

MicroRNAs (miRNAs) are short non-coding RNAs that mediate protein translation and are known to be dysregulated in diseases including diabetes, Alzheimer’s, and cancer.[1–3] With greater stability and predictive value than mRNA, this relatively small class of biomolecules has become increasingly important in determining disease diagnosis and prognosis. However, the sequence homology, wide range of abundance, and common secondary structures of miRNAs have complicated efforts to develop accurate, unbiased quantification techniques.[4,5] Applications in the discovery and clinical fields require high-throughput processing, large coding libraries for multiplexed analysis, and the flexibility to develop custom assays. Microarray approaches provide high sensitivity and multiplexing capacity, but their low-throughput, complexity, and fixed design make them less than ideal for use in a clinical setting.[6,7] PCR-based strategies suffer from similar throughput issues, yet offer highly sensitive and specific detection for genome-wide miRNA expression profiling.[8] Alternative bead-based systems provide a high sample throughput, but with reduced sensitivity,[9] dynamic range, and multiplexing capacities (luminexcorp.com). miRNA profiling by deep sequencing is emerging as a powerful tool for small RNA analysis; however, the high cost of implementation and need for large amounts of input RNA currently limit its utility.[10] The ideal system for miRNA quantification would offer the detection performance of array and PCR-based methods, the throughput of bead-based systems, and improved reproducibility with a user-friendly workflow.
We previously reported the synthesis of chemically and geometrically complex hydrogel microparticles using flow lithography.[11–13] By polymerizing across laminar co-flowing streams of monomer, multifunctional particles with distinct chemical regions can be rapidly (>104/hr) produced with high degrees of reproducibility. Separate “code” and “probe” regions are used to identify particles and capture targets, respectively.[12] The bulk-immobilization of probe molecules in the bio-inert, PEG-based gel scaffolds provides solution-like capture kinetics and high degrees of both specificity and sensitivity, leading to significant advantages over surface-based immobilization strategies employed in microarrays and existing particle systems.[14] Patterns of unpolymerized holes in the code portion of the particle serve as the basis for a graphical multiplexing barcode to identify the probe(s) in a particular particle.
Despite rapid developments in the synthesis of information-rich encoded micro- and nanoparticles,[15,16] there has been little progress in the creation of high-throughput systems for analyzing these complex entities. The emerging bead-based systems with the most promising detection performance lack a method for rapid decoding and target quantification,[16–18] while commercially available flow-through scanning systems constrain multiplexing capacity and provide only simple intensity measurements for inefficient, gate-based analysis (luminexcorp.com). Unlike bead systems that optically encode spheres and use arrays of lasers and photomultiplier tubes (PMTs), our particles have multiple distinct regions, making single-color, morphology-based scanning possible, with only one excitation source and one detector required. This setup significantly reduces the cost and complexity of analysis. Furthermore, the coding library can easily be expanded to accommodate high levels of multiplexing or parallel processing of samples. The flexibility of our synthesis method also allows for embedding living entities such as cells[19] in the probe region of particles, or for incorporating multiple probe regions on a single particle for intrabead multiplexing.[12]
Here, we describe the performance of a gel particle scanning system and an accompanying post-hybridization labeling scheme for use in high-throughput multiplexed miRNA quantification (Fig. 1A). We constructed a slit-scan system[20] that uses a thin laser illumination window to sequentially excite passing portions of precisely oriented particles,[21] thus producing a temporal fluorescent signature from which code identity and bound target amount can be determined (Fig. 1B,C). A polydimethylsiloxane (PDMS) focusing chamber bonded to a glass slide was mounted on a fluorescence microscope for analysis procedures. To align the soft gel particles, the sample and sheath streams were injected into the device with 8-psi forcing pressure, creating a single-file flow of particles traveling at velocities of ~0.5 m/s. Slit illumination was created with a 532-nm laser spatially filtered through a chrome-coated glass mask inserted into the field stop position of the microscope. As particles passed through the illumination window, fluorescent signatures were captured with a PMT and the resulting signals were processed using a homemade amplifier with a low-pass filter.
Figure 1.
