

Abstract
Volatile organic compounds (VOCs) emanating from humans have the potential to revolutionize non-invasive diagnostics. Yet, little is known about how these compounds are generated by complex biological systems, and even less is known about how these compounds are reflective of a particular physiological state. In this proof-of-concept study, we examined VOCs produced directly at the cellular level from B lymphoblastoid cells upon infection with three live influenza virus subtypes: H9N2 (avian), H6N2 (avian), and H1N1 (human). Using a single cell line helped to alleviate some of the complexity and variability when studying VOC production by an entire organism, and it allowed us to discern marked differences in VOC production upon infection of the cells. The patterns of VOCs produced in response to infection were unique for each virus subtype, while several other non-specific VOCs were produced after infections with all three strains. Also, there was a specific time course of VOC release post infection. Among emitted VOCs, production of esters and other oxygenated compounds was particularly notable, and these may be attributed to increased oxidative stress resulting from infection. Elucidating VOC signatures that result from the host cells response to infection may yield an avenue for non-invasive diagnostics and therapy of influenza and other viral infections.
Keywords: breath analysis, esters, gas chromatography, influenza, mass spectrometry, volatile organic compounds
Introduction
Influenza A viruses, a group of enveloped RNA viruses of the Orthomyxoviridae family, cause infections in humans and a wide range of animals.[1–4] In humans, influenza is a respiratory pathogen, and the most common symptoms are headache, chills, dry cough, fever, and myalgia. In a minority of cases, patients develop a severe and fatal pneumonia or sepsis, (an overwhelming systemic inflammatory response). There is a seasonal pattern to the disease, with peaks in the winter months.[5] Typically, several subtypes circulate during the seasonal months (e.g., H1N1, H3N2). Although we traditionally think of influenza as a human pathogen, there are also several natural animal reservoirs for influenza. In wild waterfowl, virus replication occurs in the gastrointestinal tract with frequent shedding in the stool, and this can lead to transmission to other birds.[1, 3, 4] In poultry, such as chicken and turkey, influenza viruses can be highly pathogenic and can cause a severe, fatal respiratory and systemic infection, thereby rapidly killing large flocks if infected with highly pathogenic avian influenza (HPAI) viruses. The avian subtypes can potentially spread to humans directly, thus causing severe pneumonia with certain subtypes (e.g., H5N1) that are distinctly different from the seasonal subtypes.[1, 3, 4] Strains from the same subtype can have different degrees of virulence. Novel, potentially pandemic, influenza subtypes or strains can emerge as a result of genetic shift, such as genomic reassortment of various influenza viruses circulating in mammals or wild birds.[5, 6]
Detection of infection-specific volatile organic compound (VOC) biomarkers would allow timely diagnosis and intervention in an early influenza infection and outbreak. In this study, we examined the response of B lymphoblastoid cells to infection with influenza viruses by using GC/MS to assess the production of VOCs. These chemicals are produced by cells, can be measured directly, and can vary depending on cell line and metabolic state. A signature VOC “fingerprint” can exist for a specific cell line. Analysis of VOCs can yield a number of diagnostic possibilities for humans depending on clinical state (e.g., infection, cancer, or response to therapy).[7–12] We have previously demonstrated that cell lines with various HLA gene expression profiles exhibit unique VOC production.[13]
Thus, we hypothesized that cells can express an immunologic “odorprint” based on exposure to a particular antigen. In particular, an immunological cell line, such as B lymphoblastoids, will likely produce unique and distinct VOCs upon infection with different influenza subtypes as a result of specific virus–cell interactions. Though several host cell types are important in influenza infection (including respiratory epithelial and dendritic cells), we chose a B lymphoblastoid model for several reasons. First, B-cells are intricately involved in the systemic inflammation associated with influenza infection in humans, and B-cells frequently encounter viral particles in the respiratory tract when initiating the adaptive immune response.[14] Secondly, B-cells are recruited quickly to respiratory tissue in a non-specific manner early in infection.[15, 16] Thirdly, B-cells can act independently of T-cells in generating an immune response against influenza,[17] thus potentially indicating a partial innate response. There is only limited data supporting direct infection of B-cells with influenza virus, but this is highly plausible given that B-cells recognize virus-specific antigens and are in close proximity to active viral replication.[18] Finally, some influenza infections cause respiratory epithelial cell apoptosis,[19] which might allow viral particle access to resident B-cells.
In this study, we used a cell line for investigating an isolated immune system component as proof-of principle. We selected three pathogenic virus strains that represent different influenza virus subtypes: human seasonal influenza (H1N1) and two low pathogenic avian influenza (LPAI) viruses, H6N2 and H9N2. A B-lymphoblastoid model is particularly suitable for this type of investigation. For example, different influenza strains have different effects on the respiratory epithelium. Specifically, H9N2 can induce epithelial apoptosis,[19] thus limiting the ability to distinguish infection-specific VOCs from VOCs released because of cell death. Avian influenza strains differ in their affinities for sialic acid receptors on human epithelial cells,[20] so respiratory cell models utilizing infection with some strains can confound VOC interpretation. Inflammatory cells such as neutrophils would be ideal to study as an isolated immune system component, as neutrophils are the first responders of the innate immune system and they migrate towards the infected airway cells. The VOC signature of this response would be diagnostically important and useful. Unfortunately, neutrophils are extremely challenging to culture, therefore they were not considered suitable for our proof-of-principle study. For these reasons, we chose lymphoblastoid C1R B cells as our model cell-line system. We aimed to use an immunological cell model that reduces the variability associated with the different viral strains. Analysis of B lymphoblastoid cells infected with these three strains allows us to identify pathogen-specific VOCs and further our understanding of the molecular mechanisms activated during viral infection. Ultimately, this line of investigation might help establish ways to elucidate novel therapeutic targets in influenza infection and vaccine development.
Results
We determined gas phase volatile biomarkers associated with influenza virus infection by measurement of the VOCs released into headspace by infected B lymphoblastoid cells. The human influenza H1N1 virus strain was found to be the most efficient at infecting human B cells, although both avian viruses also infected the cells at higher multiplicity of infection (MOI). Importantly, both avian and human influenza subtypes provided adequate infection without inducing apoptosis, although a higher MOI was required for H9N2 and H6N2. For H1N1, replication was clearly present 24 h post infection at each tested MOI, with the extent of infection increasing with higher MOI. H9N2 and H6N2 did not infect C1R cells as effectively, but they did infect the human cell line successfully. With these strains, the first signs of infection were observed at 6–8 h post inoculation; loss of membrane integrity/morphological rupture did not occur until after 48 h of incubation at the highest MOI. We employed the higher MOI (10) for the avian strains, as this provided the most pronounced infection.
