

Abstract
Chemical separation has great importance in industrial applications. Separation of xylene isomers still prevails to be one of the most important challenges in chemical industry, due to the large amount of commercial use of p-xylene in the production of beverage bottles, fibers and films. A novel Zn(II)-based dynamic coordination framework based on flexible ether-linkage, exhibiting selective adsorption of p-Xylene over its congener C8-alkyl aromatic isomers at ambient conditions is reported. Notably, no dynamic structure based MOF compound is known in the literature which shows clear preference of p-xylene over other isomers. This type of framework-breathing and guest-induced reversible solid-state structural transformations with unique adsorption selectivity can be exploited purposefully to develop smart functional host materials capable of industrially important chemical separations.
Metal–organic frameworks (MOFs) or porous coordination polymers (PCPs) are in a continuous stage of exploration targeting a vast array of prospective applications, including gas storage, separation, heterogeneous catalysis, drug delivery, sensing, proton conductivity etc. owing to their easy-accessible high surface area, tunable pore functionality and structural flexibility1,2,3,4,5,6,7,8,9,10,11,12,13,14,15. Separation is one of the most important aspects in chemical industry. Xylene isomers separation is one of them, as illustrated by the statistic that only considering the U.S., there have been as many as 130 patents awarded and applied for in the span of 2006–2009S1. Remarkably, the entire world's mixed xylene production in 2008 was about 39.2 Mt. Among all isomers, p-xylene (PX) has the most extensive commercial use with more than 80% of the world production of xylenes being used for the production of PX, because of its expansive commercial usage as a feedstock in manufacture of terephthalic acid, a monomer used in the production of polymer polyethylene terephthalate (PET), widely utilized in the production of beverage bottles, fibres and films16. In this context, separation of C8-alkylaromatic components, specifically the xylene product stream comprising of four isomers: o-xylene (OX), m-xylene (MX), p-xylene (PX), and ethylbenzene (EB) is a pressing issue, since the separation of PX (kinetic diameter ~0.58 nm) from its bulkier m- and o-isomers (MX and OX; ~0.64 nm and 0.65 nm respectively) is of extreme importance in the petrochemical industry. This is practically infeasible by distillation (closely matched boiling points; 138°C–144°C). Among the recognized methods, fractional crystallization and adsorption using zeolites are mostly employed to separate xylene isomers, where adsorption has been markedly proved to be of higher efficiency and gained broader acceptance16,17.
In recent times, few MOFs especially, MIL-47, MIL-53, MIL-125(Ti) have been found appropriate for execution of liquid-phase purifications and separations of xylene isomers18,19,20,21. These primarily focus on high-temperature vapor-phase and liquid-phase binary breakthrough chromatographic experiments, chiefly by virtue of the molecular sieving phenomena aimed at the simulated moving bed technology employed in industry at around 180°C and 9 bar. Very few rigid porous MOFs have been reported in the literature exhibiting a clear para preference, which is highly desired for an absorbent to be used for separation of PX from mixture of C8-alkylaromatic components16,17,18,19,20,21,22,23,24,25,26,27. Notably, no dynamic structure based MOF is reported which shows clear para preference. Framework flexibility and extensive breathing phenomena in MIL-53 (Al) resulted in well-defined two-step adsorption isotherms at 110°C, although showing similar uptake amounts for all the four isomeric probe adsorptives28. The aspect of framework flexibility can be exploited to achieve a clear adsorption-selectivity among xylene isomers at room temperature to develop smart functional host materials. Although the strategy of pore surface functionalization has been proficiently exploited in rigid frameworks for chemical separation and storage; analogous examples are still scarcely reported for dynamic systems29,30,31,32,33,34,35,36, perhaps due to the difficulties in proper strategic design and correlation of structural attributes coupled with dynamic behavior of such materials. The exploration of such kind of systems might serve as a significant way-out for achieving separation of small molecules with structural similarity. With this background, we have synthesized a dicarboxylate based ligand, with two ether linkages which can act as flexible nodes. Additionally, Zn(II) is highly accommodative to build dynamic frameworks, since it has versatile coordination chemistry and can readily adopt either of tetrahedral and octahedral geometries with four and six coordination numbers respectively. Construction of an inherently flexible framework essentially involves the cooperative interplay of these two factors. Herein, we report dynamic behavior of a MOF demonstrated by solid state structural transformations between porous and non-porous phases, exhibiting structural flexibility based selective sorption of p-xylene over the other xylene isomers in vapor phase at ambient temperature (Fig. 1).