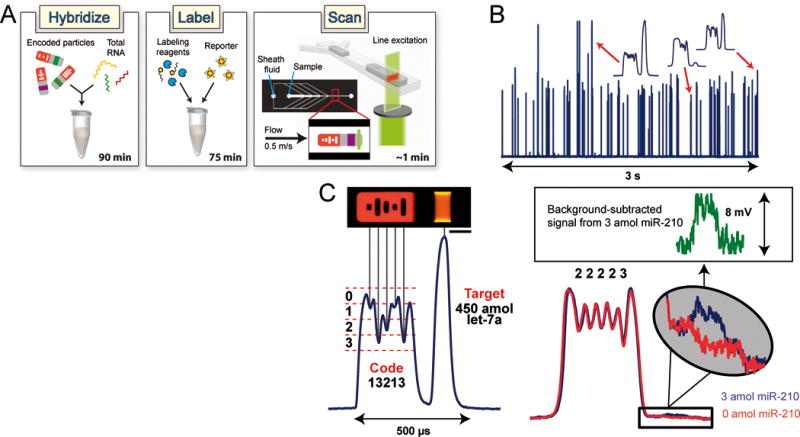
Encoded gel particle assay system. (A) Workflow of platform includes (i) hybridization of particles with target, (ii) incubation of particles with universal labeling adapter, ligation enzyme, and fluorescent reporter, and (iii) scanning of particles to determine code identity and amount of target bound. A typical particle consists of a fluorescent barcoded region and a probe-laden region flanked by two inert sections. Central-most hole has a fixed value to indicate particle orientation. (B) Actual PMT fluorescence signatures of 75 flow-aligned particles. (C) Magnified signatures of individual particles. As probe-target reaction rate is observed to be faster than target diffusion through gel matrix,[14] the increased fluorescent intensity on the sides of the particle in image can be attributed to binding of target near the side faces of probe region. Scale bar is 50 μm.
The fluorescent signal obtained along each particle is integrated across the particle width by the detector. The sizes of the holes in the code region determine the depths of the fluorescence troughs in the signature and thus indicate the particle identity. We optimized the particle architecture and hole design to find that four distinguishable coding levels (0–3) could be obtained for 70-μm wide particles, leading to 192 possible codes for a five-bar particle (Fig. 1C). Multiplexing capacity could easily be augmented to >105 by adding more bars, using multiple fluorescent levels for the code region, or incorporating multiple probes on each particle (see Supporting Information). We developed and trained a decoding algorithm to accurately decode particles and quantify targets. In this algorithm, particle orientation (code- or probe-first) and velocity are determined to analyze the coding holes and establish a first estimate of code identity. A revised assignment is calculated by checking the consistency among holes identified as the same level, and a decoding confidence score is then computed and used to accept or reject particles.
To generate signal in the probe region of particles, we employed a unique ligation-based scheme to fluorescently label bound miRNA targets. Existing approaches rely on the bulk-labeling of RNA using chemical[22] or enzymatic means.[6,23] These methods suffer from high cost, sequence bias due to secondary structure,[6] complicated protocols6, and in the case of microarrays, the need for small-RNA purification and clean-up.[22,23] To overcome these issues, we implemented a two-step method to efficiently label targets after hybridization in about one hour. We used T4 DNA ligase to link a universal oligonucleotide adapter to the 3′ end of targets captured on gel-embedded DNA probes (Fig. 2). For maximum fluorescent efficiency, we used a biotinylated adapter with phycoerythrin-conjugated streptavidin reporter (SA-PE), though it is also possible to use fluorophore-conjugated adapters directly. Importantly, this labeling method was highly efficient, had no minimal input RNA requirement, and showed no sequence bias for the targets used in this study. For each new miRNA target species, we simply incorporated a target-specific sequence into the universal probe template; complex modification and customization were not necessary.
Figure 2.