For each strain, 12 repeat experiments were conducted, with both 24 and 48h incubation (example, gas chromatogram in Figure 1). As medium composition can affect the VOCs produced by cells,[21–23] we confirmed that the volatiles did not originate from the medium (see the Experimental Section). A large amount of information is present in the chromatogram, with a mixture of both high-abundance and lower abundance trace chemicals (inset in Figure 1). Analysis of GC/MS data showed that infected and non-infected cells exhibited clear differences in their VOC profiles: a number of GC peaks were found to vary between infected and uninfected cells for the same incubation time. Distinct differences were also found between the three influenza subtypes. An example of a GC peak (identified as 2-methoxy-ethanol) is shown for three viruses (and at two MOIs for H1N1; Figure 2). This demonstrates production of an infection-specific compound, as (presumed) 2-methoxy-ethanol was absent in all control (uninfected) samples.
Figure 1.

GC/MS analysis of volatile organic compounds produced by C1R cells infected with H1N1 at MOI 10 and incubated for 24 h. A representative chromatogram from 12 replicates is shown. Inset: detail illustrates the high information content in the experimental data.
Figure 2.

Overlay of GC chromatograms differentiates between uninfected C1R cells and those infected as indicated and incubated for 48 h. Peak C1 (Tables 1, 2, S1, and S2) was identified as 2-methoxy-ethanol. Representative chromatograms of 12 replicates are shown. The p value p ≤ 0.05 was used throughout.
The VOC signature of infected cells also appeared to change with the time of infection. Several peaks were found at different levels at 24 and 48 h, and some compounds were present at 24 h incubation but absent at 48 h (Figure 3), or vice versa. For example, the compound identified as 3,7-dimethyloctan-3-ol was observed for the avian strains (H6N2, H9N2) at 48 h but not at 24 h. This compound was not observed at any time point for the human (H1N1) strain.
Figure 3.

Overlay of GC chromatograms shows abundance differences at different incubation times. C1R cells were infected and incubated as indicated. Peak C12 (Tables 1, 2, S1, and S2) was identified as 3,7-dimethyloctan-3-ol, and is evident after 48 h incubation with H9N2 and H6N2 strains, but essentially not present under all other conditions. Representative chromatograms of 12 replicates are shown. Appearance of peak C12 was consistent with observed morphological changes in cells only after 48 h incubation. The p value p≤ 0.05 was used throughout.
Compounds with statistically significant abundance differences in control and influenza-infected samples are shown for 24 and 48 h (see Table S1 and S2 in the Supporting Information). Peaks common to 24 and 48 h incubation times are given in Table 1, with suggested chemical identities for the peaks in Table 2 along with citations where the chemical was reported previously.
Table 1.
Averaged peak retention times and abundances for peaks that were common to both 24 and 48 hour infection samples, with distinguishable control and virus strains. (p < 0.05)
| Peak | Cntrl | MOI 1 H1N1 | MOI 10 H1N1 | MOI 10 H6N2 | MOI 10 H9N2 | Mutually distinguishable viral strains[a] |
Tentative compound (alternative(s)) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| tR | A. | tR | A. | tR | A. | tR | A. | tR | A. | |||
| C1 | 1.92 (0.12) |
10047 (3800) |
2.05 (0.04) |
18975 (5286) |
2.07 (0.01) |
34638 (7187) |
2.06 (0.01) |
53071 (6207) |
2.07 (0.00) |
27768 (4821) |
Cntrl/MOI 1 H1N1; Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2; MOI 10 H1N1/MOI 10 H6N2; MOI 10 H1N1/MOI 10 H9N2; MOI 10 H6N2/MOI 10 H9N2 | 2-methoxy-ethanol |
| C2 | – | – | 5.96 (0.09) |
109624 (136496) |
6.09 (0.07) |
15356 (13469) |
6.11 (0.09) |
15183 (8325) |
6.06 (0.09) |
14263 (4506) |
MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | thiirane |
| C3 | 12.62 (0.08) |
8601 (2233) |
12.62 (0.03) |
18629 (7844) |
12.71 (0.03) |
17975 (4401) |
12.69 (0.03) |
30312 (12957) |
12.71 (0.03) |
15104 (5906) |
Cntrl/MOI 1 H1N1; Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H6N2; MOI 10 H1N1/MOI 10 H6N2; MOI 10 H6N2/MOI 10 H9N2 | propanoic acid, ethyl ester |
| C4 | 19.71 (0.07) |
10925 (2735) |
19.75 (0.05) |
11214 (1939) |
19.79 (0.09) |
8924 (3672) |
19.85 (0.07) |
8271 (3269) |
19.87 (0.04) |
7127 (1269) |
Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | butanoic acid, 2-methyl-, methyl ester |
| C5 | 30.13 (0.04) |
10292 (3091) |
30.11 (0.06) |
18579 (7496) |
30.26 (0.04) |
13955 (3882) |
30.25 (0.04) |
23767 (13960) |
30.26 (0.04) |
10047 (2748) |
Cntrl/MOI 1 H1N1; Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2; MOI 10 H1N1/MOI 10 H6N2; MOI 10 H1N1/MOI 10 H9N2; MOI 10 H6N2/MOI 10 H9N2 | butanoic acid, 2-methyl-, ethyl ester |
| C6 | – | – | 35.43 (0.06) |
9945 (1756) |
35.59 (0.03) |
6493 (772) |
35.59 (0.04) |
7526 (2329) |
35.58 (0.04) |
6829 (1452) |
MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | hexan-3-one, 5-methyl- |
| C7 | – | – | 36.06 (0.11) |
7882 (842) |
36.25 (0.05) |
6463 (778) |
36.27 (0.05) |
6569 (1613) |
36.24 (0.05) |
5955 (528) |
MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | heptan-3-one |
| C8 | 48.10 (0.08) |
10895 (5148) |
48.10 (0.09) |
9703 (2285) |
48.16 (0.05) |
13743 (6077) |
