

Abstract
SNARE complex assembly constitutes a key step in exocytosis that is rendered Ca2+-dependent by interactions with synaptotagmin-1. Two putative sites for synaptotagmin binding have recently been identified in SNAP-25 using biochemical methods: one located around the center and another at the C-terminal end of the SNARE bundle. However, it is still unclear whether and how synaptotagmin-1 × SNARE interactions at these sites are involved in regulating fast neurotransmitter release. Here, we have used electrophysiological techniques with high time-resolution to directly investigate the mechanistic ramifications of proposed SNAP-25 × synaptotagmin-1 interaction in mouse chromaffin cells. We demonstrate that the postulated central binding domain surrounding layer zero covers both SNARE motifs of SNAP-25 and is essential for vesicle docking, priming, and fast fusion-triggering. Mutation of this site caused no further functional alterations in synaptotagmin-1-deficient cells, indicating that the central acidic patch indeed constitutes a mechanistically relevant synaptotagmin-1 interaction site. Moreover, our data show that the C-terminal binding interface only plays a subsidiary role in triggering but is required for the full size of the readily releasable pool. Intriguingly, we also found that mutation of synaptotagmin-1 interaction sites led to more pronounced phenotypes in the context of the adult neuronal isoform SNAP-25B than in the embryonic isoform SNAP-25A. Further experiments demonstrated that stronger synaptotagmin-1 × SNAP-25B interactions allow for the larger primed vesicle pool supported by SNAP-25 isoform B. Thus, synaptotagmin-1 × SNARE interactions are not only required for multiple mechanistic steps en route to fusion but also underlie the developmental control of the releasable vesicle pool.
Introduction
Formation of SNARE complexes between vesicle and plasma membrane is essential for exocytosis and neurotransmitter release (Südhof and Rothman, 2009; Jahn and Fasshauer, 2012). Ca2+ sensitivity is conferred to vesicle fusion by a dedicated calcium sensor, synaptotagmin-1 (syt-1) (Geppert et al., 1994; Voets et al., 2001) or a related isoform, which triggers full SNARE complex assembly, membrane merger, and formation of the fusion pore (Rizo et al., 2006; Martens et al., 2007; Chapman, 2008).
Ternary SNARE complexes (Davis et al., 1999; Gerona et al., 2000; Littleton et al., 2001; Chicka et al., 2008) and dimeric SNAP-25:syntaxin-1 complexes (Rickman and Davletov, 2003; Bai et al., 2004b; Rickman et al., 2004b; Bhalla et al., 2006; Hui et al., 2011) directly bind to syt-1. Syt-1 × SNARE association predominantly relies on electrostatic interactions (Bennett et al., 1992; Shao et al., 1997; Rickman and Davletov, 2003; Tang et al., 2006; Kuo et al., 2009; van den Bogaart et al., 2011), with some binding activity being sensitive to Ca2+ and some being apparently Ca2+-independent (Sollner et al., 1993; Chapman et al., 1995; Gerona et al., 2000; Zhang et al., 2002; Rickman and Davletov, 2003; Bai et al., 2004b; Rickman et al., 2004b; Pang et al., 2006; Lynch et al., 2008). A polybasic motif (K326, K327) within the C2B segment of syt-1 was proposed to constitute the major interaction site for SNAREs (Rickman and Davletov, 2003; Rickman et al., 2004b; Dai et al., 2007; Gaffaney et al., 2008; Xue et al., 2008). However, the same site was also implicated in phosphatidylinositol-4,5-bisphosphate binding (Bai et al., 2004a; Arac et al., 2006; Li et al., 2006; Osborne et al., 2007; van den Bogaart et al., 2012), and an alternative mode of SNARE interaction has been suggested (Choi et al., 2010).
Several groups of negatively charged residues in SNAP-25 were postulated to function as syt-1 binding sites. The very C terminus of SNAP-25 has long been suspected to mediate fusion triggering because C-terminal truncation by BoNT/A shifts the calcium dependence of release (Capogna et al., 1997; Trudeau et al., 1998; Sakaba et al., 2005; Sørensen et al., 2006). Crosslinking and pull-down experiments implicated a group of acidic amino acids (D172, D179, D186, D193) in the C-terminal half of the second SNARE motif in Ca2+-dependent syt-1 binding (Gerona et al., 2000; Zhang et al., 2002; Lynch et al., 2007). In contrast, constitutive t-SNARE binding of syt-1 was suggested to involve a group of negatively charged amino acids localized near layer 0 (D51, E52, E55) (Rickman et al., 2006; Kim et al., 2012). Mutation of either binding site dramatically reduced release in biochemical PC12-cell assays (Zhang et al., 2002; Rickman et al., 2006), but the implications of these interactions for fast neurotransmitter release remain unclear. Notably, in some systems (chromaffin cells, autaptic neurons), deletion of syt-1 predominantly slows down secretion and only mildly decreases total release, which seems inconsistent with the reported effects in PC12 cell assays. Here, we use fast measurement techniques in combination with rescue experiments to mechanistically evaluate the proposed interaction sites for syt-1 on SNAP-25.
Materials and Methods
Virus generation and biochemistry.
Mutations were introduced into SNAP-25A/B by PCR using primers with modified sequences. PCR products were restricted by BamHI/BssHII and ligated into a modified Semliki Forest Virus shuttle vector (pSFVI1, Invitrogen) carrying an internal ribosome entry site sequence for bicistronic expression of EGFP. All constructs were verified by sequencing. Replication-deficient Semliki Forest viruses for rescue experiments were produced as previously described (Sørensen et al., 2002).
For protein production, cDNAs were subcloned into a pET28a expression vector using NheI/BamHII restriction sites. His-tagged SNAP-25 constructs were expressed in Escherichia coli (BL21), and bacterial pellets were resuspended in 500 mm NaCl, 20 mm HEPES, pH 7.3, 10 mm imidazole, and protease inhibitors (Roche). Cell lysates were incubated with nickel-NTA agarose beads (QIAGEN; 90 min, 4°C). Unbound material was removed with one wash in 1 m NaCl, 20 mm HEPES, pH 7.3, 20 mm imidazole, 0.1% Triton X-100, and three washes in the same buffer but with 100 mm NaCl. Protein was eluted with 100 mm NaCl, 20 mm HEPES, and 150 mm imidazole, pH 7.0 (30 min, 4°C), and dialyzed into 100 mm NaCl, 20 mm HEPES, pH 7.3. Recombinant syntaxin-1A (aa 1–261) and VAMP-2 (aa 2–94) were prepared essentially as previously described (Connell et al., 2007).
Ternary SNARE reactions were performed using recombinant syntaxin-1A (aa 1–261), VAMP-2 (aa 2–94), and indicated SNAP-25 proteins in the presence of 0.8% β-octylglucoside with fivefold excess of synaptobrevin to drive t-SNAREs into SNARE complexes and then analyzed by SDS-PAGE followed by Coomassie staining. Binding reactions of t-SNAREs with GST-synaptobrevin-2 were performed as previously described (Rickman et al., 2004a). Purification of recombinant GST-C2AB and brain-derived syntaxin was performed as described previously (Rickman et al., 2006). For GST-C2AB binding assays, brain-purified syntaxin and bacterially produced SNAP-25 proteins were preincubated (30 min, 22°C) in 100 mm NaCl, 20 mm HEPES, pH 7.3, 1 mm EDTA, and 0.8% octylglucoside. GST-C2AB (1 μg/reaction) was first immobilized on glutathione-Sepharose beads, and the syntaxin/SNAP-25 preincubation mix (3 μg/reaction) was added in a final buffer composition of 150 mm NaCl, 20 mm HEPES, pH 7.3, 1 mm EDTA, and 0.1% Triton X-100. After incubation (90 min, 4°C) beads were washed three times, and bound protein was analyzed by SDS-PAGE followed by staining with the sensitive Sypro Ruby stain (Invitrogen). The fluorescent signal was imaged using a ChemiDoc XRS system (Bio-Rad) and quantified using Quantity One software (Bio-Rad). Statistical analysis was performed using Graph Pad Prism.
Cell culture, electrophysiological recordings, and data analysis.
Mouse chromaffin cells were isolated from adrenal glands of mouse embryos of either sex at E18/E19 and cultured for a maximum of 3 d as described previously (Sørensen et al., 2003; Schonn et al., 2010). Animals were handled in full compliance with the federal German and Danish animal welfare acts and all local regulations of the University of Saarland and University of Copenhagen. Combined capacitance measurements and amperometric recordings were performed as reported (Mohrmann et al., 2010). Release of catecholamines was triggered by UV flash photolysis of a caged-calcium compound, nitrophenyl-EGTA (Synaptic Systems), infused into the cell via the patch pipette. Before a UV flash was applied, the intracellular Ca2+ concentration was adjusted by brief illumination with the monochromator to establish a comparable starting value slightly below 1 μm. Photo artifacts were generally eliminated from amperometric recordings by subtraction of a control trace recorded after removal of the cell from the field of view. Intracellular calcium concentration was determined using two dyes with different Ca2+ affinity (fura-4F and furaptra, Invitrogen) (Voets, 2000; Sørensen et al., 2002). To prevent flash-induced damage to fura dyes, vitamin C was added to the intracellular solution. For the ratiometric determination of calcium concentration, fura dyes were alternatingly excited at 340 and 370 nm. The intracellular solution contained the following (in mm): 100 Cs-gluconate, 8 NaCl, 4 CaCl2, 32 Cs-HEPES, 2 Mg-ATP, 0.3 GTP, 5 nitrophenyl-EGTA, 0.4 fura-4F, 0.4 furaptra, and 1 vitamin C, pH 7.2 (osmolarity adjusted to 290 mOsm). The extracellular solution was composed of the following (in mm): 145 NaCl, 2.8 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 11.1 glucose, pH 7.2 (osmolarity adjusted to 300 mOsm).
To obtain information about pool sizes and fusion rates, capacitance traces were fitted with a sum of three exponential functions using custom macros written in IGOR Pro software (Wavemetrics). The amplitudes and time constants of two burst components, “fast burst” and “slow burst,” were estimated together with the linear rate of the sustained component (Mohrmann et al., 2010). If the fit resulted in a negative amplitude for at least one component, or if two time constants were not separated by a minimal factor of 2, the fitting was done with a sum of two exponential functions (one for the burst, one for sustained component). Time constants <50 ms were considered to be associated with secretion from the Readily-Releasable Pool (RRP), whereas time constants in the range of 50–500 ms represent release from the Slowly-Releasable Pool (SRP). However, a clear distinction of both burst components by these criteria is not always possible. Data are presented as mean ± SEM. Statistical analysis was done in InStat (Graph Pad Software).
Immunocytochemistry.