Figure 1. Schematic illustration of selective guest-responsive framework flexibility.

Schematic representation of the framework flexibility with selective guest-accommodation.
Results
The compound {[Zn4O(L)3(DMF)2].xG}n(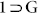 ) was synthesized by solvothermal reaction of Zn(NO3)2·6H2O with newly designed flexible dicarboxylic acid ligand H2L (Fig. 2, S1) in a solvent mixture of DMF/1-butanol (1:1). Single-crystal X-ray diffraction (SC-XRD) studies revealed that the compound
) was synthesized by solvothermal reaction of Zn(NO3)2·6H2O with newly designed flexible dicarboxylic acid ligand H2L (Fig. 2, S1) in a solvent mixture of DMF/1-butanol (1:1). Single-crystal X-ray diffraction (SC-XRD) studies revealed that the compound  is a porous 2D sheet structure (Fig. S10–13), which crystallized in monoclinic centrosymmetric space group P21/c (Table S2). Adsym subroutine of PLATON was applied to check that no additional symmetry could be applied to the model. The asymmetric unit comprises of four Zn(II) units, one central O2−, three L2−, and two N,N'-dimethyl formamide (DMF) molecules, both bound particularly to one of the metal centers (Zn4 centre as shown in Fig. S8). Therefore, a {Zn4O(CO2)6(DMF)2} secondary building unit (SBU) is formed by involving six carboxylates from six different L2− ligand units and two O-atoms of two DMF molecules. The SBU cluster core consists of a single O atom bonded to four Zn atoms, consequently forming regular tetrahedrons for three zinc centers and octahedron for the other zinc. Each Zn4 cluster is connected to another Zn4 cluster though two L2− ligand to form a one dimensional (1D) chain. This 1D chain is again connected to similar another two chains on both side via L2− to form the resultant 2D framework. This porous framework comprises of free disordered guest solvent molecules (G). PLATON analysis revealed that the compound
is a porous 2D sheet structure (Fig. S10–13), which crystallized in monoclinic centrosymmetric space group P21/c (Table S2). Adsym subroutine of PLATON was applied to check that no additional symmetry could be applied to the model. The asymmetric unit comprises of four Zn(II) units, one central O2−, three L2−, and two N,N'-dimethyl formamide (DMF) molecules, both bound particularly to one of the metal centers (Zn4 centre as shown in Fig. S8). Therefore, a {Zn4O(CO2)6(DMF)2} secondary building unit (SBU) is formed by involving six carboxylates from six different L2− ligand units and two O-atoms of two DMF molecules. The SBU cluster core consists of a single O atom bonded to four Zn atoms, consequently forming regular tetrahedrons for three zinc centers and octahedron for the other zinc. Each Zn4 cluster is connected to another Zn4 cluster though two L2− ligand to form a one dimensional (1D) chain. This 1D chain is again connected to similar another two chains on both side via L2− to form the resultant 2D framework. This porous framework comprises of free disordered guest solvent molecules (G). PLATON analysis revealed that the compound 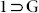 comprises of large voids of 3,441.2 Å3, which represent 34.2% per unit cell volume. PXRD for the bulk phase of this compound
comprises of large voids of 3,441.2 Å3, which represent 34.2% per unit cell volume. PXRD for the bulk phase of this compound 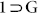 and its simulated pattern matched precisely, indicative of the phase purity (Fig. S19), which is also supported by SC-XRD analysis of randomly selected crystals.
and its simulated pattern matched precisely, indicative of the phase purity (Fig. S19), which is also supported by SC-XRD analysis of randomly selected crystals.
Figure 2. Strategically designed flexible ligand.