48.17 (0.07) |
13705 (4113) |
48.15 (0.04) |
13617 (3724) |
MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | octan-2-one |
| C9 | 67.17 (0.05) |
22904 (3975) |
67.20 (0.06) |
22780 (3323) |
67.34 (0.04) |
14834 (4705) |
67.35 (0.04) |
14875 (3667) |
67.33 (0.04) |
13788 (2653) |
Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | not identified |
| C10 | 68.59 (0.06) |
18440 (4064) |
68.59 (0.06) |
17319 (3374) |
68.76 (0.04) |
11818 (3835) |
68.77 (0.05) |
11820 (2142) |
68.76 (0.03) |
11314 (2118) |
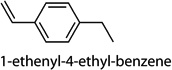
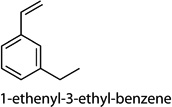
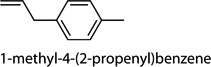
Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | 1-phenylbut-1-ene (1-ethenyl-4-ethyl-ben-zene, 1-ethenyl-3-ethyl-benzene, 1-methyl-4-(prop-2-enyl)-benzene) |
| C11 | 70.92 (0.06) |
9138 (2726) |
70.92 (0.07) |
8959 (1387) |
71.11 (0.03) |
7200 (1726) |
71.12 (0.04) |
7978 (1823) |
71.11 (0.05) |
7729 (1855) |
Cntrl/MOI 10 H1N1; MOI 1 H1N1/ MOI 10 H1N1 | 1-phenylbut-1-ene (1-ethenyl-4-ethyl-ben-zene, 1-ethenyl-3-ethyl-benzene, 1-methyl-4-(prop-2-enyl)-benzene) |
| C12 | 78.22 (0.07) |
133058 (594423) |
78.23 (0.07) |
1231871 (331605) |
78.41 (0.03) |
541882 (84018) |
78.41 (0.03) |
607416 (206791) |
78.40 (0.03) |
631543 (202320) |
Cntrl/MOI 10 H1N1; Cntrl/MOI 10 H6N2; Cntrl/MOI 10 H9N2; MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | 3,7-dimethyloctan-3-ol |
| C13 | 79.81 (0.06) |
15112 (4098) |
79.82 (0.07) |
13800 (2201) |
80.02 (0.04) |
11344 (8837) |
80.02 (0.03) |
9559 (1669) |
80.00 (0.03) |
9119 (1420) |
MOI 1 H1N1/MOI 10 H1N1; MOI 1 H1N1/MOI 10 H6N2; MOI 1 H1N1/MOI 10 H9N2 | 4-ethylbenzaldehyde |
| C14 | 88.63 (0.07) |
15356 (23190) |
88.61 (0.04) |
7988 (2512) |
88.78 (0.14) |
28348 (15442) |
88.82 (0.10) |
23635 (13835) |
88.87 (0.04) |
18743 (6348) |
MOI 1 H1N1/MOI 10 H1N1; MOI1 H1N1/MOI 10 H6N2; MOI1 H1N1/ MOI 10 H9N2 | decanal (isomers, homologues) |
tR: retention time [min]; A: abundance (total ion count); standard deviation in parentheses.
Student’s t-test (p ≤ 0.05).
Table 2.
Compound assignment and references for peaks listed in Table 1.
| Peak | Proposed structure(s) | Ref. | Biological status |
|---|---|---|---|
| c1[a] | 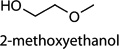 |
[48] | exhaled breath |
| c2 | [49] | bacterial activity | |
| c3 | 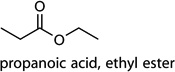 |
[50,51] | microbial activity; exposure |
| c4 | 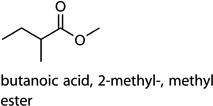 |
[52,53] | bacterial activity |
| c5 | 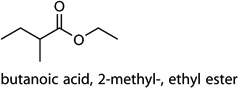 |
[54,55] | bacterial activity |
| c6[a] | 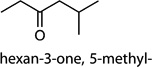 |
[54–57] | bacterial activity; malignancy |
| c7[a] | 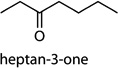 |
[55,58,59] | bacterial activity; malignancy; tuberculosis |
| c8[a] | 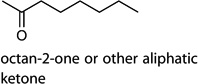 |
[7] | malignancy |
| c9 | not identified | ||
| c10[a] c11[a] |
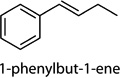 |
[60] | malignancy |
 |
|||
 |
|||
 |
|||
| c12[a] | 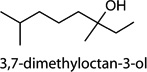 |
[28] | influenza |
| c13 | 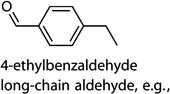 |
||
| c14[a] |
Tentative structural assignment; most-likely structures are shown.
Discussion
Infection of the influenza virus in mammalian cells is a multistep process. It involves the virus binding to and entering the cell, followed by delivery of its genome to the nucleus and subsequent production of viral proteins and new copies of its RNA, and assembly of these components into new viral particles to exit the host cell.[2] The biochemistry of the infection process is quite complex and involves multiple chemical reactions in the host cell. During the infection process, the viral envelope fuses with a vacuole membrane, and viral RNA replication takes place in the nucleus. The viral RNA and nucleoprotein (NP) are released into the cytoplasm for the RNA-dependent RNA transcription of the complementary positive-sense cRNA. The fusion is achieved by influx of protons into the viral envelope and decreasing pH due to acidic conditions in the endosome.[2] The biochemical transformations accompanying infection result in specific changes, such as altered protein synthesis. Even minor alterations to a cell’s genome can lead to differences in its VOC production.[13] In the case of cell infection, many signal transduction and protein expression pathways are affected simultaneously, so an even larger downstream VOC production effect is expected from this cascade. In the multiple steps of virus proliferation, many small molecules are involved as cofactors, reactants, and as products or side products of these processes. A number of these compounds can cross the cell membrane and thus can be detected in the headspace of infected cell culture. This is consistent with our experimental observations in the present work.
We postulate that the VOCs measured with our cell culture model reflect global metabolic changes in the cell. Such VOC changes are not necessarily limited to viral infection and might be present in other cases (e.g., bacterial infection or malignancy, see Table 2). A possible reason for VOC change is induction of apoptotic pathways. However, we carefully controlled for cell viability in our experiments, and we saw no evidence of cell death over the reported times (beyond the timeline of our experiments, cell death might occur).