Chromaffin cells used for immunohistochemistry were cultured on coverslips coated with poly-l-lysine (Sigma, P-1524). Cells were fixated in 4% PFA for 20 min (25°C), permeabilized in 0.1% Triton X-100 for 10 min, and blocked for half an hour in PBS containing 3% BSA Fraction V (Sigma). Cells were incubated with primary antibody (mouse anti-SNAP-25 71.1 at 1:500, Synaptic Systems) for 16 h (4°C), washed, and incubated with secondary antibodies (Alexa Fluor 546-conjugated goat anti-mouse antibody; dilution 1:400, Invitrogen) for 1 h (25°C). Coverslips were mounted on slides using Mowiol 4–88 (Carl Roth)-containing mounting medium. Samples were analyzed at room temperature using a confocal laser scanning microscope (LSM 710, Carl Zeiss Microimaging). Infected cells were identified based on EGFP fluorescence. Pictures were obtained using a 63×/1.4 oil-objective (Zeiss Plan-Apochromat). z-stacks of ∼20 confocal sections (0.35 μm) were acquired to reconstruct whole chromaffin cells (ZEN 2008 software, Carl Zeiss Microimaging). Optimal detector gain settings were determined in preparatory experiments and kept constant for all subsequent experiments. Sum projections of z-stacks were generated using ImageJ (National Institutes of Health), and the summed fluorescence intensity was corrected for background.
Electron microscopy of cultured chromaffin cells.
Chromaffin cells from Snap-25−/− (E18) mice were cultivated on coverslips coated with rat tail type 1 collagen (Cellocate, Eppendorf). Infection with Semliki Forest viruses was done on the second day of cultivation. After incubation for 6–8 h, cells were fixed for 45 min (25°C) with 2.5% glutaraldehyde in 0.1 m cacodylate buffer, pH 7.4. Cells were washed, embedded, and analyzed as described previously (Toonen et al., 2006). Analysis was done in a blinded fashion for the experimental condition. Chromaffin granules were defined as docked if there was no measurable distance between granule and plasma membrane.
Results
Disruption of Ca2+-independent syt-1 binding
To investigate the mechanistic involvement of syt1 × SNARE interactions in fusion triggering, we have mutated and functionally characterized three different groups of acidic amino acids in SNAP-25 that may act as syt-1 binding interfaces: D51, E52, and E55 in the N-terminal SNARE motif (S1) near the zero layer, D166 and E170 in the C-terminal SNARE motif (S2) near layers −1/−2, and finally D172, D179, D186, and D193 that form a charged surface spanning the C-terminal half of S2 (Fig. 1A).
Figure 1.
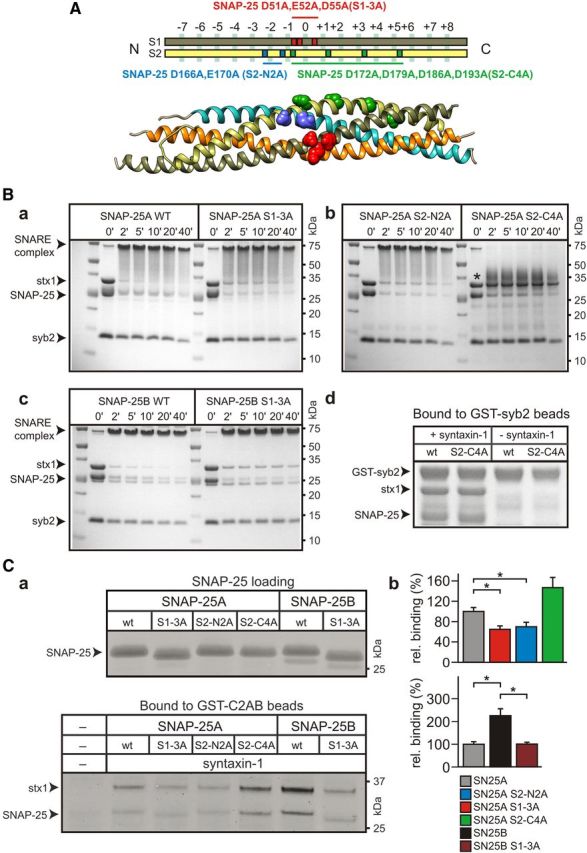
Syt-1 binding of t-SNARE dimers containing SNAP-25A/B and mutants. A, Localization of the different groups of acidic residues mutated in this study: S1–3A (SNARE helix 1 D51A, E52A, E55A) shown in red, S2–N2A (SNARE helix 2 D166A, E170A) in blue, and S2–C4A (D172A, D179A, D186A, D193A) in green. S1 of SNAP-25 is displayed in olive green, and S2 in yellow. Bottom, Crystal structure of the SNARE complex (PDB ID: 1SFC) (Sutton et al., 1998), rendered with UCSF Chimera 1.7, highlighting the mutated residues. B, Formation of the ternary SNARE complex. SNAP25 proteins were incubated with syntaxin-1A (stx1) and fivefold excess of VAMP-2 (syb2) for indicated times, and subsequently complex assembly was stopped by the addition of SDS. The complexes were visualized in Coomassie-stained SDS-PAGE gels. Low levels of syntaxin dimers caused faint bands at 75 kDa in all samples at zero incubation time (0′). In the case of SN25A S1–3A (Ba), SN25A S2–N2A (Bb), and SN25B S1–3A (Bc), SNARE complex formation did not exhibit major differences compared with the corresponding SNAP-25 wild-type (WT) isoforms. For SN25A S2–C4A (Bb), SNARE complex exhibited markedly different physicochemical properties and ran as a “smear” just above the syntaxin band (*). When tested in a GST-syb2 pull-down assay, SN25A S2–C4A supported syntaxin-dependent complex formation as well as the wild-type SNAP-25A protein (Bd). C, Normalized mutant and wild-type SNAP-25 protein (Ca; loading control in top) was incubated with brain-purified syntaxin-1A to allow for formation of t-SNARE dimers, which were then incubated in a threefold molar excess with Sepharose-immobilized GST-syt-1C2AB for 30 min. Bound protein was analyzed by SDS-PAGE followed by Sypro Ruby quantitative protein staining (Ca; bottom). Cb, Quantification by densitometry. There is a pronounced binding of SNAP-25B-containing dimers to GST-syt-1C2AB. Binding data are shown as a percentage of maximum binding relative to wild-type SNAP-25A. Error bars indicate SEM (n = 3 each). Pairwise comparisons (Student's t test, paired, two-tailed) were made between wild-type SNAP-25A and SNAP-25B, or between the wild-type protein and its corresponding mutants. *p < 0.05.
In the first part of our analysis, we focused on the central acidic motif (D51, E52, E55), which has been suggested to mediate Ca2+-independent syt-1 binding (Rickman et al., 2006). The substitution of the corresponding amino acids (D51K, E52K, E55K) has previously been reported to not affect SNARE complex formation, to nearly abolish syt-1 binding, and to suppress release in biochemical assays using permeabilized PC12 cells (Rickman et al., 2006). To avoid the charge inversion of the original mutation, we generated an alanine-substituted variant, SNAP-25A D51A, E52A, E55A (denoted “SN25A S1-3A”). As expected, bacterially expressed SN25A S1-3A could enter SNARE complexes with similar efficiency as wild-type SNAP-25A (Fig. 1B). Coincubation of SN25A S1-3A with syntaxin-1A (aa 1–261) in the presence of fivefold molar excess of VAMP-2 (aa 2–94) and 0.8% β-octylglucoside led to almost complete engagement of t-SNAREs in SNARE complexes within 2 min (i.e., within the time frame necessary for protein sample analysis) (Fig. 1Ba). The formation of the SNARE complex was only marginally affected by the mutation. To investigate Ca2+-independent binding of this variant to the cytoplasmic domain of syt-1 (C2AB), we performed a standard protein pull-down assay using SNAP-25:syntaxin dimers (Rickman et al., 2006). For this purpose, wild-type and mutant SNAP-25 were first mixed with brain-purified syntaxin in the presence of 0.8% β-octylglucoside to form t-SNARE dimers, and these complexes were consecutively incubated for 30 min with immobilized GST-C2AB, the former being in threefold molar excess to saturate C2AB. Bound material was analyzed by Sypro Ruby staining. We detected a moderate but significant decrease in the fraction of retained t-SNARE complexes on GST-C2AB for SN25A S1-3A compared with wild-type protein (n = 3; paired t test: p < 0.05) (Fig. 1Ca,b), confirming that this mutation interferes with syt-1 interactions without abolishing them.
Physiological measurements were performed in Snap-25−/− chromaffin cells that virally expressed mutant or wild-type protein for 5–6 h (Semliki Forest virus system). Quantitative immunostaining showed that all SNAP-25 variants studied under these conditions were overexpressed >10-fold over endogenous levels (Fig. 2). The high overexpression level is an intrinsic feature of the used expression system, which should be kept in mind when interpreting the results. However, we note that overexpression of SNAP-25A under these conditions fully reconstitutes secretion in KO cells (Sørensen et al., 2003) and does not change release properties in wild-type cells (see Fig. 6Aa) (Sørensen et al., 2003), indicating that the higher expression level does not affect secretion for the wild-type version of the protein.
Figure 2.

Analysis of virally driven SNAP-25 expression. A, Example microphotographs of chromaffin cells stained against SNAP-25. To test expression levels, Snap-25−/− cells were infected with Semliki Forest viruses and stained 5–6 h after infection. Displayed images represent single confocal slices acquired near the midline of the cell body in z-dimension. Endogenous SNAP-25 expression in wild-type control cells (leftmost column) was on average 15 times lower than virally expressed levels; the image of the wild-type cells was accordingly scaled to demonstrate the native distribution pattern of SNAP-25. Scale bar, 5 μm. B, Quantification of total SNAP-25 immunofluorescence intensity (per cell). Intensity throughout all confocal slices was summed up in a region of interest enclosing the whole-cell body. Total intensity was corrected for background fluorescence using an identically shaped, blank region as reference. Immunofluorescence in wild-type cells and infected cells was compared using ANOVA and Dunnett test, yielding p < 0.01 for all comparisons, except wild-type versus SN25B S2-C4A, which resulted in p < 0.05. The number of analyzed cells is indicated above each bar. Error bars indicate SEM. *p < 0.05. **p < 0.01.
Figure 6.

Syt-1 interactions are essential for maintenance of an expanded primed vesicle pool in SNAP-25B-expressing cells. A, Functional analysis of secretion in uninfected wild-type cells (black, n = 14) and cells overexpressing SNAP-25A (yellow, n = 13) or SNAP-25B (orange, n = 14). Aa, Averaged traces of [Ca2+]i (top), capacitance traces (middle), amperometric currents, and cumulative charge (bottom) after uncaging flash (arrow). Ab, Secretion measured 0–1 s after uncaging (burst) and 1–5 s after uncaging (sustained) was quantified and compared. Only SNAP-25B overexpression caused an increase in burst release. B, Analysis of the effects of SNAP-25A (purple, n = 17) and SNAP-25B (blue, n = 18) overexpression in syt-1−/− cells (uninfected, red, n = 18). Isoform-specific alterations of secretion were abolished, but burst size was slightly increased. C, Characterization of secretion in syt-1−/−; syt-7−/− chromaffin cells (uninfected, red, n = 23) either overexpressing SNAP-25A (purple, n = 23) or SNAP-25B (blue, n = 24). Error bars indicate SEM. Statistical comparisons were done using Student's t test (unpaired, two-tailed). *p < 0.05. **p < 0.01. n.s., not significant.