Chemical structure of the designed ligand (H2L) having two ether linkages as its flexible nodes.
Thermogravimetric analyses (TGA) and variable temperature powder X-ray diffraction (VT-PXRD) measurements (Fig. S14, S18) were carried out to examine the thermal stability of this framework. The TGA curve for  shows that all the guest molecules present in the voids as well as the coordinated DMF molecules inside the shiny single crystalline phase of
shows that all the guest molecules present in the voids as well as the coordinated DMF molecules inside the shiny single crystalline phase of 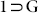 can be removed by heating at 160°C, to obtain the slightly pale colorless desolvated crystals of compound 1 (Fig. S9). Variable temperature PXRD patterns for the compound
can be removed by heating at 160°C, to obtain the slightly pale colorless desolvated crystals of compound 1 (Fig. S9). Variable temperature PXRD patterns for the compound 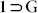 also validated this phase transformation. The thermal stability and phase purity of 1 were also confirmed by TGA and PXRD-analysis respectively (Fig. S14, S19). Furthermore, TGA plot showed that the sample is stable up to 410°C. The release of free guest molecules along with the coordinated DMF molecules led to a drastic structural transformation, giving rise to squeezed non-porous framework [Zn4O(L)3]n (1), from porous
also validated this phase transformation. The thermal stability and phase purity of 1 were also confirmed by TGA and PXRD-analysis respectively (Fig. S14, S19). Furthermore, TGA plot showed that the sample is stable up to 410°C. The release of free guest molecules along with the coordinated DMF molecules led to a drastic structural transformation, giving rise to squeezed non-porous framework [Zn4O(L)3]n (1), from porous 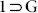 . As evidenced from the SC-XRD analysis of both the phases, this single-crystal to single-crystal (SCSC) solid state structural transformation (Fig. 3) is due to the slippage of 2D layers with respect to each other upon guest removal. PLATON analysis for 1 revealed that the ensuing 2D framework [space group P21/c (Table S3)] underwent considerable squeezing (void volume of 915.8 Å3, 12.8 % per unit cell volume) from
. As evidenced from the SC-XRD analysis of both the phases, this single-crystal to single-crystal (SCSC) solid state structural transformation (Fig. 3) is due to the slippage of 2D layers with respect to each other upon guest removal. PLATON analysis for 1 revealed that the ensuing 2D framework [space group P21/c (Table S3)] underwent considerable squeezing (void volume of 915.8 Å3, 12.8 % per unit cell volume) from  (void volume ~ 3,441.2 Å3, 34.2% per unit cell volume). The coordination environment for all the four Zn-centers has now become same with tetrahedral geometry (Fig. S8). Each of the edges of clusters is again connected to generate the resultant 2D framework. To examine the reversibility aspect of the structural transformation, the bulk crystalline phase 1 was kept immersed in a solution of 1-butanol (1 mL) and DMF (1 mL) for 24 h without any disturbance. PXRD analysis (Fig. S19) and SC-XRD analysis of arbitrarily chosen crystals of this resolvated phase
(void volume ~ 3,441.2 Å3, 34.2% per unit cell volume). The coordination environment for all the four Zn-centers has now become same with tetrahedral geometry (Fig. S8). Each of the edges of clusters is again connected to generate the resultant 2D framework. To examine the reversibility aspect of the structural transformation, the bulk crystalline phase 1 was kept immersed in a solution of 1-butanol (1 mL) and DMF (1 mL) for 24 h without any disturbance. PXRD analysis (Fig. S19) and SC-XRD analysis of arbitrarily chosen crystals of this resolvated phase  confirmed about the reversible structural transformations of this dynamic framework.
confirmed about the reversible structural transformations of this dynamic framework.
Figure 3. Reversible solid state structural transformations in SCSC fashion: Breathing phenomenon.

Reversible structural transformation from porous compound  to non-porous 1 viewed along a-axis (hydrogen atoms and free solvent molecules are omitted for clarity.
to non-porous 1 viewed along a-axis (hydrogen atoms and free solvent molecules are omitted for clarity.