We selected a B-lymphoblastoid model to reduce the influence of respiratory cell apoptosis on VOC production. Thus the cells emit a specific and unique odorprint VOC pattern that corresponds to alterations of cellular pathways due to the specific infection process. As a result of such alterations, the intraand extracellular levels of certain chemicals fluctuates throughout the infection process, and might indicate the stage along the infection timeline. The data presented in Tables S1 and S2 indeed support this: a number of peaks disappear or appear between the two infection times, or they change in abundance. The peaks observed throughout the infection (i.e., common to 24 and 48 h; Tables S1 and S2), are of greater importance, as the corresponding compounds can be linked to the specific viral infection more directly. Thus, the compounds listed in Table 2 can be considered as biomarkers for specific influenza strains (note: structural assignments in Table 2 are putative).
Our data show that in the virus-induced VOC profiles, some compounds appeared upon infection with all strains, whereas others were specific to one virus strain (Tables 1, S1, and S2, and Figures 2 and 3). A number of volatiles were observed exclusively for infected cells, whereas others were detected at different abundances between uninfected and infected cells, and/or between different virus strains. Indeed, infection with human and avian strains of influenza is known to differ at the cellular level. Because of differences in the types of glycoprotein and glycolipid composition of sialic acid receptors in cells of different species (such as humans and birds), the influenza A virus subtypes preferentially bind to specific host cells. This is most notable in cells of the respiratory epithelium, but can be seen in other cell types as well. The human influenza A virus preferentially binds to sialyloligosaccharides containing N-acetylneuraminic acid a2,6-galactose (NeuAc α2,6 Gal) by a2,6-galactose linkage, whereas avian influenza viruses (e.g., H9N2 and H6N2) preferentially bind by α2,3-galactose linkage to oligosaccharides containing N-acetylneuraminic acid and N-glycolylneuraminic acid α2,3-galactose, respectively.[24–26] The influenza virus HA glycoprotein was shown to be viral lectin with lymphocyte-activating properties.[27]
Viral replication fitness within host cells can also vary, with avian strains replicating and altering cellular metabolism to varying degrees. This was clearly seen in our replication studies: H9N2 and H6N2 had lower replication fitness than (human-specific) H1N1. Finally, some avian strains can induce apoptosis in human cells, rather than increasing viral replication (commonly seen with H9N2). As each subtype produces a unique VOC, this can lead to important rapid diagnostic possibilities. However, the origin and predictability of the VOCs are yet to be established in humans.
Further study will be needed to determine the specific biochemical origins of VOCs differentially expressed for different virus strains in a systemic response during the infection. Identifying and detecting these chemicals will require analysis of several host cell lines, including respiratory epithelial cells and antigen-presenting cells other than B-cells. Ultimately, in vitro co-culture models and and in vivo human studies might allow determination of biomarkers specific to viral infection, specific virus identification, and time of infection.
A limitation of our model is that MOI 10 (used with the avian viruses) is extremely high and not necessarily physiologically relevant. As the goal of this study was to explore a general concept for detecting changes in cellular volatiles, we employed a model system that is based on physiological processes but with enhanced detection of biomarkers. The use of high MOI ensures a high infection rate (almost complete infection of cells, although not biologically realistic). This high infection rate results in more discernible VOC signatures of infection, which in turn might allow detection of low abundance biomarkers that would be difficult to detect otherwise. This might lead to the development of sensors with ultra-sensitive detection limits, specific for low-abundant compounds and VOCs unique to individual influenza strains. Studies to explore volatile production at lower MOI are warranted. This will allow establishing whether there are qualitative changes in cell VOC production at different viral loads, and whether these changes have phenomenological significance. Experiments at physiological MOI values would be very interesting, but are extremely challenging because of the expected low VOC production.
VOC differences with different viral subtypes will yield information on varying metabolic conditions, from viral replication fitness to specific cellular defense mechanisms. The majority of the tentatively identified compounds have been previously observed, sometimes linked to infection (not necessarily viral), or to malignancy (Table 2). Further examination of the compounds (Tables 2, S1, and S2) reveals striking structural similarities. For instance, a majority of the discovered biomarkers are homologous esters or other carbonyl compounds. The formation of structurally related compounds might point to a common origin in certain biological pathways associated with cell infection. In other words, we might be observing “snapshots” at various stages of pathways that are substantially altered by infection. However, as the identity of reported biomarkers is only tentative, no specific conclusions regarding the metabolic pathways can be made.
Interestingly, there is a substantial degree of overlap between our data and VOC biomarkers reported in a clinical study to assess the effect of live attenuated influenza vaccination (LAIV) on volatile products in human exhaled breath.[28] We believe this provides further evidence of phenomenological significance of the observed VOCs in our cell culture infection model: specifically, that influenza infection engenders production of a specific pattern of VOCs. The authors of the LAIV study reported an immediate and sustained increase in breath biomarkers, such as alkane derivatives. In our study, we also observed structurally very similar alkane and arene derivatives. The appearance of the alkane derivative compounds in our model was expected, based on the results of previous studies,[28, 29] as infection with influenza results in increased oxidative stress.[30] This, in turn, might lead to increased excretion of VOC biomarkers in breath, including alkanes and methylated alkanes.[31–33] The hydrocarbons are believed to be produced as a result of free radical oxidative fragmentation of lipids.[33] Given the great variety of lipid compounds and the possibility of multiple affected reaction pathways (because of the nonspecific nature of oxidation and other processes) in free radical reactions, it is very likely that a unique distribution of reaction products would be formed for each case. It is also likely that observed chemical structures of lipids oxidative degradation products will vary to some extent among different studies. Elevated excretion of hydrocarbons might be associated with other processes such as malignancy, specifically in lung cancer.[7] The observation of structurally related compounds in our study is certainly encouraging.