Exocytosis was stimulated by calcium uncaging and was monitored by parallel measurements of membrane capacitance and amperometric recordings at the same cells. Because amperometric measurements only collect data from a small part of the cell, we performed the quantitative analysis only on capacitance measurements. In control recordings, expression of wild-type SNAP-25A in Snap-25−/− cells robustly reconstituted secretion: the mean membrane capacitance change (ΔCm) reached 333 ± 33 fF 5 s after a step-like increase in [Ca2+]i (n = 28) (Fig. 3A). In comparison, uninfected Snap-25−/− cells only support 30–50 fF secretion (Sørensen et al., 2003; Mohrmann et al., 2010). Expression of SN25A S1-3A partly reestablished exocytosis in Snap-25−/− cells (216 ± 26 fF, n = 30, 65% of control, unpaired t test: p = 0.0064 compared with SNAP-25A), and resulted in a dramatic slowdown of secretion (Fig. 3A). The kinetic alterations were best visible in normalized plots of the secretory burst (Fig. 3Ab). Our analysis of flash-induced membrane capacitance changes revealed a significant increase in the time constants of both the fast and slow component (SN25A S1-3A, τfast = 34.0 ± 1.4 ms and τslow = 387.7 ± 34.0 ms; controls, τfast = 13.9 ± 4.7 ms and τslow = 236.1 ± 46.1 ms; unpaired t test: p = 1.5 × 10−5 and p = 0.0094, respectively) (Fig. 3Ac). In addition, the amplitude of the fast burst component was strongly depressed (SN25A S1-3A, 22.7 ± 6.5 fF; controls, 107.6 ± 13.4 fF; unpaired t test: p = 2.8 × 10−7), whereas the amplitude of the slow burst component was increased (SN25A S1-3A, 118.3 ± 19.2 fF; controls, 70.7 ± 10.7 fF; unpaired t test: p = 0.0381) (Fig. 3Ad). The fast and slow burst and the underlying functional vesicle pools (RRP and SRP) are distinguished by fitting a sum of exponential functions to the traces. In case of substantial changes of both pool sizes and fusion rates, the assignment of release phases can become ambiguous. It is therefore possible that the apparent increase in SRP size results from an overestimation of the slow burst component because of blending with either the fast burst of release or the sustained component, which were both changed because of this mutation. Regardless of this, it is clear that release was considerably slower in SN25A S1-3A-expressing cells, indicating impaired fusion triggering. Further, SN25A S1-3A cells exhibited a decreased sustained rate of release (SN25A S1-3A, 15.0 ± 1.9 fF/s; controls, 30.9 ± 4.0 fF/s; unpaired t test: p = 0.0005) (Fig. 3Ad), indicating that upstream steps in the secretory pathway might also be affected by the S1-3A mutation.
Figure 3.
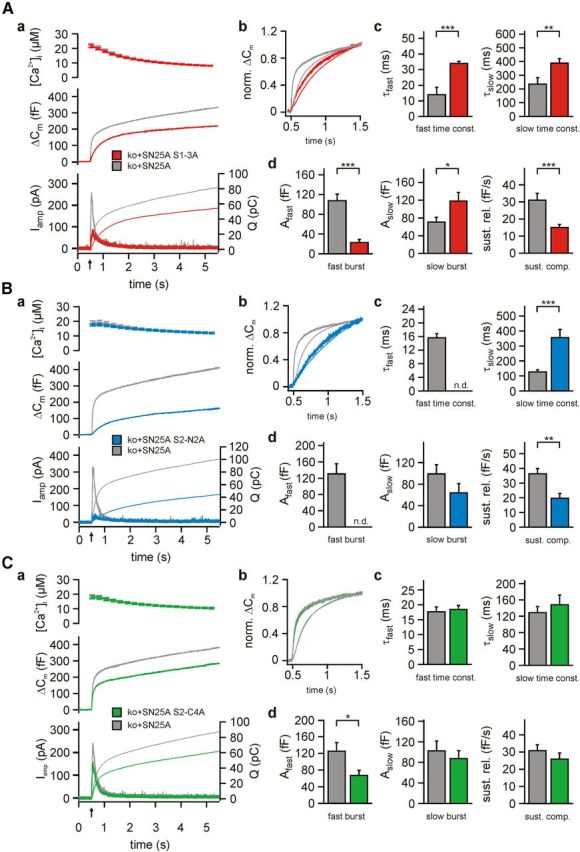
Comparative functional characterization of secretion properties in cells expressing SN25A S1–3A, SN25A S2-N2A, or SN25A S2-C4A. A, Electrophysiological measurement of secretion in Snap-25−/− chromaffin cells expressing SN25A S1–3A. Aa, Averaged traces of [Ca2+]i (top), capacitance measurements (middle), and amperometric recordings (bottom) in Snap-25−/− cells expressing either wild-type protein (gray; n = 28) or SN25A S1–3A (red; n = 30). Uncaging flash was applied at 0.5 s (arrow). Ab, Averaged capacitance changes (bold lines) and amperometric charge traces (thin lines) were normalized to their respective values 1 s after the uncaging flash to compare the kinetics of burst release. Ac, Ad, For the burst release component, mean values for release amplitudes Afast and Aslow as well as the corresponding time constants τfast and τslow are shown. Further, the mean release rate for the linear, sustained component of secretion is shown (lower right). B, Characterization of secretion in Snap-25−/− chromaffin cells expressing SN25A S2-N2A (blue, n = 25; control: gray, n = 28). Panel organization as in A. There is a more prominent decrease in burst release compared with SN25A S1–3A. C, Secretion properties of Snap-25−/− cells expressing either wild-type protein (gray; n = 32) or SN25A S2-C4A (green; n = 26). Panel organization as in A. No alterations in time constants could be observed, but the fast burst component was significantly reduced. Error bars indicate SEM. Statistical comparisons were done using Student's t test (unpaired, two-tailed). *p < 0.05. **p < 0.01. ***p < 0.001. n.d., not detectable.
If syt-1 binding to the central acidic motif in SNAP-25 is involved in exocytosis triggering, the SN25A S1-3A mutation is expected to also alter the calcium dependency of release. To investigate this notion, we monitored release while [Ca2+]i was slowly increased by repetitive brief illumination (using a monochromator as light source) to simultaneously photorelease Ca2+ and measure [Ca2+]i, generating a calcium ramp within the cell (Sørensen et al., 2002) (Fig. 4A). We defined the secretion threshold as the [Ca2+]i at the maximum of the second derivative of the release waveform that was formed by averaging the integrated amperometric signal with the corresponding ΔCm trace. Interestingly, we found a near doubling of the secretion threshold in SNAP-25A S1-3A-expressing cells, suggesting a general shift of the Ca2+ dependency of release (SN25A S1-3A, 4.28 ± 0.36 μm, n = 17; controls, 2.12 ± 0.14 μm, n = 21; unpaired t test: p = 5.6 × 10−7) (Fig. 4Ab).
Figure 4.
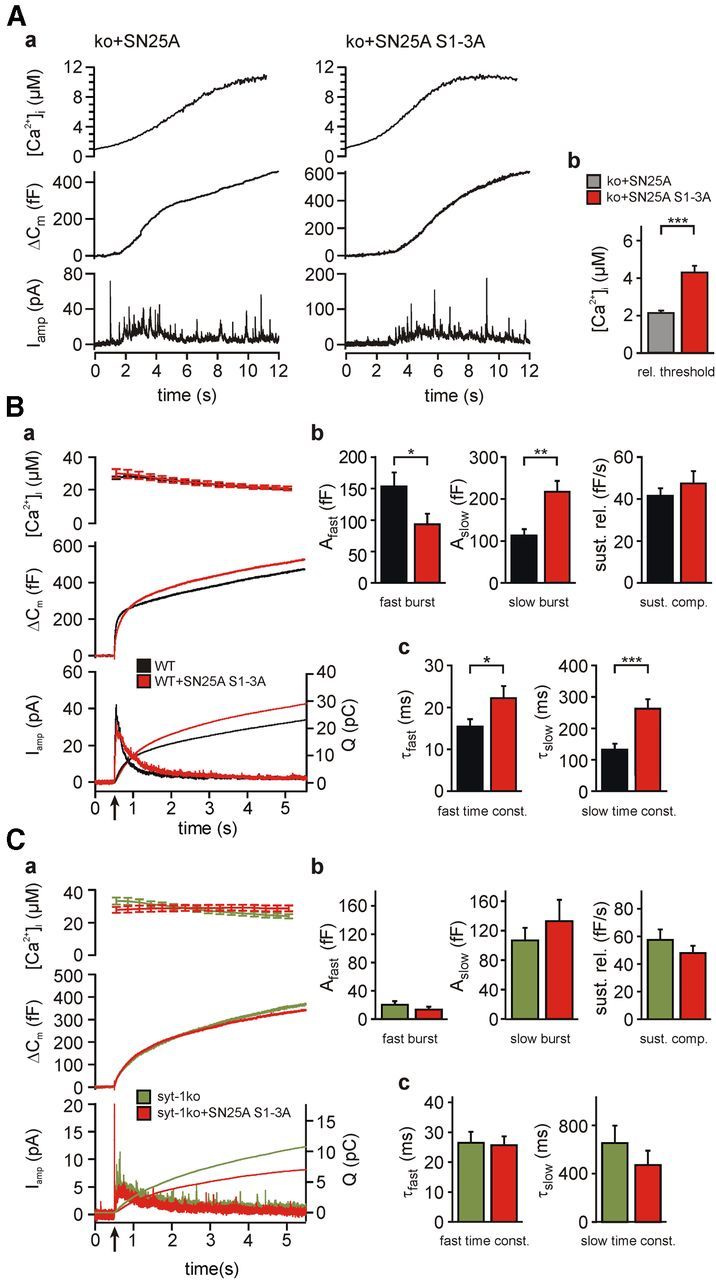
Extended functional characterization of SNAP-25 S1–3A variants. Aa, Expression of SN25A S1–3A increased the Ca2+-threshold of release. A prolonged train of weak light flashes was applied to cells loaded with a calcium cage, generating a slow increase in [Ca2+]i (top). Release was studied by capacitance measurements and amperometric recordings to determine the threshold Ca2+ level that induces a steep increase in secretion (middle, bottom). Ab, Quantification of Ca2+-ramp experiments (ko+SNAP-25A, n = 21; ko+SN25A S1–3A, n = 17). B, Overexpression of SN25A S1–3A in wild-type cells slows down secretion. Ba, Averaged traces of [Ca2+]i (top), capacitance measurements (middle), and amperometric recordings (bottom) in uninfected wild-type cells (black, n = 20) or wild-type cells expressing additional SN25A S1–3A (red; n = 21). Bb, Bc, Quantitative analysis demonstrates functional alterations similar to the rescue phenotype (compare Fig. 3A), albeit without a reduction in total release. C, Analysis of the secretion properties of syt-1−/− cells overexpressing SN25A S1–3A (red, n = 22) compared with uninfected syt-1−/− cells (green, n = 22). Depicted are the intracellular Ca2+ elevation (top), the average capacitance change (middle), and amperometry (bottom). Cb, Cc, Kinetic analysis confirmed the absence of kinetic changes upon SN25A S1–3A overexpression. Error bars indicate SEM. Statistical comparisons were done using Student's t test (unpaired, two-tailed). *p < 0.05. **p < 0.01. ***p < 0.001.