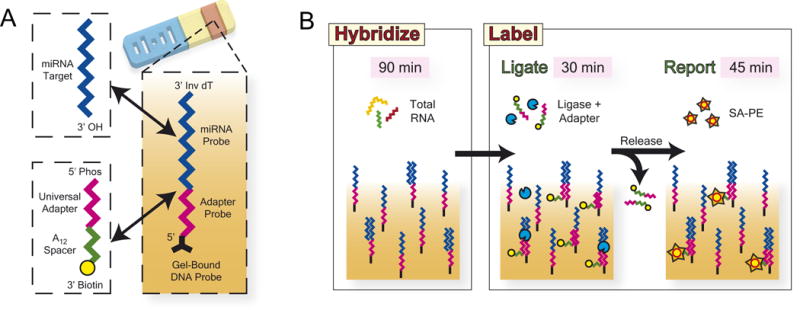
Post-hybridization miRNA labeling via ligation to a universal adapter. (A) DNA probes, linked at their 5′ end throughout the probe region of encoded hydrogel particles, contain a miRNA-specific sequence adjacent to a universal adapter sequence such that the 3′ end of a captured target would abut the 5′ end of a captured adapter oligonucleotide. The probe is capped with an inverted dT to mitigate incidental ligation and the adapter has a poly(A) spacer to extend its biotinylated 3′ end away from the hydrogel backbone for efficient reporting. (B) After particles are hybridized with total RNA, T4 DNA ligase is used to link the universal adapters to the 3′ end of captured targets, unligated adapters are released using a low-salt rinse, and streptavidin-phycoerythrin (SA-PE) is used as a fluorescent reporter.
We investigated the dynamic range, sensitivity, and specificity of the platform in the context of a 12-plex assay featuring ten clinically relevant miRNA targets. Because of its relative invariance across tissue types and disease states, RNU6B was used as an internal control for normalization purposes. We also used 100 amol of miSpike (a synthetic 21-mer) as an external control to validate the consistency of the labeling and scanning processes. We synthesized twelve batches of single-probe particles for this study. To compensate for discrepancies in target hybridization rates, we implemented a coarse rate-matching by tuning the probe concentration for each target using previously determined scaling laws (see Supporting Information)[14]. To fully demonstrate the versatility of the scanner, five separate codes were correlated to particles of each probe type, thereby simulating a 60-plex assay.
To assess sensitivity and dynamic range, we simultaneously spiked four of the twelve targets into 50-μl incubation mixes at amounts ranging from 1 to 2187 amol. We observed a linear detector response over four logs, with sub-attomole sensitivity achieved for three of the four targets (Fig. 3A) and strong agreement between neat samples and those spiked with 200 ng of E. coli total RNA to add complexity (see Supporting Information). By comparison, existing bead-based approaches have a 200-amol limit of detection and only one log of range (luminexcorp.com). To assess specificity, we performed assays with let-7a particles and four members of the let-7 family spiked separately at 200 amol into samples containing 200 ng E. coli total RNA. Scans revealed a maximum cross-reactivity of 27% (Fig. 3B), which is lower than other systems[6] (microarray ~50%) and can be dramatically improved with lower hybridization salt concentrations (see Supporting Information). These assays were very reproducible, with intra- and inter-run CV’s of 2–7%. Due to limitations in detection and particle preparation, it is common for users of current bead-based systems to employ 4,500 copies of each type of bead in an assay for high-confidence estimates of target level.[24] By contrast, we found it sufficient to analyze only 10–15 hydrogel particles for each probe type.
Figure 3.