However, in contrast to the LAIV study,[28] the compounds observed in our study (Tables 2, S1, and S2) are predominantly aliphatic alcohols and carbonyls, specifically homologous esters. A LAIV was cold adapted (to 25°C) and designed for viral replication in the cooler temperature of the nasopharynx.[34] The VOCs induced in that case[28] might not fully represent the VOC production seen in other upper respiratory tract infections with non-attenuated native viruses. The production of oxidized compounds such as alcohols and carbonyls found in this study are consistent with induced oxidative stress and low intracellular pH conditions reported during viral infection. In a mechanism described for influenza viral pneumonia, free radicals are produced by reactions of various intracellular molecules with oxygen radicals and reactive oxygen intermediates.[35] These free radicals, such as nitrogen oxide species (e.g., peroxynitrite), are powerful oxidizing agents and will inevitably lead to increased levels of oxygenated compounds. It has been reported that compounds such as aldehydes, ketones, and organic acids found in human breath[36, 37] and body fluids might signal the presence of radical oxygen species caused by infections and metabolic disorders.[38] Increased aldehyde excretion as a result of lipid peroxidation during a pathological process of cancer has been described.[7] In fact, aldehydes are postulated to be secondary messengers in signal transduction events during such processes.[39] Esters have not been commonly reported as oxidation stress biomarkers. In humans, specific enzymes (esterases) hydrolyze esters into alcohol and acid below 408C. As the rate of hydrolysis is extremely high, the observation of esters in breath or other body fluid would be challenging. Therefore such biomarkers could only be detected in model systems such as single cell cultures, as in this study. This underscores the potential of such model systems as a link between a mechanistic understanding of underlying biological processes and real-world clinical applications.
The greatest value of studying VOC production lies in the potential of defined biomarkers for rapid monitoring of infection, provided biomarkers have been established across various cell model systems during infection. The recent outbreaks of avian and swine flu have demonstrated the need for fast and non-invasive tools for viral infection monitoring. This is particularly necessary when both seasonal and pandemic strains are mixing as noted in the 2009 swine flu outbreak.[5, 6] Association of certain VOCs with the timeline of the infection could significantly enhance diagnosis as well as aid in determining treatment options, particularly with multiple circulating strains of influenza and with variable antiviral resistance (seen in H1N1).[5, 6] This timeline might also assist government health agencies in identifying regional infection outbreaks, and provide important forensic information about the spread of infection.
Significant obstacles remain before such detection is viable on large scale, or even in a laboratory setting. The complexity of VOCs detection at the organism level coupled with the low abundance of chemicals of interest poses a formidable challenge for biomarker detection, even if the biomarkers are known. Development of suitable bed-side analytical instrumentation tailored for volatile biomarker detection in breath (or other body emanation) is necessary to advance the field. Another important development would be a concerted study of metabolomics profiling and infection pathways, if links between the two can be established. Cataloging VOCs that are specific to certain types of infection and pathogen (as opposed to those produced in response to all pathogens), might allow better elucidation of the biochemical origin of these compounds. Studies of influenza and other infections (not only viral) in murine models will help establish whether the biomarkers determined in cell-line studies are discernible at the organism level. For cellular studies, further advances are also needed. In particular, baseline VOCs will need to be established for each cell type that is affected during the infection process, to determine how VOCs differ during infection. For influenza, such cell lines would be nasopharyngeal cells, bronchial epithelial cells, alveolar pneumocytes, and a range of other innate and adaptive immune cells. Furthermore, VOC baselines would be helpful for co-culture systems that mimic the functionality of tissues and organs. Such studies will allow expanding and cross-verifying the panels of infection-specific biomarkers, and will provide a rational platform for animal and human clinical studies. Ultimately, it is important to bridge the gap between in vivo systems (where complex cellular immunological interactions take place) and single-culture models. Although the simple approach of single cell-line study does not reproduce all virus–cell and cell–cell interactions in a complex biological system, it is still very useful to understand isolated aspects.
Conclusions
This study lays the foundation for single cell-line biomarker libraries for influenza A viral infection of B cells. Our results from a model single cell-line system in conjunction with published data suggest that alkanes and oxygenated compounds such as esters can be considered as biomarkers of influenza A infection. Their increased production might have diagnostic value. Some of these biomarkers are consistent with the general oxidative stress associated with viral infection. Based on the tentative identification of the compounds in this study, future studies are needed to understand and compile biomarker lists, including for other cell lines and viruses. Additional studies are needed to establish the diagnostic benefit of these compounds. The data from our model is in agreement with those of the LAIV study, and we predict that breath biomarkers from different virus infections will be observed in well-controlled studies. An important implication of this work and similar studies is the generation of a list of relevant biomarkers of influenza infection. This will inform and advance future clinical studies, by allowing a targeted rather than untargeted search for biomarkers, and this will greatly increase the chance of success.
Experimental Section
Inoculation of B-lymphoblastoid cells with influenza virus strains and assessment of inoculation efficiency
We used the human B-lymphoblastoid cell line “C1R”, which is HLA class I HLA-A and HLA-B.[40] The cells were a generous gift from Dr. Peter Cresswell (Yale University, New Haven, CT). The C1R cell lines are available from commercial sources, for example, CR1-neo (ATCC CRL-2369; [41]).
C1R cells were infected with three influenza viruses: avian H9N2 and H6N2, and human H1N1.[42] The following virus strains were used (available from commercial sources, e.g., ref. [43]): H1N1A/ Puerto Rico/8/1934; H6N2A/chicken/CA/1772/2002; H9N2A/pheasant/CA/2373/1998 (http://www.atcc.org/Products/CellsandMicroorganisms/Viruses/Influenza_Research_Materials.aspx). Avian and human virus isolates were passaged by inoculation into the allantoic cavity of 9-day-old specific pathogen free (SPF) embryonated chicken eggs (SPAFAS, Charles River) and incubating at 37°C for 48 h, followed by overnight incubation at 4°C. Allantoic fluid samples were harvested, batched, and centrifuged (8000g, 15 min at 4°C). Supernatants were aliquoted, snap-frozen on dry ice and stored at − 80°C. Allantoic fluid samples were also tested for hemagglutination by using 0.5% chicken blood (Colorado Serum Company, Denver, CO),[42] and infectious virus titers were determined by a standard plaque assay by MDCK cell titration.[44]
Cells were infected with influenza virus at different MOI in serumfree RPMI 1640 medium containing l-[(toluene-4-sulfonamido)-2-phenyl]ethylchloromethylketone (TPCK) trypsin (1 µgmL−1) and incubated at 37°C with 5% CO2 (three culture vials, 24 and 48 h).