The overall slowdown of release kinetics is the predominant defect caused by the SN25A S1-3A variant, and a similar kinetic phenotype was induced by overexpressing the SN25A S1-3A variant in wild-type cells (WT+SN25A S1-3A, τfast = 22.2 ± 2.9 ms and τslow = 262.6 ± 30.6 ms, n = 19; WT, τfast = 15.45 ± 1.78 ms and τslow = 132.5 ± 18.6 ms, n = 21; unpaired t test: p = 0.0471 and p = 0.0006, respectively) (Fig. 4B). This observation confirms that SN25A S1-3A is incorporated into fusion complexes and competes with endogenous wild-type protein. Interestingly, the kinetic phenotype of SN25A S1-3A-expressing cells is reminiscent of the functional alterations observed in syt-1−/− chromaffin cells (Voets et al., 2001; Nagy et al., 2006), which exhibit strongly depressed or absent fast burst release (Fig. 4B). To test whether the SN25A S1-3A phenotype is indeed caused by the elimination of a SNAP-25 × syt-1 interaction, we overexpressed the mutant variant in syt-1−/− cells. If SN25A S1-3A-induced release defects were unrelated to a loss of syt-1, a composite secretion phenotype would be expected. Strikingly, we did not detect any significant release changes upon overexpression of SNAP-25A S1-3A in syt-1−/− cells (SN25A S1-3A, τfast = 25.7 ± 2.9 ms and τslow = 471.3 ± 118.6 ms, n = 22; syt1 ko, τfast = 26.5 ± 3.6 ms and τslow = 652.1 ± 145.2 ms, n = 22; unpaired t test: p = 0.8696 and p = 0.3405, respectively) (Fig. 4C), which confirms that the observed deficits are the result of the abolishment of syt-1 × SNARE interactions.
Thus, our findings indicate that the central acidic motif in the S1 domain of SNAP-25 is intimately involved in fusion triggering and that a SNAP-25 × syt-1 interaction, most likely in the form of direct physical binding, is necessary for fast vesicle fusion.
The syt-1 interaction site extends to S2
According to the crystal structure of the SNARE complex (Sutton et al., 1998), two acidic residues in the S2 domain (D166 and E170) are located in the vicinity of the syt-1 interaction site in S1, as described above (Fig. 1A). In the crystal, polar residues of this central region coordinate enclosed divalent Sr2+ ions (Fasshauer et al., 1998; Sutton et al., 1998), and the possibility has been entertained that these residues might bind either Ca2+ or accessory factors (Sørensen et al., 2002). As a later study was unable to detect Ca2+-binding to SNAREs (Chen et al., 2005), we have investigated here whether D166 and E170 contribute to the central syt-1 binding interface in SNAP-25.
Like the SN25A S1-3A, SNAP-25A D166A, E170A (denoted “SN25A S2-N2A”) only marginally affected SNARE complex formation (Fig. 1Bb, left part of the gel), and pull-down experiments showed that this variant decreased Ca2+-independent binding to t-SNARE dimers to a similar degree as the SN25A S1-3A mutation (n = 3; paired t test: p < 0.05) (Fig. 1Ca,b). Hence, we analyzed the release properties of Snap-25−/− cells expressing SN25A S2-N2A. In Ca2+-uncaging experiments, SN25A S2-N2A expression only reconstituted total ΔCm to 39% of controls (SN25A S2-N2A, 162.1 ± 27.9 fF, n = 25; controls, 411.2 ± 49.4 fF, n = 28; unpaired t test: p = 8.9 × 10−5) (Fig. 3B). A detailed kinetic analysis demonstrated a complete loss of the RRP component upon SN25A S2-N2A expression (Fig. 3Bd). Moreover, we observed a significant increase in the time constant for the remaining SRP (SN25A S2-N2A, 354.7 ± 54.8 ms; controls, 127.2 ± 14.6 ms; unpaired t test p = 3.9 × 10−5) (Fig. 3Bc,d). In addition, the sustained rate was significantly decreased (SN25A S2-N2A, 19.6 ± 3.2 fF/s; controls, 36.3 ± 3.6 fF/s; unpaired t test: p = 0.0013) (Fig. 3Bd). Having demonstrated a shift in the threshold for release in SN25A S1-3A-expressing cells, we also performed calcium ramp experiments in SN25A S2-N2A-expressing cells and indeed found a significant increase in the threshold calcium concentration (SN25A S2-N2A, 2.75 ± 0.29 μm, n = 19; control, 2.06 ± 0.13 μm, n = 27; unpaired t test: p = 0.0204). This increase is smaller than in SN25A S1-3A-expressing cells, which is probably the result of the smaller overall amount of secretion that is only about twice that found in SNAP-25 KO cells. With small release amplitudes, the maximum point of acceleration tends to get buried in the noise of the recording. In addition, the threshold might partly depend on release not driven by SNAP-25 (SNAP-25 KO release), but by other available Qbc-SNAREs (e.g., SNAP-23, which has been reported to participate in release at lower calcium concentrations; [Chieregatti et al., 2004]).
In summary, substitution of D166 and E170 by alanine residues caused a release phenotype that is reminiscent of the functional deficits caused by the SN25A S1-3A variant (Fig. 3A) and correlates with biochemical data showing a decreased binding between syt-1 and t-SNARE dimers. We therefore suggest that D166 and E170 contribute to a critical negative charge density in the central region of the SNARE complex, which allows syt-1 to interact with SNAP-25 to mediate fusion triggering. However, we cannot exclude a secondary function of these residues that could explain the stronger secretion phenotype compared with SN25A S1-3A.
Negative charges in the C-terminal half of S2 stabilize the RRP
Zhang et al. (2002) proposed that a quartet of acidic amino acids (D172, D179, D186, D193) in S2 may interact with the C2A domain of syt-1 in a Ca2+-dependent fashion. Using SNAP-25 isoform B, whose expression is developmentally upregulated in postnatal brain (Bark et al., 1995), the authors showed that mutation of this acidic motif reduced release in PC12 cells. To investigate the involvement of these residues in fast exocytosis, we generated and functionally characterized an alanine substitution mutant (D172A, D179A, D186A, D193A) in Snap-25−/− chromaffin cells. We introduced this mutation in SNAP-25 isoform A (denoted “SN25A S2-C4A”) (Fig. 1A), as this splice variant constitutes the endogenous isoform in chromaffin cells (Bark et al., 1995). The SN25A S2-C4A produced aberrant SNARE complexes, which in the presence of SDS ran as a smear above and possibly intermixed with the syntaxin-1 band (Fig. 1Bb, right part of gel, asterisk). To demonstrate that SN25A S2-C4A nevertheless forms a ternary SNARE complex, we performed a pull-down experiment with GST-synaptobrevin-2. Indeed, SN25A S2-C4A formed a complex with synaptobrevin-2 in the presence but not in the absence of syntaxin-1 (Fig. 1Bd). Thus, SN25A S2-C4A forms a SNARE complex, which is less SDS-resistant because of a change in physicochemical properties (Fig. 1Bb). In pull-down experiments, acceptor complexes containing SN25A S2-C4A showed strong Ca2+-independent association with GST-C2AB (Fig. 1C), suggesting that constitutive syt-1 × t-SNARE interactions persists with this mutant.
Upon expression in Snap-25−/− chromaffin cells, SN25A S2-C4A considerably reconstituted secretion, resulting in a total ΔCm of 284 ± 37 fF (n = 26; controls, 382 ± 45 fF, n = 32) (Fig. 3C). A kinetic analysis yielded unchanged time constants indicating that fusion triggering is largely unaffected by the mutation (SN25A S2-C4A, τfast = 18.5 ± 1.4 ms and τslow = 148.0 ± 24.1 ms; controls, τfast = 17.6 ± 1.7 ms and τslow = 128.7 ± 14.8 ms; unpaired Student's t test: p = 0.7129 and p = 0.4871) (Fig. 3Cc). However, we found a significant drop in the size of the RRP (SN25A S2-C4A, 66.9 ± 12.5 fF; controls, 125.2 ± 20.7 fF; unpaired Student's t test: p = 0.0267), whereas the SRP remained unaffected (SN25 S2-C4A, 87.7 ± 15.3 fF; controls, 102.7 ± 18.9 fF; p = 0.5526) (Fig. 3Cd). The sustained rate was unchanged (SN25 S2-C4A, 26.0 ± 3.6 fF/s; controls, 30.9 ± 3.5 fF/s; unpaired t test: p = 0.3314) (Fig. 3Cd). These data show that the quartet of acidic amino acids is not critically required for triggering of release but may rather be involved in stabilizing RRP vesicles.
Isoform-dependent effects of disrupting syt-1 × SNARE interactions
The minor release deficits in SN25A S2-C4A-expressing cells were unexpected given that previous data from PC12 cells indicated a crucial requirement for a C-terminal SNAP-25 × syt-1 interaction in fusion triggering (Zhang et al., 2002). In the case of the SN25A S1-3A variant, despite a dramatic effect on fusion kinetics, total ΔCm was reduced by only 35%, much less than expected from previous PC12 cell experiments (Rickman et al., 2006). These inconsistencies might be partially explained by different time scales of release measurements in PC12 and chromaffin cells. However, we also realized that the two putative syt-1 interaction sites were originally identified in isoform B of SNAP-25 (Zhang et al., 2002; Rickman et al., 2006), which differs by nine amino acid substitutions from isoform A (Bark and Wilson, 1994). In chromaffin cells, SNAP-25B was reported to support 2–3 times more secretion than SNAP-25A without causing any kinetic changes (Sørensen et al., 2003), which indicated an isoform-dependent control of the size of the primed vesicle pool through interaction with a yet unknown factor (Nagy et al., 2005). To investigate whether the phenotypic deviations observed in this study were caused by isoform-dependent functional differences, we also characterized the release properties of the mutations in isoform B.
