar]
- 20.Huber P, Ronchetti E. Robust Statstics. Hoboken, NJ: Wiley; 2009. [Google Scholar]
- 21.Glaser N, Marcin J, Wooton-Gorges S, Buonocore M, Rewers A, Strain J, DiCarlo J, Neely E, Barnes P, Kuppermann N. Correlation of Clinical and Biochemical Findings with DKA-related Cerebral Edema in Children using Magnetic Resonance Diffusion Weighted Imaging. J Pediatr. 2008;153:541–546. doi: 10.1016/j.jpeds.2008.04.048. [DOI] [PubMed] [Google Scholar]
- 22.Assal J, Aoki T, Manzano F, Kozak G. Metabolic effects of sodium bicarbonate in management of diabetic ketoacidosis. Diabetes. 1973;23:405–411. doi: 10.2337/diab.23.5.405. [DOI] [PubMed] [Google Scholar]
- 23.Ohman J, Marliss E, Aoki T, Munichoodappa C, Khanna V, Kozak G. The cerebrospinal fluid in diabetic ketoacidosis. N Engl J Med. 1971;284:283–290. doi: 10.1056/NEJM197102112840601. [DOI] [PubMed] [Google Scholar]
- 24.Tasker R, Lutman D, Peters M. Hyperventilation in severe diabetic ketoacidosis. Pediatr Crit Care Med. 2005;6:405–411. doi: 10.1097/01.PCC.0000164343.20418.37. [DOI] [PubMed] [Google Scholar]
- 25.Skippen P, Seear M, Poskitt K, Kestle J, Cochrane D, Annich G, Handel J. Effect of hyperventilation on regional cerebral blood flow in head-injured children. Crit Care Med. 1997;25:1402–1409. doi: 10.1097/00003246-199708000-00031. [DOI] [PubMed] [Google Scholar]
- 26.Marion D, Firlik A, McLaughlin M. Hyperventilation therapy for severe traumatic brain injury. New Horizons. 1995;3:439–447. [PubMed] [Google Scholar]
- 27.Greisen G, Munck H, Lou H. Severe hypocarbia in preterm infants and neurodevelopmental deficit. Acta Paediatr Scand. 1987;76:401–404. doi: 10.1111/j.1651-2227.1987.tb10489.x. [DOI] [PubMed] [Google Scholar]
- 28.Wolman S, Orkin L. Postoperative human reaction time and hypocarbia during anaesthesia. Brit J Anaesth. 1968;40:920–926. doi: 10.1093/bja/40.12.920. [DOI] [PubMed] [Google Scholar]
- 29.Hovorka J. Carbon dioxide homeostasis and recovery after general anesthesia. Acta Anaesth Scand. 1982;26:498–504. doi: 10.1111/j.1399-6576.1982.tb01806.x. [DOI] [PubMed] [Google Scholar]
- 30.Hornbein T, Townes B, Schoene R, Sutton J, Houston C. The cost to the central nervous system of climbing to extremely high altitude. N Engl J Med. 1989;321:1714–1719. doi: 10.1056/NEJM198912213212505. [DOI] [PubMed] [Google Scholar]
- 31.Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J, Kaufman F, Quayle K, Roback M, Malley R, Kuppermann N. Risk Factors for Cerebral Edema in Children with Diabetic Ketoacidosis. N Engl J Med. 2001;344:264–269. doi: 10.1056/NEJM200101253440404. [DOI] [PubMed] [Google Scholar]
- 32.Mahoney C, Vlcek B, Del Aguila M. Risk factors for developing brain herniation during diabetic ketoacidosis. Pediatr Neurol. 1999;21:721–727. doi: 10.1016/s0887-8994(99)00079-x. [DOI] [PubMed] [Google Scholar]
- 33.Gleason C, Short B, Jones M. Cerebral blood flow and metabolism during and after prolonged hypocapnia in newborn lambs. J Pediatr. 1989;115:309–314. doi: 10.1016/s0022-3476(89)80091-5. [DOI] [PubMed] [Google Scholar]