Inoculation efficiency was determined by infecting C1R cells at three MOIs (1, 5 and 10). Cells were seeded in 12-well plates (~ 800000 cells per well) and incubated with 0.5 mL of virus (at MOI 0, 1, 5, and 10); virus absorption was carried out for 7 h in order to ensure infection. After replacing the virus inoculum with fresh RPMI 1640 medium, cells were incubated for a further 24 or 48 h under standard cell-culture conditions. Then, microscopic cell examination, Trypan Blue exclusion, and hemagglutination assay of the cell-culture supernatant were used to confirm viral proliferation.[45] Inoculation with MOI 10 was employed in studies for VOC detection with the two avian strains, because this yielded the most pronounced infection (see Results). MOI 1 and 10 were used for H1N1
Headspace sampling of cell cultures
We have previously reported our method for measuring VOC production from mammalian cell cultures in suspension.[13] We altered this method to allow headspace VOC measures both pre- and post-infection of B lymphoblastoid cells, and this is briefly described here. For headspace volatiles collection, C1R cells were infected with H9N2, H6N2 (MOI 10) or H1N1 (MOI 10 and MOI 1). The same method was followed for all viral strains (Figure 4). Prior to inoculation, cell viability and cell concentration were assessed with a Trypan Blue exclusion assay at the beginning of each experiment. Cells (400000 in 0.5 mL of RPMI 1640 medium with FBS (10%) and gentamicin (50 µgmL−1) were placed in uncapped 10 mL Supelco clear glass headspace vials (Sigma–Aldrich); 12 replicate vials were prepared for each experiment; vials were tightly sealed with rubber caps, and cells were incubated overnight in a cell culture incubator (37°C, 5% CO2). Cells in nine vials were then infected with virus for 1 h (n = 3 H9N2, n = 3 H6N2, n=3 H1N1; MOI 10). The three remaining vials were treated identically but without virus in the added medium (controls). The cells were then re-suspended in serum and antibiotic-free RPMI 1640 medium. The cell vials were then capped, and the tightly sealed vials were further incubated for either 24 or 48 h (separate experiments) at 37°C on a gently rotating shaker (130 rpm). Vials were then transferred to a 4°C chill tray on a GC/MS instrument for VOC analysis. The procedure was repeated 4 times (total: 12 replicates for each virus strain at MOI 10, for the two incubation times). In addition, an independent experiment with virus strain H1N1 was performed without a control as the control chromatograms were similar and highly conserved from the previous experiments.
Figure 4.

Work flow: C1R cells were placed into 12 vials and incubated. Vials were infected with influenza virus (n = 9 total, gray; n = 3 H9N2, n = 3 H6N2, and n = 3 H1N1, MOI 10) or untreated (controls; n = 3, white). After re-suspension in medium and further incubation, vials were removed at either 24 h (n = 9 gray and n = 3 white) or 48 h (n = 9 gray and n = 3 white). All vials underwent the same VOC sampling. All experiments were repeated four times. The H1N1 experiment at MOI 1 was conducted independently (n = 12 at 24-hours, n = 12 at 48-hours). Totals: virus-infected 96; controls 24; VOC chromatograms 120.
GC/MS analysis was performed as previously described.[13] The equipment comprised a model CP-3800 GC (VF 5 ms; Varian), with a phenol (5%)/polydimethylsiloxane (PDMS, 95%) column, and a model 4000 Ion Trap MS (Varian) equipped with an EI source. For sampling, the solid-phase microextraction (SPME) fiber was inserted into the GC inlet and the adsorbed headspace chemicals were desorbed for 6 min 50 s at 240°C. The GC cycle was as follows: 1 °Cmin−1 (5°C to 50 °C to 75 °C to 100°C to 125°C to 140°C) then 10°Cmin−1 to 200 °C, with 5 min holds at 75 °C, 100°C, 125°C, 140°C, and 200°C for a complete run time of 176 min. The mass range scanned was 0–1000 Th. The headspace was automatically sampled by using a Supelco gray hub divinylbenzene/carboxen/ polydimethylsiloxane (DVB/CAR/PDMS) SPME fiber (Sigma–Aldrich). To minimize sampling errors, the virus-infected and control (uninfected) vials were alternated. In total, 120 GC/MS profiles were collected: 12 replicates for control and for each of the three virus strains at MOI 10, and additional 12 replicates for infection with H1N1 at MOI 1, each at 24 and 48 h post-infection (Figure 4). Peak detection and matching was carried out as previously described,[13] by locating the peaks in GC profiles that belong to the same compound, as verified by MS spectra matching. Peaks that were significantly different among compared groups (infected versus control, different infection times, different virus strains) were determined by a Student’s t-test (p = 0.05). We also collected VOC data by the same procedure from RPMI 1640 medium and allantoic fluid, and confirmed that none of the peaks had originated from these sources. Both allantoic fluid and medium introduce negligible amounts of volatiles (virus particles do not produce volatile compounds in isolation).[46] The RPMI 1640 formulation is given in.[47] The peaks with statistically different abundances among different groups were recorded, and compounds corresponding to these peaks were identified by using spectra matching with NIST Mass Spectral Search Software v. 2.0 with NIST 2005 and Wiley 2009 MS libraries. Peaks with unacceptably low S/N ratio, SPME/septa/column bleed peaks (siloxanes) and environmental contaminants (e.g., phthalates) were excluded. The matches with probability ≥80% were assumed to be the compounds in question; otherwise, the matching results were verified, and the most likely candidate compounds were suggested as a tentative identification.
Supplementary Material
Acknowledgements
This work was partially supported by Defense Advanced Research Projects Agency (DARPA), the Army Research Office [W911NF-06-1-0272], and The Hartwell Foundation, the National Institutes of Health [#T32-HL007013] and [#T32-ES007059], UC Davis School of Medicine and NIH #8KL2TR000134-07K12 mentored training award. Partial support was also provided by the National Center for Advancing Translational Sciences (NCATS) of the NIH, through grant #UL1 TR000002.