Indeed, the S1-3A mutation caused a much stronger release defect when tested in the background of isoform B (Fig. 5A; compare Fig. 3A). Expression of wild-type SNAP-25B allowed for a total ΔCm of 490 ± 75 fF (n = 32), in agreement with previous data showing that SNAP-25B supports a larger secretory burst (Sørensen et al., 2003; Nagy et al., 2005). In contrast, SN25B S1-3A only reconstituted release to an overall ΔCm of 73 ± 17 fF (n = 30; 15% of control amplitude; unpaired t test: p = 1.7 × 10−6), barely exceeding the secretion level of uninfected knock-out cells (Sørensen et al., 2003; Mohrmann et al., 2010). Thus, total release of the SN25B S1-3A was much more reduced than in cells expressing the corresponding SN25A mutant (SN25A S1-3A, 216 ± 26 fF, n = 30; unpaired t test: p = 1.9 × 10−5) despite the higher secretion level normally supported by isoform B. This effect was not caused by a difference in viral SNAP-25 expression, as all SNAP-25 variants were expressed at comparable levels in infected cells (ANOVA: p = 0.56) (Fig. 2).
Figure 5.
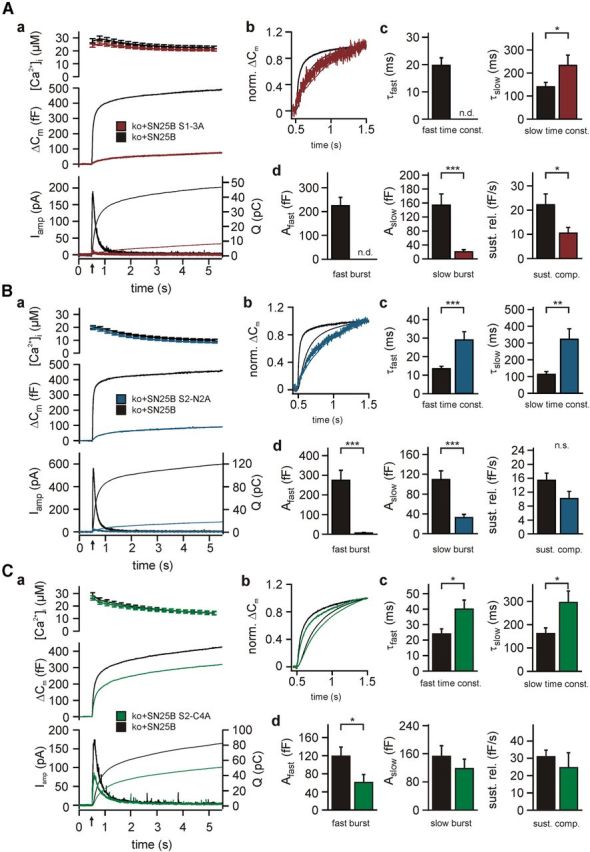
Isoform-specific alterations of phenotypes in the context of the SNAP-25B isoform. A, Electrophysiological characterization of secretion in Snap-25−/− chromaffin cells expressing SN25B S1–3A. Aa, Averaged traces of [Ca2+]i (top), capacitance measurements (middle), and amperometric recordings (bottom) in Snap-25−/− cells expressing either wild-type protein (black; n = 32) or SN25B S1–3A (dark red, n = 30). Ab, Averaged capacitance changes (bold lines) and amperometric charge traces (thin lines) were normalized to their respective values at 1 s after the flash for a kinetic comparison of burst release. Ac, Ad, Kinetic analysis presenting the mean amplitudes and time constants, as well as the average rate of sustained release. There is a dramatic loss of total release compared with the moderate phenotype of the SN25A S1–3A variant. B, Electrophysiological characterization of secretion in Snap-25−/− cells expressing SN25B S2-N2A (dark blue, n = 27; control: black, n = 21). Panel organization as in A. This mutant variant largely exhibited the same phenotypic features as SN25A S2-N2A. C, Analysis of secretion in SN25B S2-C4A-expressing cells (dark green, n = 31; control: black, n = 27). Panel organization as in A. There is a significant slowdown of secretion indicated by the increase of both time constants and a decrease in Afast, which suggest an isoform-specific effect on fusion triggering. Error bars indicate SEM. Statistical comparisons were done using Student's t test (unpaired, two-tailed). *p < 0.05. **p < 0.01. ***p < 0.001. n.s., Not significant; n.d., not detectable.
A kinetic analysis revealed that both pools of primed vesicles were basically empty in the case of the SN25B S1-3A mutant (SN25B S1-3A, fast burst not detectable, Aslow = 20.4 ± 6.1 fF; control, Afast = 225.0 ± 35.0 fF, Aslow = 153.9 ± 32.3 fF; unpaired t test: p = 0.0002) (Fig. 5Ad). Thus, the SNAP-25B mutation results in a significant reduction of the slow burst, which was not seen in the SN25A S1-3A (Fig. 3Ad). The strong phenotype of SN25B S1-3A might be attributed to a difference in vesicle priming and/or unpriming. Alternatively, it might reflect a depletion of release-ready vesicles resulting from increased spontaneous fusion. To investigate this point, we recorded spontaneous release in resting chromaffin cells by amperometry. The average frequency of spontaneous events was not significantly different in cells expressing wild-type SNAP-25B (SNAP-25B, 0.082 ± 0.026 Hz, n = 39) or SN25B S1-3A (SN25B S1–3A, 0.043 ± 0.009 Hz, n = 39), or in cells expressing wild-type SNAP-25A (SNAP-25A, 0.103 ± 0.020 Hz, n = 39; ANOVA: p = 0.106). These experiments argue against a depletion of vesicle pools in SN25B S1-3A-expressing cells but point to a difference in the priming process as main causative.
Next, we tested the S2-N2A mutation in the context of the SNAP-25B isoform. Again, we found that total release was more suppressed in cells expressing the mutant variant of isoform B (Fig. 5B; compare Fig. 3B), with ΔCm reaching only 90 ± 16 fF (n = 27; p = 0.0263 compared with SN25A S2-N2A, unpaired t test), 19% of the control value (461 ± 68 fF, n = 21; unpaired t test: p = 3.6 × 10−7). However, in other respects, the phenotype of SN25B S2-N2A mirrored the results found for the corresponding SNAP-25A variant, including a pronounced drop in RRP and SRP size (SN25B S2-N2A, Afast = 6.4 ± 3.0 fF, Aslow = 32.6 ± 6.4 fF, n = 27; controls, Afast = 274.9 ± 50.4 fF, Aslow = 109.3 ± 17.4 fF, n = 21; unpaired t test: p = 2.5 × 10−7 and p = 0.0004, respectively) (Fig. 5Bd) and increases in the time constants of release components (SN25B S2-N2A, τfast = 29.0 ± 4.3 ms, τslow = 323.3 ± 62.2 ms; controls, τfast = 13.4 ± 1.3 ms, τslow = 113.1 ± 16.2 ms; unpaired t test: p = 7.4 × 10−5 and p = 0.016, respectively) (Fig. 5Bc).
We also characterized the S2-C4A mutation in the context of the B isoform and again found isoform-specific phenotypic changes, although the overall reduction of release was similar to SN25A S2-C4A (75% of control) (Fig. 5C; compare Fig. 3C). Like for the mutant SNAP-25A variant, the size of the RRP was significantly reduced (SN25B S2-C4A, Afast = 60.8 ± 17.5 fF, n = 31; controls, Afast = 119.0 ± 20.1 fF, n = 27; unpaired t test: p = 0.0323), whereas the SRP and sustained component were unaffected (SN25B S2-C4A, Aslow = 117.5 ± 27.2 fF, Rsust = 24.4 ± 8.7 fF/s; controls, Aslow = 152.4 ± 30.3 fF, Rsust = 30.9 ± 3.9 fF/s; unpaired t test: p = 0.394 and p = 0.522, respectively) (Fig. 5Cd). However, the kinetic analysis demonstrated a slight, but significant, slowdown of both time constants for vesicle release (SN25B S2-C4A, τfast = 40.0 ± 5.9 ms and τslow = 295.2 ± 48.8 ms; controls, τfast = 24.0 ± 3.3 ms and τslow = 162.1 ± 24.2 ms; unpaired t test: p = 0.015 and p = 0.016, respectively) (Fig. 5Cc), which would indicate moderately delayed fusion triggering in cells expressing SN25B S2-C4A.
Our data demonstrate that amino acid changes interfering with syt-1 binding generally cause more severe phenotypes in the context of SNAP-25B, which suggests that the two isoforms engage syt-1 to a different degree. In support of this idea, our biochemical experiments showed that immobilized GST-C2AB bound SNAP-25B:syntaxin-1 complexes with almost doubled efficiency compared with assemblies containing isoform A (n = 3; paired t test: p < 0.05) (Fig. 1Ca,b). Furthermore, SN25B S1-3A displayed a stronger relative reduction in binding than SN25A S1-3A (Fig. 1Ca,b), which correlates well with the stronger physiological effect of the S1-3A mutation in the context of isoform B.
Isoform-dependent SNAP-25 × syt-1 interactions determine primed vesicle pool size
SNAP-25B developmentally succeeds SNAP-25A as the predominant Qbc-SNARE in neurons (Bark et al., 1995), which results in a developmental reshaping of synaptic release properties (Delgado-Martinez et al., 2007; Scullin et al., 2012). As mentioned above, SNAP-25B expression increases the size of the primed vesicle pool in chromaffin cells (Sørensen et al., 2003). Having demonstrated that syt-1 binding to SNAP-25B is enhanced in biochemical assays and that mutations interfering with syt-1 interactions exhibit exacerbated phenotypes in the context of isoform B, we hypothesized that stronger or altered syt-1 × SNAP-25B interactions could potentially account for the increased size of the primed vesicle pools.
To test this idea, we changed the prevalent SNAP-25 isoform in syt-1−/− cells, syt-1−/−;syt-7−/− cells, or wild-type chromaffin cells by virally overexpressing SNAP-25B. In line with earlier findings (Sørensen et al., 2003), expression of SNAP-25B significantly increased burst size, whereas overexpression of SNAP-25A was without effect (WT, 225.0 ± 25.3 fF, n = 14; WT+SNAP-25A, 225.6 ± 44.7 fF, n = 13; p = 0.9905 vs WT; WT+SNAP-25B, 390.9 ± 50.2 fF, n = 14; p = 0.0066 vs WT; unpaired t test) (Fig. 6A). Strikingly, however, both isoforms supported indistinguishable levels of secretion when overexpressed in syt-1−/− cells (Fig. 6B); burst size was only slightly increased regardless of the expressed isoform type (syt-1ko, 130.3 ± 12.9 fF, n = 18; syt-1ko+SNAP-25A, 191.9 ± 21.4 fF, n = 17; syt-1ko+SNAP-25B, 195.8 ± 32.3 fF, n = 18; unpaired t test: p = 0.0166 and p = 0.0119, respectively). Because syt-7 has been shown to mediate slow burst release in syt-1−/− chromaffin cells (Schonn et al., 2008; Segovia et al., 2010), we also investigated its role in regulating pool size. In syt-1−/−; syt-7−/− (syt-1/syt-7dko) cells, neither SNAP-25A nor SNAP-25B changed residual burst release (syt-1/syt-7dko, 52.8 ± 5.9 fF, n = 23; syt-1/syt-7dko+SNAP-25B, 52.5 ± 8.3 fF, n = 24; p = 0.9728 vs dko; syt-1/syt-7dko+SNAP-25A, 45.2 ± 4.2 fF, n = 23; p = 04464 vs SNAP-25B; unpaired t test) (Fig. 6C). However, the sustained rate was significantly increased by expression of SNAP-25A for yet unknown reasons (syt-1/syt-7dko, 14.0 ± 2.1 fF/s, n = 23; syt-1/syt-7dko+SNAP-25A, 27.6 ± 4.4 fF/s, n = 23; p = 0.0075 vs dko; syt-1/syt-7dko+SNAP-25B, 12.7 ± 2.3 fF/s, n = 24; p = 0.004 vs SNAP-25B; unpaired t test).