To examine the permanent porosity and sorption performance for the compound 1, gas adsorption property of compound 1 was comprehensively investigated. N2, H2, Ar, CH4 and CO2 sorption experiments were carried out in a relative pressure range from 10−4 to 1 atm at 77 K (N2, H2 and Ar) and 195 K (CH4 and CO2). Compound 1 exhibited differential adsorption behaviour towards CO2 (195 K) as compared to those for N2 (77 K), H2 (77 K), Ar (77 K), CH4 (195 K) (Fig. S24–26). Gas adsorption measurements were done volumetrically using activated compound 1. Adsorption analysis of N2 (77 K), H2 (77 K), Ar (77 K) and CH4 (195 K) showed only negligible uptake (23, 14, 6.7, 27.65 mLg−1 for N2, H2, Ar and CH4 respectively), but notably, significant uptake was observed for CO2 (114 mLg−1) with a typical Type-1 sorption profile. The H-K (Haworth-Kawazoe) plot for CO2 sorption at 195 K (Fig. S28) revealed an effective pore diameter of ~5.1 Å, which is relatively larger than all the probe adsorptive-gases, indicative of the fact that size-selectivity between different probe adsorptive species is not the only deciding factor. This also evidently suggests that the larger quadruple moment of CO2 (1.34 × 10−39 Cm2) plays the most crucial role behind the low-temperature CO2-selective adsorption phenomena37,38,39, since the selective uptake of CO2 by the dynamic framework 1 can be attributed to the favorable electrostatic interactions with the polar pore surface. Since the adsorption amounts of other gases (H2, O2, N2, Ar, and CH4) are much negligible compared to that for CO2, the compound can prove expedient for separation of CO2 at low temperature.
Microporous and flexible nature of the activated phase 1 with pore diameter close to the kinetic diameter of the xylenes (~0.6 nm), prompted us to investigate the adsorption characteristics for compound 1 systematically, for achieving separation of xylenes having similar physical properties and comparable molecular sizes. The vapor sorption experiments for the π-electron rich solvents like benzene (BZ) and toluene (TL), p-xylene (PX), m-xylene (MX) and o-xylene (OX) were measured along with nonaromatic solvent cyclohexane (CY) at 298 K. As an important observation, significant amounts of π-e− rich guests like benzene, and toluene were adsorbed (98 mLg−1 and 73 mLg−1 respectively), while as a simultaneous feature, analogous aliphatic guest cyclohexane parallely recorded a high uptake amount (74 mLg−1) at 298 K (Fig. S29). These sorption profiles reinforced the coexistence of favorable π-π interactions between the π-electron rich polar framework (operative for aromatic guests namely, benzene and toluene) and CH-π interactions (functional for aliphatic solvent cyclohexane) as the dual crucial influences playing their parts for the respective host-guest interactions. Remarkably, the adsorption isotherms recorded for the three isomers of xylene, i.e. PX, MX, OX along with EB at 298 K presented selective adsorption of PX over the other three isomers. While the adsorption amount for the p-isomer was found to be 64 mLg−1, the corresponding uptake amounts for the m- and o-isomers were surprisingly much lower, 5 mLg−1 and 1.15 mLg−1 respectively (Fig. S31), corresponding to 4 molecules of PX-uptake per formula unit, while only 0.34 and 0.08 molecules uptake for MX and OX respectively (Fig. 4). EB also recorded a very marginal uptake of only 9.4 mLg−1 (0.63 molecules per formula unit). This differential adsorption behavior towards the four constitutional isomers consolidated that the framework flexibility of desolvated phase 1 allow the entry of the linear guest PX but not the bent ones, namely MX and OX, due to steric hindrance that originates in the case of the latter two. Meanwhile, EB possessing the ethyl side group has a minimum dimension of 5.285 Å (‘MIN-1', Table S1)40, much higher than the congeners making its entry improbable. ‘MIN-1' for all the other three isomers being very similar, the second minimum dimension for molecular orientations that enable a molecule to enter the channel, ‘MIN-2' (Table S1) comes into play, which is considerably lower for p-xylene (6.618 Å) than the other two (7.269Å and 7.258 Å for m- and o-isomers respectively). Interestingly, the effective pore diameter of 5.1Å, as obtained from HK-pore size distribution plot for the microporous adsorbent host material 1, is lower than all the three ‘MIN-2' values for each of the xylene isomers. This evidently indicates that the strategically achieved framework flexibility feature arising out of the ether-linkage based ligands, make possible the allowance of smaller PX but not the other two, due to limiting restricted flexibility which cannot let the bigger isomers enter inside the porous channels. The small variation in the molecular dimensions between these three isomeric guests might be the only crucial reason in deciding the selective entry of PX, while refusing the flexible framework 1 to open for OX and MX, owing to steric hindrance. Hence, the aspect of size-selectivity proves decisive in determining the observed adsorption-selectivity via guest-inclusion in case of xylene isomers.