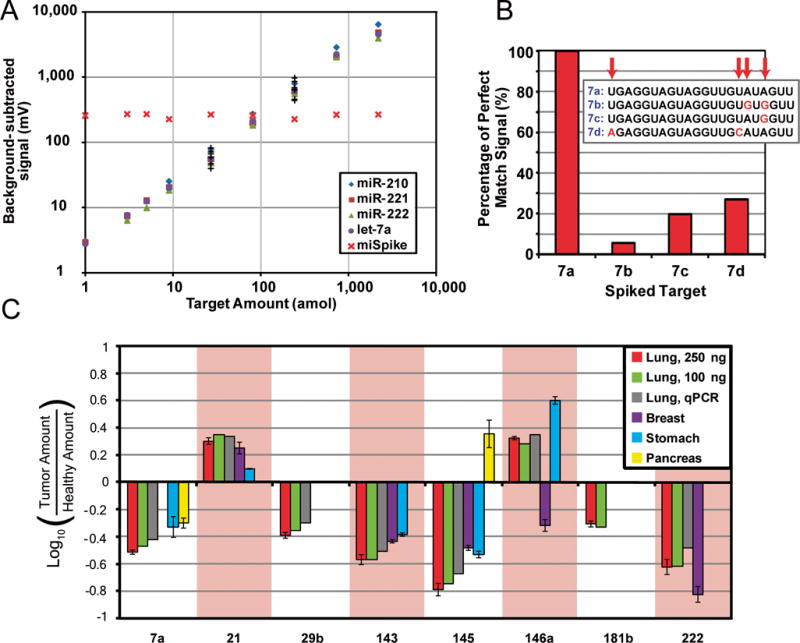
System performance in 12-plex assay. (A) Calibration curves for particle batches, with background-subtracted signal plotted against spiked target amount. miR-210, −221, −222 and let-7a were spiked into the same incubation mixes at the indicated amounts. The remaining seven naturally-occurring targets (‘+’ symbols) were spiked into the 27- and 243-amol trials to validate performance. For all trials, 200 ng of E. coli total RNA was also spiked in for complexity. Mean CV of target level is 6.35% when considering target levels greater than 5 amol. Each point represents, on average, 19 particles from a single run. (B) Specificity of let-7a probe in the presence of sequences closely related to intended target. (C) Cancer profiling results for dysregulated targets in four human tissue types. Error bars represent standard deviation in triplicate measurements on aliquots of the same single-patient sample. Amount of total RNA used in gel-particle assays is 250 ng, unless otherwise noted. See supporting information for details of dysregulation analysis.
As a further validation of the platform, we performed expression profiling across tumor and adjacent normal tissue for several cancer types. As anticipated, we observed the dysregulation of miRNA targets in all of the diseases investigated (Fig. 3C). Although we used 250 ng of total RNA for these samples, similar profiling results were obtained for lung samples using only 100 ng, suggesting that less input RNA would be sufficient. Aliquots of the lung samples were profiled with qPCR by the Dana Farber Cancer Institute’s Molecular Diagnostics Laboratory to independently confirm our results. The two platforms exhibited strong agreement for the seven targets available for comparison (Fig. 3C). With a simple workflow and a total assay time of only 3 h, the profiling is far more efficient than microarray approaches[6] (~24 h) and recent amplification schemes[25] (~9 h), and it exhibits sensitivity and reproducibility superior to that of existing bead-based methods[9] (luminexcorp.com).
In past work, gel particles were imaged with a CCD camera and then analyzed at rates <1 particle/min. For the mock 60-plex assay demonstrated here (i.e., five codes per probe type), the scanner provided a decoding accuracy of ~98%, with only ~10% score-based rejection at throughputs up to 25 particles/s. This represents a 1,500-fold increase in analysis rate. With manual loading from Eppendorf tubes (see Supporting Information), eight 50-μl samples could be scanned and analyzed in 30 min, leading to a projected throughput of ~125 samples per 8-h workday. In future applications of this technology, it is expected that automation of the particle-loading and rinsing processes using well-plates and a liquid handling system will greatly augment efficiency to levels that match or exceed state-of-the-art particle analysis systems used for genotyping (>500 samples/day) (illumina.com).
In summary, we demonstrate high-performance miRNA profiling using a platform that combines a microfluidic scanner and post-hybridization labeling with graphically-encoded hydrogel microparticles. The system’s unprecedented combination of sensitivity, flexibility, and throughput offers exciting possibilities for discovery and clinical applications, particularly in the rapid quantification of low-abundance miRNA and other biomolecules for cancer diagnostics.
Experimental Section
Details of particle synthesis, assay protocols, particle scanning, multi-probe particles, profiling analysis, and labeling optimization studies are given in Supporting Information.
Supplementary Material
Footnotes
We gratefully acknowledge support from the National Institutes of Health and the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the views of the National Institutes of Health or the National Cancer Institute. The authors D.C.P. and P.S.D. have an equity stake in Firefly BioWorks, Inc.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Stephen C. Chapin, Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 (USA)
David C. Appleyard, Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 (USA)
Daniel C. Pregibon, Firefly BioWorks, Inc., 1 Kendall Sq., Building 1400W, 3rd Floor, Cambridge, MA 02139 (USA), pregibon@fireflybio.com
Patrick S. Doyle, Department of Chemical Engineering, Massachusetts Institute of Technology, 77 Massachusetts Avenue, Cambridge, MA 02139 (USA).