References
- 1.Adams S, Sandrock C. Med. Princ. Pract. 2010;19:421–432. doi: 10.1159/000320299. [DOI] [PubMed] [Google Scholar]
- 2.Bouvier NM, Palese P. Vaccine. 2008;26:D49–D53. doi: 10.1016/j.vaccine.2008.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cardona CJ, Xing Z, Sandrock CE, Davis CE. Comp. Immunol. Microbiol. Infect. Dis. 2009;32:255–273. doi: 10.1016/j.cimid.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 4.Sandrock C. Comp. Immunol. Microbiol. Infect. Dis. 2009;32:253–254. doi: 10.1016/j.cimid.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Uyeki TM. N. Engl. J. Med. 2010;362:2221–2223. doi: 10.1056/NEJMe1004468. [DOI] [PubMed] [Google Scholar]
- 6.SteelFisher GK, Blendon RJ, Bekheit MM, Lubell K. N. Engl. J. Med. 2010;362:65. doi: 10.1056/NEJMp1005102. [DOI] [PubMed] [Google Scholar]
- 7.Hakim M, Broza YY, Barash O, Peled N, Phillips M, Amann A, Haick H. Chem. Rev. 2012;112:5949–5966. doi: 10.1021/cr300174a. [DOI] [PubMed] [Google Scholar]
- 8.Filipiak W, Sponring A, Baur MM, Filipiak A, Ager C, Wiesenhofer H, Nagl M, Troppmair J, Amann A. BMC Microbiol. 2012;12:113. doi: 10.1186/1471-2180-12-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu J, Bean HD, Wargo MJ, Leclair LW, E Hill J. J. Breath Res. 2013;7:016003. doi: 10.1088/1752-7155/7/1/016003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chambers ST, Bhandari S, Scott-Thomas A, Syhre M. Med. Mycol. 2011;49:S54–S61. doi: 10.3109/13693786.2010.508187. [DOI] [PubMed] [Google Scholar]
- 11.Erhart S, Amann A, Haberlandt E, Edlinger G, Schmid A, Filipiak W, Schwarz K, Mochalski P, Rostasy K, Karall D, Scholl-Burgi S. J. Breath Res. 2009;3:016004. doi: 10.1088/1752-7155/3/1/016004. [DOI] [PubMed] [Google Scholar]
- 12.Filipiak W, Sponring A, Baur MM, Ager C, A Filpiak, Weisenhofer H, Nagl M, Troppmair J, Amann A. Microbiology-Sgm. 2012;158:3044–3053. doi: 10.1099/mic.0.062687-0. [DOI] [PubMed] [Google Scholar]
- 13.Aksenov AA, Gojova A, Zhao W, Morgan JT, Sankaran S, Sandrock CE, Davis CE. ChemBioChem. 2012;13:1053–1059. doi: 10.1002/cbic.201200011. [DOI] [PubMed] [Google Scholar]
- 14.Waffarn EE, Baumgarth N. J. Immunol. 2011;186:3823–3829. doi: 10.4049/jimmunol.1002090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumgarth N, Choi YS, Rotheausler K, Yang Y, Herzenberg LA. In: Curr. Top. Microbiol. Immunol. Manser T, editor. Vol. 319. Berlin: Springer; 2008. pp. 41–61. [DOI] [PubMed] [Google Scholar]
- 16.Choi YS, Baumgarth N. J. Exp. Med. 2008;205:3053–3064. doi: 10.1084/jem.20080979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Priest SO, Baumgarth N. Front. Biosci. 2013;S5:105–117. doi: 10.2741/s360. [DOI] [PubMed] [Google Scholar]
- 18.Takahashi Y, Onodera T, Kobayashi K, Kurosaki T. Infect. Disord. Drug Targets. 2012;12:232–240. doi: 10.2174/187152612800564400. [DOI] [PubMed] [Google Scholar]
- 19.Xing Z, Harper R, Anunciacion J, Yang Z, Gao W, Qu B, Guan Y, Cardona CJ. Am. J. Respir. Cell Mol. Biol. 2011;44:24–33. doi: 10.1165/rcmb.2009-0120OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stollenwerk N, Harper RW, Sandrock CE. Crit. Care. 2008;12:219. doi: 10.1186/cc6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barash O, Peled N, Hirsch FR, Haick H. Small. 2009;5:2618–2624. doi: 10.1002/smll.200900937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barash O, Peled N, Tisch U, Bunn PA, Hirsch FR, Haick H. Nanomedicine. 2012;8:580–589. doi: 10.1016/j.nano.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peled N, Barash O, Tisch U, Ionescu R, Broza YY, Ilouze M, Mattei J, Bunn PA, Jr, Hirsch FR, Haick H. Nanomedicine. 2013;9:758–766. doi: 10.1016/j.nano.2013.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gambaryan A, Yamnikova S, Lvov D, Tuzikov A, Chinarev A, Pazynina G, Webster R, Matrosovich M, Bovin N. Virology. 2005;334:276–283. doi: 10.1016/j.virol.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 25.Rogers GN, D’Souza BL. Virology. 1989;173:317–322. doi: 10.1016/0042-6822(89)90249-3. [DOI] [PubMed] [Google Scholar]
- 26.Sriwilaijaroen N, Kondo S, Yagi H, Wilairat P, Hiramatsu H, Ito M, Ito Y, Kato K, Suzuki Y. Glycoconjugate J. 2009;26:433–443. doi: 10.1007/s10719-008-9193-x. [DOI] [PubMed] [Google Scholar]
- 27.Rott O, Charreire J, Cash E. Med. Microbiol. Immunol. 1996;184:185–193. doi: 10.1007/BF02456134. [DOI] [PubMed] [Google Scholar]
- 28.Phillips M, Cataneo RN, Saunders C, Hope P, Schmitt P, Wai J. J. Breath Res. 2010;4:026003. doi: 10.1088/1752-7155/4/2/026003. [DOI] [PubMed] [Google Scholar]
- 29.Phillips M, Herrera J, Krishnan S, Zain M, Greenberg J, Cataneo RN. J. Chromatogr. B. 1999;729:75–88. doi: 10.1016/s0378-4347(99)00127-9. [DOI] [PubMed] [Google Scholar]
- 30.Schwarz KB. Free Radical Biol. Med. 1996;21:641–649. doi: 10.1016/0891-5849(96)00131-1. [DOI] [PubMed] [Google Scholar]