Figure 4. p-Xylene selective Sorption isotherm.

Solvent sorption isotherms for compound 1 at 298 K for three of the xylene isomers along with isomeric ethylbenzene. Closed and open symbols denote adsorption and desorption respectively.
To confirm the structural transformations occurring on the interplay of host-guest interactions, TGA plots and PXRD profiles for each of the guest-exposed phases were recorded (Fig. 5, S15–17, S20–21) which conclusively reaffirmed the results suggested from solvent sorption studies, as MX and OX could do no change to the characteristic PXRD pattern for 1, while the other solvents were able to mediate the dynamic transformation from 1 to 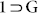 phases, having similar PXRD forms as for
phases, having similar PXRD forms as for 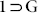 . TGA results also strengthened this observation since no significant weight loss was observed for the phases
. TGA results also strengthened this observation since no significant weight loss was observed for the phases 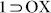 and
and 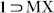 , but all the other guest solvent vapor-exposed phases revealed considerable loss in %weight within 100°C, indicative of loss of trapped solvent- guests. To further reinforce the selective interplay of PX with the flexible framework 1 as compared to OX and MX, PXRD profiles for binary and ternary solvent vapor-exposed phases were recorded (Fig. S23), which was reiterated by 13C NMR experiments with the DCl/DMSO-d6 digested samples after vapor exposure to three different binary mixtures (1:1) of the xylene isomers (Fig. S32).
, but all the other guest solvent vapor-exposed phases revealed considerable loss in %weight within 100°C, indicative of loss of trapped solvent- guests. To further reinforce the selective interplay of PX with the flexible framework 1 as compared to OX and MX, PXRD profiles for binary and ternary solvent vapor-exposed phases were recorded (Fig. S23), which was reiterated by 13C NMR experiments with the DCl/DMSO-d6 digested samples after vapor exposure to three different binary mixtures (1:1) of the xylene isomers (Fig. S32).
Figure 5. Powder X-Ray Diffraction studies.

Experimental PXRD patterns of 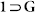 , 1, 1
, 1, 1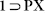 PX, 1
PX, 1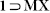 MX, 1
MX, 1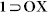 OX and
OX and 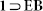 compounds.
compounds.