In summary, our results show that syt-1 is essential for the SNAP-25B-mediated expansion of RRP and SRP. Removing both calcium sensors for catecholamine secretion abolished the effects of SNAP-25B overexpression on burst release. Taking our biochemical findings into account, these data suggest that modulation of syt-1 × SNARE interactions by usage of different SNAP-25 splice isoforms constitutes a physiologically important mechanism to developmentally regulate release properties.
Loss of acidic surface charges causes defects in vesicle docking
We recently showed that syt-1 mediates docking of chromaffin granules via SNAP-25 interaction, likely by associating with the t-SNARE acceptor complex (de Wit et al., 2009). Accordingly, one might expect that docking should mainly rely on calcium-independent SNAP-25 × syt-1 interactions. Indeed, we reported that expression of the charge-reversal SNAP-25A D51K, E52K, E55K mutation in munc18-1 null cells could not facilitate docking, in contrast to wild-type SNAP-25A under the same conditions (de Wit et al., 2009). To more directly investigate the role of the negative surface charges in docking, we expressed SN25A S1-3A, S2-C4A, S2-N2A, and SNAP-25B S1-3A in Snap-25−/− cells and analyzed vesicle distributions by electron microscopy. In line with earlier work, uninfected Snap-25−/− cells exhibited a strongly reduced density of chromaffin granules near the plasma membrane (Fig. 7A–C). This defect could be fully rescued by viral expression of wild-type SNAP-25A and SNAP-25B. Although vesicle docking was unaffected by the type of expressed SNAP-25 isoform (Fig. 7C), all three mutant variants were unable to fully reconstitute docking in Snap-25−/− cells. Cells expressing the SN25A S1-3A variants displayed very prominent docking defects that were indistinguishable from the deficits observed in Snap-25−/− cells (SN25A S1-3A, 7.8 ± 0.7 vesicles/section, n = 20; SN25B S1–3A, 11.0 ± 1.3 vesicles/section, n = 20; ko, 5.8 ± 1.1, n = 12; ANOVA/Tukey-Kramer test: p[SN25A S1–3A vs ko] = 0.996; p[SN25B S1–3A vs ko] = 0.652) (Fig. 7C). Our analysis also showed severe docking defects in SN25A S2-C4A- and SN25A S2-N2A-expressing Snap-25−/− cells (Fig. 7C), but the reconstituted level of docking was on average significantly higher than in cells expressing SN25A S1–3A (SN25A S2-C4A, 19.0 ± 1.8 vesicles/section, n = 20; SN25A S2-N2A, 17.0 ± 2.2 vesicles/section, n = 20; p[SN25A S1–3A vs SN25A S2-C4A] = 0.001; p[SN25A S1–3A vs SN25A S2-N2A] = 0.015). Thus, all three groups of acidic surface charges play a role in the vesicle docking mechanism. Because vesicles that fuse upon physiological stimuli only amount to a fraction of the docked pool, a strict correlation between morphological and functional deficits cannot be expected. Nevertheless, the absence of isoform-dependent morphological differences (both between native and mutated proteins) confirms that deviating release deficits reflect downstream differences in priming and fusion.
Figure 7.
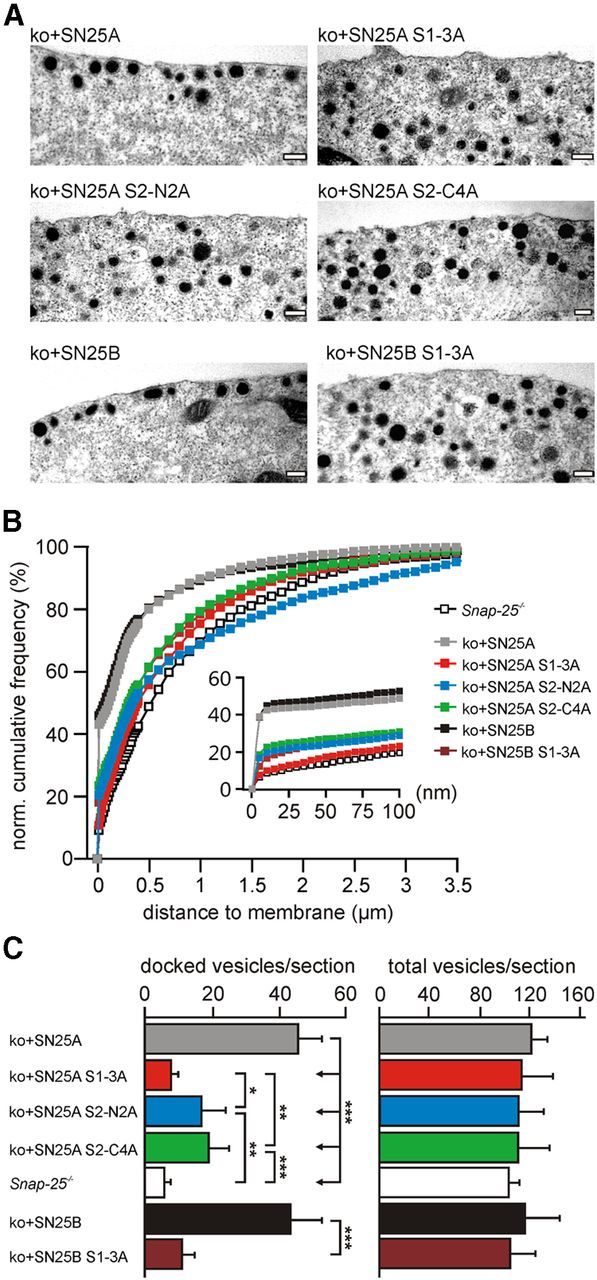
Mutant SNAP-25 variants do not restore docking of secretory granules in Snap-25−/− chromaffin cells. A, Electron micrographs of Snap-25−/− chromaffin cells expressing syt-1-binding mutants. Scale bar, 200 nm. B, Normalized cumulative vesicle distribution as a function of distance to the plasma membrane. Inset, The cumulative vesicle distribution in a membrane-proximal region of 0–100 nm. C, For each section, the number of docked vesicles and the total vesicle number were quantified. Vesicles were considered “docked” if there was no detectable separation from the plasma membrane. For each condition, 20 infected Snap-25−/− cells were analyzed in a blind fashion; 12 uninfected cells were used as controls. Error bars indicate SEM. Data were statistically compared using ANOVA and post hoc test (Tukey-Kramer). *p < 0.05. **p < 0.01. ***p < 0.001.
Discussion
Syt-1 interaction sites in SNAP-25
We used rescue experiments and fast measurement techniques to evaluate the mechanistic role of putative syt-1 interaction sites in SNAP-25. This approach is instrumental in verifying whether elimination of possible interaction sites results in the expected secretion phenotype in living cells (i.e., a slowdown of release), which mimics and occludes the effect of removing syt-1. Our experiments provided evidence that a central group of acidic amino acids (D51, E52, E55) in SNAP-25A is essential for syt-1-mediated fusion triggering. This conclusion is supported by several findings: (1) GST-pull-down experiments indicated less binding of the syt-1 C2AB-domain to t-SNARE complexes harboring the central layer mutations; (2) rescue experiments with the mutant protein profoundly decreased release rates for burst secretion phenocopying a syt-1 deficiency and (3) increased the threshold for Ca2+-dependent release; and (4) overexpression of SN25A S1–3A in syt-1−/− cells did not exacerbate the secretion phenotype.
Our results are in agreement with work by Rickman et al. (2006) who used lysine substitutions to disable the same motif. Here, we used alanine substitutions to avoid unspecific effects of a local charge inversion. Pull-down experiments with the SN25A S1–3A variant demonstrated a significant reduction of retained t-SNARE dimers on immobilized C2AB-domains, albeit to a lesser extent than for the corresponding lysine substitutions. Obviously, cooperativity effects might contribute to the observed clear-cut secretion phenotype, or a fraction of syntaxin:SN25A S1–3A dimers might engage in mechanistically unproductive syt-1 binding in vitro. Finally, it should be noted that, although SNARE complex assembly with these mutants was almost normal in vitro (Fig. 1Ba), SNARE complex assembly in vivo occurs in the presence of Munc18–1, Munc13/CAPS, and complexins, which could involve other interactions with the central charged region.
Our data indicate that the central syt-1 interface also extends to S2 of SNAP-25, specifically to residues D166 and E170, which are located close to layer 0. In support of this notion, mutation of D166 and E170 resulted in the following: (1) a weakened binding of t-SNARE dimers to syt-1 in pull-down experiments, (2) loss of the fast burst of secretion, (3) an increased Ca2+ threshold for secretion, and (4) an isoform-dependent effect, with a stronger phenotype in the context of SNAP-25 isoform B. However, as with the S1–3A mutation, we cannot exclude that the acidic residues might mediate additional syt-1-independent functions. The comparably strong decrease in total primed pool size in SN25A S2-N2A-expressing cells may indicate such a further role. Indeed, Chen et al. (2005) proposed that E170 is required for normal core complex assembly by interacting with Q174 in the polar layer 0, and interference with assembly of layer 0 is known to compromise vesicle priming (Sørensen et al., 2006). Nevertheless, we found almost normal SNARE complex formation with the SN25A N2A mutant (Fig. 1Bb). Although other cellular factors may also functionally interact with the acidic motif, the existence of a larger central syt-1 binding interface formed by residues in S1 and S2 would be in accord with recent single molecule FRET measurements (Choi et al., 2010).
Alanine substitutions of acidic residues in the C-terminal motif (D172, D179, D186, D193) of SNAP-25A did not significantly delay fusion triggering. Rather, the SNAP-25A mutation caused a moderate reduction of RRP size, which would be attributed to a defective SRP-to-RRP conversion (Sørensen, 2004). This might in principle be caused by a defective syt-1 interaction, but the mutation did not compromise syt-1 binding in C2AB pull-down reactions (Fig. 1C). However, the different properties of the SNARE complex containing this variant (Fig. 1Bb) might cause association with C2AB or other cellular factors in an abnormal configuration. When introduced into SNAP-25 isoform B, the quadruple mutation caused mild kinetic deficits in addition to a RRP reduction. Overall, our data suggest that the C-terminal acidic residues only serve a subsidiary role in the triggering mechanism.