References
- 1.Lu J, et al. Nature. 2005;435:834. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- 2.Volinia S, et al. Proc Natl Acad Sci USA. 2006;103:2257. [Google Scholar]
- 3.Esquela-Kerscher A, Slack FJ. Nat Rev Cancer. 2006;6:259. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 4.Wark AW, Lee HJ, Corn RM. Angew Chem Int Ed. 2008;47:644. doi: 10.1002/anie.200702450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baker M. Nat Methods. 2010;7:687. doi: 10.1038/nmeth0910-687. [DOI] [PubMed] [Google Scholar]
- 6.Wang H, Ach RA, Curry B. RNA. 2007;13:151. doi: 10.1261/rna.234507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Husale S, Persson HHJ, Sahin O. Nature. 2009;462:1075. doi: 10.1038/nature08626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen C, et al. Nuc Acids Res. 2005;33:e179. doi: 10.1093/nar/gni178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen J, Lozrach J, Garcia EW, Barnes B, Luo S, Mikoulitch I, Zhou L, Schroth G, Fan JB. Nuc Acids Res. 2008;36:e87. doi: 10.1093/nar/gkn387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Creighton CJ, Reid JG, Gunaratne PH. Brief Bioinform. 2009;10:490. doi: 10.1093/bib/bbp019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dendukuri D, Pregibon DC, Collins J, Hatton TA, Doyle PS. Nat Mater. 2006;5:365. doi: 10.1038/nmat1617. [DOI] [PubMed] [Google Scholar]
- 12.Pregibon DC, Toner M, Doyle PS. Science. 2007;315:1393. doi: 10.1126/science.1134929. [DOI] [PubMed] [Google Scholar]
- 13.Bong KW, Bong KT, Pregibon DC, Doyle PS. Angew Chem Int Ed. 2009;49:87. doi: 10.1002/anie.200905229. [DOI] [PubMed] [Google Scholar]
- 14.Pregibon DC, Doyle PS. Anal Chem. 2009;81:4873. doi: 10.1021/ac9005292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finkel NH, Lou XH, Wang CY, He L. Anal Chem. 2004;76:353A. doi: 10.1021/ac0416463. [DOI] [PubMed] [Google Scholar]
- 16.Birtwell S, Morgan H. Integr Biol. 2009;1:345. doi: 10.1039/b905502a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brunker SE, Cederquist KB, Keating CD. Nanomedicine. 2007;2:695. doi: 10.2217/17435889.2.5.695. [DOI] [PubMed] [Google Scholar]
- 18.Broder GR, et al. Anal Chem. 2008;80:1902. doi: 10.1021/ac7018574. [DOI] [PubMed] [Google Scholar]
- 19.Panda P, Ali S, Lo E, Chung BG, Hatton TA, Khademhosseini A, Doyle PS. Lab Chip. 2008;8:1056. doi: 10.1039/b804234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horan PK, Wheeless JLL. Science. 1977;198:149. doi: 10.1126/science.905822. [DOI] [PubMed] [Google Scholar]
- 21.Chapin SC, Pregibon DC, Doyle PS. Lab Chip. 2009;9:3100. doi: 10.1039/b909959j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brazas RM, Enos JM, Duzeski JL, Schifreen RS, Watt MV. J Mol Diagn. 2006;8:671. [Google Scholar]
- 23.Shingara J, Keiger K, Shelton J, Laosinchai-Wolf W, Powers P, Conrad R, Brown D, Labourier E. RNA. 2005;11:1461. doi: 10.1261/rna.2610405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J. Genome Biol. 2006;7:R61. doi: 10.1186/gb-2006-7-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Y, Zhang X, Li Z, Jiao X, Wang Y, Zhang Y. Angew Chem Int Ed. 2009;48:3268. doi: 10.1002/anie.200805665. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.