Although the crystals of 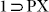 did not diffract while repeated attempts were made to get the crystal structure of the phase
did not diffract while repeated attempts were made to get the crystal structure of the phase 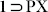 , a similar resolvated phase
, a similar resolvated phase 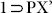 (as indicated from the similar PXRD patterns of both) (Fig. S20) was obtained on exposing the crystals of 1 to the vapor of a solution of p-Xylene (2 mL) and DMF (1 mL) for 72 h. SC-XRD analysis of this new phase
(as indicated from the similar PXRD patterns of both) (Fig. S20) was obtained on exposing the crystals of 1 to the vapor of a solution of p-Xylene (2 mL) and DMF (1 mL) for 72 h. SC-XRD analysis of this new phase 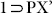 (formula: {[Zn4O(L)3(DMF)2].(C8H10)}n) revealed that the compound has again transformed to the open phase, having unit cell parameters very similar to the crystals of
(formula: {[Zn4O(L)3(DMF)2].(C8H10)}n) revealed that the compound has again transformed to the open phase, having unit cell parameters very similar to the crystals of 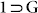 and crystallized in monoclinic centrosymmetric space group P21/c (Table S4). p-xylene molecules could actually be located in the SC-XRD structure for this
and crystallized in monoclinic centrosymmetric space group P21/c (Table S4). p-xylene molecules could actually be located in the SC-XRD structure for this 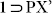 phase crystals, sitting inside the porous channels of the host framework (Fig. 6), after it could open up the shrinked windows of desolvated phase 1 to get accommodated inside the hydrophobic channels, owing to the inherent dynamism of the framework, arising out of the highly flexible ether nodes. 13CNMR experiment also proved the inclusion of PX (Fig. S33). After confirming the phase purity for the new phase
phase crystals, sitting inside the porous channels of the host framework (Fig. 6), after it could open up the shrinked windows of desolvated phase 1 to get accommodated inside the hydrophobic channels, owing to the inherent dynamism of the framework, arising out of the highly flexible ether nodes. 13CNMR experiment also proved the inclusion of PX (Fig. S33). After confirming the phase purity for the new phase 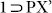 from PXRD, both TGA and PXRD analysis for
from PXRD, both TGA and PXRD analysis for 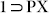 and
and 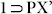 phases confirmed their similar nature (Fig. S17, S20). To check reversibility of this PX-inclusion, the crystals of
phases confirmed their similar nature (Fig. S17, S20). To check reversibility of this PX-inclusion, the crystals of 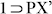 were heated at 160°C under reduced pressure for 4 h, to obtain the desolvated phase 1'. TGA and PXRD profiles (Fig. S17, S22) established the resemblance with the parent desolvated phase 1, confirming the PX-inclusion reversibility.
were heated at 160°C under reduced pressure for 4 h, to obtain the desolvated phase 1'. TGA and PXRD profiles (Fig. S17, S22) established the resemblance with the parent desolvated phase 1, confirming the PX-inclusion reversibility.
Figure 6. Resolvated framework on p-xylene accommodation mediated breathing.

Overall structure of resolvated phase 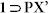 showing free PX molecules in the channels, along a-axis (PX molecules shown in spacefill model for clarity).
showing free PX molecules in the channels, along a-axis (PX molecules shown in spacefill model for clarity).
To ensure the reproducibility of the xylene adsorbents, reproducing the same isotherm on unchanged desolvated sample 1 was checked ten consecutive times for PX, and three times for each of the other xylene isomers (Figs S34–S38). The results reflect excellent repeatability in each case. PXRD patterns recorded for the adsorbent MOF after each of the adsorption-desorption cycles are depicted in Figs S39–S40. These patterns are markedly identical to the desolvated adsorbent MOF 1, which implies that the sample remains intact after each of the sorption cycles.
To check the practical importance of separation, phase 1 was immersed into ternary and quaternary mixture solution of xylenes for 1 h and the amounts of the non-adsorbed isomers were analyzed by GC in intermediate time intervals. Crystalline phase 1 (100 mg) was immersed into mixture solution of xylenes for 1 h according to the following three different combinations, and GC chromatogram were recorded by pipetting out the supernatant solutions (0.123 mL each time) at regular time intervals, and preparing the GC samples in 1.3 mL MeCN in each of the occurrences (GC parameters employed are mentioned in SI). Setup A: Quaternary xylene mixture immersion test: 100 mg of 1 was kept immersed in a mixture solution of OX (1 mL), MX (1 mL), PX (1 mL) and EB (1 mL) for 1 h, while in the intermediate time intervals 0.123 mL of the supernatant solution were pipetted out for recording the GC chromatogram at those respective times (Fig. 7a). As per the literature reported standard GC chromatogram for xylene mixtures41, it was found that EB and OX were giving rise to their characteristically separate GC signals, although the retention times for MX and PX coincided to give a mixed intensity signal, owing to their very close characteristic retention times. To check whether the observed decrease in the mixed (PX + MX) signal intensity (for setup A) occurs due to the sole effect or PX only, and not MX, the following two experiments were also performed. Setup B: Ternary xylene mixture immersion test (excluding PX): 100 mg of 1 was kept immersed in a mixture solution of OX (1 mL), MX (1 mL), MX (1 mL) and EB (1 mL) for 1 h, while in the intermediate time intervals 0.123 mL of the supernatant solution were pipetted out for recording the GC chromatogram at those respective times (Fig. 7b). Setup C: Ternary xylene mixture immersion test (excluding MX): 100 mg of 1 was kept immersed in a mixture solution of OX (1 mL), PX (1 mL) and EB (1 mL) for 1 h, while in the intermediate time intervals 0.123 mL of the supernatant solution were pipetted out for recording the GC chromatogram at those respective times (Fig. 7c). The results (Fig. 7) confirm that the observed decline in particularly one of the signal intensity is solely in presence of PX.