SNAP-25 × syt-1 interactions depend on isoform type and determine priming stability
The two putative syt-1 interaction sites were first identified in SNAP-25 isoform B (Zhang et al., 2002; Rickman et al., 2006), which prompted us to test the effects of the mutations in both isoforms. Surprisingly, we found that, although SNAP-25B supports a larger secretory burst than SNAP-25A, the mutations invert the potency rank order, so that the SNAP-25B mutations support even less and/or slower secretion than mutant SNAP-25A. Thus, we also investigated the effect of overexpressing SNAP-25B in syt1−/− cells. Strikingly, we found that the larger secretion amplitude supported by SNAP-25B is confined to syt-1-expressing cells and that SNAP-25B interacts stronger with syt-1 in pull-down experiments, indicating that SNAP-25B owes its larger secretion amplitude to syt-1 interactions.
How can SNAP-25B × syt-1 interactions mechanistically regulate the size of the primed vesicle pool? The loss of RRP and SRP in cells expressing SN25B S1–3A indicates that central syt-1 × SNARE interactions are crucial for the control of pool size. As the SN25B S1–3A phenotype is not caused by chronic pool depletion, the defect likely reflects the loss of a syt-1 × SNAP-25B-dependent stabilizing effect on primed vesicles. Moreover, both SNAP-25 isoforms support a similar sustained rate of release (Fig. 6A), indicating similar forward priming rates (at high [Ca2+]i). Therefore, the difference likely is in the unpriming rate, which must be higher for SNAP-25A. This aligns with the increased syt-1 × SNAP-25B association seen in biochemical experiments, indicating that syt-1 × SNAP-25 interactions play a decisive role for determining priming stability and thereby the size of the primed vesicle pool. The reason for the stronger syt-1 × SNAP-25B interaction might either be an additional interaction of syt-1 with the side chains of the SNAP-25B-specific residues or a slightly higher intrinsic stability of SNAP-25B-containing SNARE complexes (Nagy et al., 2005), which could stabilize the interaction surface. These options need to be addressed in future studies.
Syt-1 × SNAP-25 interactions in vesicle docking
All mutants exhibited a general inability to rescue vesicle docking. The S1–3A mutation caused the most severe docking phenotype and was unable to reconstitute docking over the level found in Snap-25−/− cells, whereas SN25A S2-N2A and SN25A S2-C4A restored docking at intermediate levels. Previous experiments have demonstrated that SNAP-25-mediated docking (in Munc18-1−/− cells) is impaired by mutations in the central hydrophobic layers or deletion of the C-terminal 26 amino acids (de Wit et al., 2009). Thus, docking deficits are more widespread than effects on fusion triggering. Indeed, the relationship between docking and functional priming is complicated: some “dead-end” docked vesicles are unable to prime and fuse (Verhage and Sørensen, 2008), whereas other vesicles fuse without traversing through a detectable docked state (Kishimoto et al., 2005; Degtyar et al., 2007). In general, however, docking represents a mechanistic step preceding fusion, and a ternary complex consisting of syntaxin, SNAP-25, and syt-1 seems to be required for docking in chromaffin cells (Toonen et al., 2006; de Wit et al., 2009).
Because mutation of all three groups of negative charges compromised docking, it might be concluded that syt-1 × SNARE interactions during docking are less strictly defined than those in later mechanistic steps and even involve weak interactions not detectable in pull-down assays. If docking is mostly based on electrostatic interaction, overall charge density of the t-SNARE complex might be a critical determinant for syt-1 interactions. This would suggest a model where initial transient and promiscuous interactions between relatively large numbers of SNAREs and synaptotagmins govern attachment to the plasma membrane (Knowles et al., 2010), leading gradually to more specific interaction modes, and concluding in a highly defined primed vesicle state, which might involve only a few ternary SNARE complexes (Mohrmann and Sørensen, 2012). This idea of dynamic interactions is supported by single-molecule FRET data and electron paramagnetic resonance experiments, which showed multiple interaction modes between syt-1 and SNAREs (Choi et al., 2010; Vrljic et al., 2010; Lai et al., 2011).
The secretion mechanism: how to couple a calcium sensor onto SNAREs
A comparison of mutations in the inner layers of the SNARE complex (Sørensen et al., 2006; Walter et al., 2010) to mutations of acidic surface motifs allows inferences about the mechanism of calcium coupling. Generally, mutating the inner layers in the N-terminal part or the middle of the SNARE complex only affects vesicle priming, but not triggering. However, mutations of negative surface charges around the middle of the complex delay fusion kinetics (this study), much like mutations within the last couple of C-terminal layers (Sørensen et al., 2006; Walter et al., 2010). This pattern of effects is consistent with a mechanism, in which the N-terminal part of the SNARE complex forms a relatively stiff rod during priming. Triggering would involve the interaction of syt-1 with this structure to allow for subsequent C-terminal assembly. This is in agreement with the notion that syt-1 and complexin arrest the SNARE complex in a configuration that is incompatible with C-terminal assembly (Giraudo et al., 2009; Maximov et al., 2009; Kaeser-Woo et al., 2012). The clamp will be lifted by Ca2+-binding to syt-1, allowing for C-terminal SNARE complex assembly. During this process, syt-1 might dissociate from the SNARE complex or, alternatively, it might stay bound, producing a quaternary complex (Dai et al., 2007), wherein syt-1 and SNAREs cooperate to fuse the membranes.
Footnotes
The work was supported by the Lundbeck Foundation (Junior Group Leader Fellowship), the Lundbeck Foundation Center for Biomembranes in Nanomedicine, the Novo Nordisk Foundation, and the Danish Medical Research Council (all J.B.S.), the University of Saarland (HOMFOR 2010) to R.M., Deutsche Forschungsgemeinschaft (SFB 894) to D.B., The Netherlands Organization for Scientific Research, NWO (VENI 916-36-043) to H.d.W., the Medical Research Council United Kingdom (U10578791) to B.D., and the European Union Seventh Framework Programme Grant agreements FP7-People-ITN-2008-238055 (BrainTrain) to H.d.W., FP7-People-2009-IEF-254647 (Exosyts) to P.S.P., and HEALTH-F2-2009-242167 (SynSys) to J.B.S. and M.V. We thank I. Herfort, D. Reuter, A.M.N. Petersen, and W. Frisch for excellent technical assistance.
The authors declare no competing financial interests.
References
- Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J. Close membrane-membrane proximity induced by Ca(2+)-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol. 2004a;11:36–44. doi: 10.1038/nsmb709. [DOI] [PubMed] [Google Scholar]
- Bai J, Wang CT, Richards DA, Jackson MB, Chapman ER. Fusion pore dynamics are regulated by synaptotagmin × t-SNARE interactions. Neuron. 2004b;41:929–942. doi: 10.1016/S0896-6273(04)00117-5. [DOI] [PubMed] [Google Scholar]
- Bark IC, Wilson MC. Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene. 1994;139:291–292. doi: 10.1016/0378-1119(94)90773-0. [DOI] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, Wilson MC. Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci U S A. 1995;92:1510–1514. doi: 10.1073/pnas.92.5.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett MK, Calakos N, Scheller RH. Syntaxin: a synaptic protein implicated in docking of synaptic vesicles at presynaptic active zones. Science. 1992;257:255–259. doi: 10.1126/science.1321498. [DOI] [PubMed] [Google Scholar]
- Bhalla A, Chicka MC, Tucker WC, Chapman ER. Ca(2+)-synaptotagmin directly regulates t-SNARE function during reconstituted membrane fusion. Nat Struct Mol Biol. 2006;13:323–330. doi: 10.1038/nsmb1076. [DOI] [PubMed] [Google Scholar]
- Capogna M, McKinney RA, O'Connor V, Gähwiler BH, Thompson SM. Ca2+ or Sr2+ partially rescues synaptic transmission in hippocampal cultures treated with botulinum toxin A and C, but not tetanus toxin. J Neurosci. 1997;17:7190–7202. doi: 10.1523/JNEUROSCI.17-19-07190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER. How does synaptotagmin trigger neurotransmitter release? Annu Rev Biochem. 2008;77:615–641. doi: 10.1146/annurev.biochem.77.062005.101135. [DOI] [PubMed] [Google Scholar]
- Chapman ER, Hanson PI, An S, Jahn R. Ca2+ regulates the interaction between synaptotagmin and syntaxin 1. J Biol Chem. 1995;270:23667–23671. doi: 10.1074/jbc.270.40.23667. [DOI] [PubMed] [Google Scholar]
- Chen X, Tang J, Südhof TC, Rizo J. Are neuronal SNARE proteins Ca2+ sensors? J Mol Biol. 2005;347:145–158. doi: 10.1016/j.jmb.2005.01.024. [DOI] [PubMed] [Google Scholar]
- Chicka MC, Hui E, Liu H, Chapman ER. Synaptotagmin arrests the SNARE complex before triggering fast, efficient membrane fusion in response to Ca2+ Nat Struct Mol Biol. 2008;15:827–835. doi: 10.1038/nsmb.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieregatti E, Chicka MC, Chapman ER, Baldini G. SNAP-23 functions in docking/fusion of granules at low Ca2+ Mol Biol Cell. 2004;15:1918–1930. doi: 10.1091/mbc.E03-09-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR. Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell E, Darios F, Broersen K, Gatsby N, Peak-Chew SY, Rickman C, Davletov B. Mechanism of arachidonic acid action on syntaxin-Munc18. EMBO Rep. 2007;8:414–419. doi: 10.1038/sj.embor.7400935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai H, Shen N, Araç D, Rizo J. A quaternary SNARE-synaptotagmin-Ca2+-phospholipid complex in neurotransmitter release. J Mol Biol. 2007;367:848–863. doi: 10.1016/j.jmb.2007.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AF, Bai J, Fasshauer D, Wolowick MJ, Lewis JL, Chapman ER. Kinetics of synaptotagmin responses to Ca2+ and assembly with the core SNARE complex onto membranes. Neuron. 1999;24:363–376. doi: 10.1016/S0896-6273(00)80850-8. [DOI] [PubMed] [Google Scholar]
- Degtyar VE, Allersma MW, Axelrod D, Holz RW. Increased motion and travel, rather than stable docking, characterize the last moments before secretory granule fusion. Proc Natl Acad Sci U S A. 2007;104:15929–15934. doi: 10.1073/pnas.0705406104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Martínez I, Nehring RB, Sørensen JB. Differential abilities of SNAP-25 homologs to support neuronal function. J Neurosci. 2007;27:9380–9391. doi: 10.1523/JNEUROSCI.5092-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit H, Walter AM, Milosevic I, Gulyás-Kovács A, Riedel D, Sørensen JB, Verhage M. Synaptotagmin-1 docks secretory vesicles to syntaxin-1/SNAP-25 acceptor complexes. Cell. 2009;138:935–946. doi: 10.1016/j.cell.2009.07.027. [DOI] [PubMed] [Google Scholar]