- 31.Phillips M, Cataneo RN, Cummin ARC, Gagliardi AJ, Gleeson K, Greenberg J, Maxfield RA, Rom WN. Chest. 2003;123:2115–2123. doi: 10.1378/chest.123.6.2115. [DOI] [PubMed] [Google Scholar]
- 32.Phillips M, Cataneo RN, Greenberg J, Grodman R, Gunawardena R, Naidu A. Eur. Respir. J. 2003;21:48–51. doi: 10.1183/09031936.02.00053402. [DOI] [PubMed] [Google Scholar]
- 33.Phillips M, Cataneo RN, Greenberg J, Gunawardena R, Naidu A, Rahbari-Oskoui F. J. Lab. Clin. Med. 2000;136:243–249. doi: 10.1067/mlc.2000.108943. [DOI] [PubMed] [Google Scholar]
- 34.Maassab HF, Bryant ML. Rev. Med. Virol. 1999;9:237–244. doi: 10.1002/(sici)1099-1654(199910/12)9:4<237::aid-rmv252>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 35.Akaike T. Rev. Med. Virol. 2001;11:87–101. doi: 10.1002/rmv.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalton P, Gelperin A, Preti G. Diabetes Technol. Ther. 2004;6:534–544. doi: 10.1089/1520915041705992. [DOI] [PubMed] [Google Scholar]
- 37.Preti G, Clark L, Cowart BJ, Feldman RS, Lowry LD, Weber E, Young IM. J. Periodontol. 1992;63:790–796. doi: 10.1902/jop.1992.63.9.790. [DOI] [PubMed] [Google Scholar]
- 38.Castro L, Freeman BA. Nutrition. 2001;17:161–165. doi: 10.1016/s0899-9007(00)00570-0. [DOI] [PubMed] [Google Scholar]
- 39.Forman HJ. Ann. N. Y. Acad. Sci. 2010;1203:35–44. doi: 10.1111/j.1749-6632.2010.05551.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Storkus WJ, Alexander J, Payne JA, Dawson JR, Cresswell P. Proc. Natl. Acad. Sci. USA. 1989;86:2361–2364. doi: 10.1073/pnas.86.7.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. http://www.atcc.org/products/all/CRL-2369.aspx. [Google Scholar]
- 42.Xing Z, Cardona CJ, Li J, Dao N, Tran T, Andrada J. J. Gen. Virol. 2008;89:1288–1299. doi: 10.1099/vir.0.83362-0. [DOI] [PubMed] [Google Scholar]
- 43. http://www.atcc.org/products/all/VR-95.aspx. [Google Scholar]
- 44.Hirst GK. J. Exp. Med. 1942;75:49–64. doi: 10.1084/jem.75.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Curr. Protoc. Immunol. 1997:A.3B.1–A.3B.2. [Google Scholar]
- 46.Schivo M, Aksenov AA, Linderholm A, Passamontes A, Peirano DJ, Harper RW, Davis CE. A45 Diagnostic Techniques, Monitoring and Technology. American Thoracic Society International Conference; 2013. p. A1552. [Google Scholar]
- 47. http://www.sigmaaldrich.com/life-science/cell-culture/learning-center/media-formulations/rpmi-1640.html. [Google Scholar]
- 48.Ligor T, Ligor M, Amann A, Ager C, Bachler M, Dzien A, Buszewski B. J. Breath Res. 2008;2:046006. doi: 10.1088/1752-7155/2/4/046006. [DOI] [PubMed] [Google Scholar]
- 49.Intarapichet K, Bailey ME. Thai J. Agric. Sci. 1992;25:299–326. [Google Scholar]
- 50.Rasanen I, Viinamäki J, Vuori E, Ojanperä I. J. Anal. Toxicol. 2010;34:113–121. doi: 10.1093/jat/34.3.113. [DOI] [PubMed] [Google Scholar]
- 51.Matysik S, Herbarth O, Mueller A. Chemosphere. 2009;76:114–119. doi: 10.1016/j.chemosphere.2009.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Trevejo JM, Hoenigman S, Kirby J. Rapid Detection of Volatile Organic Compounds for Identification of Bacteria in a Sample. The Charles Stark Draper Laboratory, Inc., USA, Beth Israel Deaconess Medical Center; 2009. p. 114. WO 2009054913 A1 20090430. [Google Scholar]
- 53.McNerney R, Turner C. Volatile Organic Compounds as Markers for Presence of Mycobacteria. London School of Hygiene & Tropical Medicine, UK, Cranfield University; 2009. p. 14. WO 2009037492 A2 20090326. [Google Scholar]
- 54.Hettinga KA, van Valenberg HJF, Lam TJGMJ, van Hooijdonk ACM. Dairy Sci. 2008;91:3834–3839. doi: 10.3168/jds.2007-0941. [DOI] [PubMed] [Google Scholar]
- 55.Roze LV, Chanda A, Laivenieks M, Beaudry RM, Artymovich KA, Koptina AV, Awad DW, Valeeva D, Jones AD, E Linz J. BMC Bio-chem. 2010;11:33. doi: 10.1186/1471-2091-11-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Citron CA, Rabe P, Dickschat JS. J. Nat. Prod. 2012;75:1765–1776. doi: 10.1021/np300468h. [DOI] [PubMed] [Google Scholar]
- 57.Hakim M, Billan S, Tisch U, Peng G, Dvrokind I, Marom O, AbdahBortnyak R, Kuten A, Haick H. Br. J. Cancer. 2011;104:1649–1655. doi: 10.1038/bjc.2011.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Matsumura K, Matsumura K, Opiekun M, Oka H, Vachani A, Alelda SM, Yamazaki K, Beauchamp GK. PLoS One. 2010;5:e8819. doi: 10.1371/journal.pone.0008819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Phillips M, Cataneo RN, Condos R, Ring Erickson GA, Greenberg J, La Bombardi V, Munawar MI, Tietje O. Tuberculosis. 2007;87:44–52. doi: 10.1016/j.tube.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 60.Ligor M, Ligor T, Bajtarevic A, Ager C, Pienz M, Klieber M, Denz H, Fiegl M, Hilbe W, Weiss W, Lukas P, Jamnig H, Hackl M, Buszewski B, Miekisch W, Schubert J. A. Amann Clin. Chem. Lab. Med. 2009;47:550–560. doi: 10.1515/CCLM.2009.133. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.