- Fasshauer D, Sutton RB, Brunger AT, Jahn R. Conserved structural features of the synaptic fusion complex: SNARE proteins reclassified as Q- and R-SNAREs. Proc Natl Acad Sci U S A. 1998;95:15781–15786. doi: 10.1073/pnas.95.26.15781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER. Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem. 2008;283:31763–31775. doi: 10.1074/jbc.M803355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–727. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Gerona RR, Larsen EC, Kowalchyk JA, Martin TF. The C terminus of SNAP25 is essential for Ca(2+)-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Garcia-Diaz A, Eng WS, Chen Y, Hendrickson WA, Melia TJ, Rothman JE. Alternative zippering as an on-off switch for SNARE-mediated fusion. Science. 2009;323:512–516. doi: 10.1126/science.1166500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Gaffaney JD, Wang Z, Johnson CP, Evans CS, Chapman ER. Mechanism and function of synaptotagmin-mediated membrane apposition. Nat Struct Mol Biol. 2011;18:813–821. doi: 10.1038/nsmb.2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Fasshauer D. Molecular machines governing exocytosis of synaptic vesicles. Nature. 2012;490:201–207. doi: 10.1038/nature11320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Südhof TC. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci. 2012;32:2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Choi BK, Choi MG, Kim SA, Lai Y, Shin YK, Lee NK. Solution single-vesicle assay reveals PIP2-mediated sequential actions of synaptotagmin-1 on SNAREs. EMBO J. 2012;31:2144–2155. doi: 10.1038/emboj.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, Liu TT, Hatakeyama H, Nemoto T, Takahashi N, Kasai H. Sequential compound exocytosis of large dense-core vesicles in PC12 cells studied with TEPIQ (two-photon extracellular polar-tracer imaging-based quantification) analysis. J Physiol. 2005;568:905–915. doi: 10.1113/jphysiol.2005.094003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles MK, Barg S, Wan L, Midorikawa M, Chen X, Almers W. Single secretory granules of live cells recruit syntaxin-1 and synaptosomal associated protein 25 (SNAP-25) in large copy numbers. Proc Natl Acad Sci U S A. 2010;107:20810–20815. doi: 10.1073/pnas.1014840107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo W, Herrick DZ, Ellena JF, Cafiso DS. The calcium-dependent and calcium-independent membrane binding of synaptotagmin 1: two modes of C2B binding. J Mol Biol. 2009;387:284–294. doi: 10.1016/j.jmb.2009.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai AL, Huang H, Herrick DZ, Epp N, Cafiso DS. Synaptotagmin 1 and SNAREs form a complex that is structurally heterogeneous. J Mol Biol. 2011;405:696–706. doi: 10.1016/j.jmb.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Shin OH, Rhee JS, Araç D, Rah JC, Rizo J, Südhof T, Rosenmund C. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. J Biol Chem. 2006;281:15845–15852. doi: 10.1074/jbc.M600888200. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Bai J, Vyas B, Desai R, Baltus AE, Garment MB, Carlson SD, Ganetzky B, Chapman ER. Synaptotagmin mutants reveal essential functions for the C2B domain in Ca2+-triggered fusion and recycling of synaptic vesicles in vivo. J Neurosci. 2001;21:1421–1433. doi: 10.1523/JNEUROSCI.21-05-01421.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Larsen EC, Marcia RF, Mitchell JC, Martin TF. Synaptotagmin C2A loop 2 mediates Ca2+-dependent SNARE interactions essential for Ca2+-triggered vesicle exocytosis. Mol Biol Cell. 2007;18:4957–4968. doi: 10.1091/mbc.E07-04-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Mol Biol Cell. 2008;19:5093–5103. doi: 10.1091/mbc.E08-03-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science. 2007;316:1205–1208. doi: 10.1126/science.1142614. [DOI] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrmann R, Sørensen JB. SNARE requirements en route to exocytosis: from many to few. J Mol Neurosci. 2012;48:387–394. doi: 10.1007/s12031-012-9744-2. [DOI] [PubMed] [Google Scholar]
- Mohrmann R, de Wit H, Verhage M, Neher E, Sørensen JB. Fast vesicle fusion in living cells requires at least three SNARE complexes. Science. 2010;330:502–505. doi: 10.1126/science.1193134. [DOI] [PubMed] [Google Scholar]
- Nagy G, Milosevic I, Fasshauer D, Müller EM, de Groot BL, Lang T, Wilson MC, Sørensen JB. Alternative splicing of SNAP-25 regulates secretion through nonconservative substitutions in the SNARE domain. Mol Biol Cell. 2005;16:5675–5685. doi: 10.1091/mbc.E05-07-0595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sørensen JB. Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne SL, Wallis TP, Jimenez JL, Gorman JJ, Meunier FA. Identification of secretory granule phosphatidylinositol 4,5-bisphosphate-interacting proteins using an affinity pulldown strategy. Mol Cell Proteomics. 2007;6:1158–1169. doi: 10.1074/mcp.M600430-MCP200. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Shin OH, Meyer AC, Rosenmund C, Südhof TC. A gain-of-function mutation in synaptotagmin-1 reveals a critical role of Ca2+-dependent soluble N-ethylmaleimide-sensitive factor attachment protein receptor complex binding in synaptic exocytosis. J Neurosci. 2006;26:12556–12565. doi: 10.1523/JNEUROSCI.3804-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Davletov B. Mechanism of calcium-independent synaptotagmin binding to target SNAREs. J Biol Chem. 2003;278:5501–5504. doi: 10.1074/jbc.C200692200. [DOI] [PubMed] [Google Scholar]
- Rickman C, Meunier FA, Binz T, Davletov B. High affinity interaction of syntaxin and SNAP-25 on the plasma membrane is abolished by botulinum toxin E. J Biol Chem. 2004a;279:644–651. doi: 10.1074/jbc.M310879200. [DOI] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B. Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem. 2004b;279:12574–12579. doi: 10.1074/jbc.M310710200. [DOI] [PubMed] [Google Scholar]
- Rickman C, Jiménez JL, Graham ME, Archer DA, Soloviev M, Burgoyne RD, Davletov B. Conserved prefusion protein assembly in regulated exocytosis. Mol Biol Cell. 2006;17:283–294. doi: 10.1091/mbc.E05-07-0620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizo J, Chen X, Araç D. Unraveling the mechanisms of synaptotagmin and SNARE function in neurotransmitter release. Trends Cell Biol. 2006;16:339–350. doi: 10.1016/j.tcb.2006.04.006. [DOI] [PubMed] [Google Scholar]
- Sakaba T, Stein A, Jahn R, Neher E. Distinct kinetic changes in neurotransmitter release after SNARE protein cleavage. Science. 2005;309:491–494. doi: 10.1126/science.1112645. [DOI] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A. 2008;105:3998–4003. doi: 10.1073/pnas.0712373105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonn JS, van Weering JR, Mohrmann R, Schlüter OM, Südhof TC, De Wit H, Verhage M, Sørensen JB. Rab3 proteins involved in vesicle biogenesis and priming in embryonic mouse chromaffin cells. Traffic. 2010;11:1415–1428. doi: 10.1111/j.1600-0854.2010.01107.x. [DOI] [PubMed] [Google Scholar]
- Scullin CS, Tafoya LC, Wilson MC, Partridge LD. Presynaptic residual calcium and synaptic facilitation at hippocampal synapses of mice with altered expression of SNAP-25. Brain Res. 2012;1431:1–12. doi: 10.1016/j.brainres.2011.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia M, Alés E, Montes MA, Bonifas I, Jemal I, Lindau M, Maximov A, Südhof TC, Alvarez de Toledo G. Push-and-pull regulation of the fusion pore by synaptotagmin-7. Proc Natl Acad Sci U S A. 2010;107:19032–19037. doi: 10.1073/pnas.1014070107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao X, Li C, Fernandez I, Zhang X, Südhof TC, Rizo J. Synaptotagmin-syntaxin interaction: the C2 domain as a Ca2+-dependent electrostatic switch. Neuron. 1997;18:133–142. doi: 10.1016/S0896-6273(01)80052-0. [DOI] [PubMed] [Google Scholar]
- Söllner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, Rothman JE. SNAP receptors implicated in vesicle targeting and fusion. Nature. 1993;362:318–324. doi: 10.1038/362318a0. [DOI] [PubMed] [Google Scholar]
- Sørensen JB. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 2004;448:347–362. doi: 10.1007/s00424-004-1247-8. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Matti U, Wei SH, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci U S A. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/S0092-8674(03)00477-X. [DOI] [PubMed] [Google Scholar]
- Sørensen JB, Wiederhold K, Müller EM, Milosevic I, Nagy G, de Groot BL, Grubmüller H, Fasshauer D. Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J. 2006;25:955–966. doi: 10.1038/sj.emboj.7601003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, Brunger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Südhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sørensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau LE, Fang Y, Haydon PG. Modulation of an early step in the secretory machinery in hippocampal nerve terminals. Proc Natl Acad Sci U S A. 1998;95:7163–7168. doi: 10.1073/pnas.95.12.7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Thutupalli S, Risselada JH, Meyenberg K, Holt M, Riedel D, Diederichsen U, Herminghaus S, Grubmüller H, Jahn R. Synaptotagmin-1 may be a distance regulator acting upstream of SNARE nucleation. Nat Struct Mol Biol. 2011;18:805–812. doi: 10.1038/nsmb.2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bogaart G, Meyenberg K, Diederichsen U, Jahn R. Phosphatidylinositol 4,5-bisphosphate increases Ca2+ affinity of synaptotagmin-1 by 40-fold. J Biol Chem. 2012;287:16447–16453. doi: 10.1074/jbc.M112.343418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhage M, Sørensen JB. Vesicle docking in regulated exocytosis. Traffic. 2008;9:1414–1424. doi: 10.1111/j.1600-0854.2008.00759.x. [DOI] [PubMed] [Google Scholar]
- Voets T. Dissection of three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/S0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Südhof TC, Neher E. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A. 2001;98:11680–11685. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrljic M, Strop P, Ernst JA, Sutton RB, Chu S, Brunger AT. Molecular mechanism of the synaptotagmin-SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol. 2010;17:325–331. doi: 10.1038/nsmb.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter AM, Wiederhold K, Bruns D, Fasshauer D, Sørensen JB. Synaptobrevin N-terminally bound to syntaxin-SNAP-25 defines the primed vesicle state in regulated exocytosis. J Cell Biol. 2010;188:401–413. doi: 10.1083/jcb.200907018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Ma C, Craig TK, Rosenmund C, Rizo J. The Janus-faced nature of the C(2)B domain is fundamental for synaptotagmin-1 function. Nat Struct Mol Biol. 2008;15:1160–1168. doi: 10.1038/nsmb.1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TF. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/S0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]