Figure 7. GC experiment for comparison of xylene-selectivity.
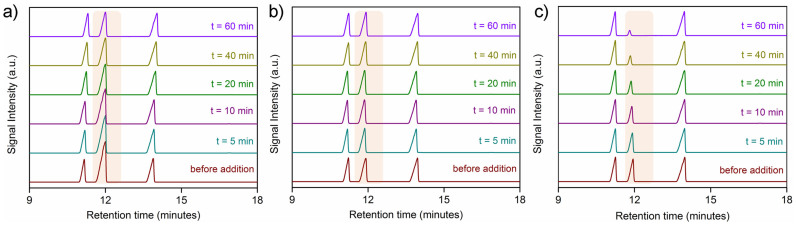
a) GC chromatogram of the supernatant solutions recorded at the specified time intervals in the setup A (Quaternary xylene mixture immersion test). Highlighted signals denote the mixed signal for the combined contributions of both MX and PX, intensity of which is getting steadily diminished with increasing time of immersion with MOF, b) GC chromatogram of the supernatant solutions recorded at the specified time intervals in the setup B (Ternary xylene mixture immersion test; excluding PX). Highlighted signals denote the individual signal for the contribution of MX only, intensity of which is remaining unchanged with increasing time of immersion with MOF, c) GC chromatogram of the supernatant solutions recorded at the specified time intervals in the setup C (Ternary xylene mixture immersion test; excluding MX). Highlighted signals denote the individual signal for the contribution of PX only, intensity of which is getting steadily diminished with increasing time of immersion with MOF.
Discussion
It is indeed a fascinating observation, that only those guest solvent species which could mediate the SCSC transformation of the desolvated PCP 1 could selectively get adsorbed inside the framework as evidenced from both the PXRD and TGA experiments (also the conclusive SC-XRD structure of resolvated 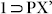 ); but the ones which were not capable to affect the PXRD of phase 1 on exposure, did not get adsorbed at all. This conclusively proved the riveting effect of framework flexibility for the present microporous material, allowing the multiple SCSC transformations on guest inclusion due to favorable host-guest interactions. This in effect developed a size-selective adsorption based separation property for p-xylene over m- and o-xylene along with ethyl benzene, by virtue of the restricted limiting allowance principle exhibited by the MOF material, making it a unique dynamic framework functioning as a molecular sieve for xylene isomers.
); but the ones which were not capable to affect the PXRD of phase 1 on exposure, did not get adsorbed at all. This conclusively proved the riveting effect of framework flexibility for the present microporous material, allowing the multiple SCSC transformations on guest inclusion due to favorable host-guest interactions. This in effect developed a size-selective adsorption based separation property for p-xylene over m- and o-xylene along with ethyl benzene, by virtue of the restricted limiting allowance principle exhibited by the MOF material, making it a unique dynamic framework functioning as a molecular sieve for xylene isomers.
In conclusion, a novel dynamic MOF has been successfully synthesized from a strategically designed ligand bearing two ether linkages as its flexible nodes. The resultant MOF showed high degree of framework flexibility owing to the ether linkages actually functioning as the adjustable nodes, accompanying the guest-inclusion mediated solid-state structural transformations. The MOF material presented selective adsorption of p-xylene over the other two constitutional isomers, invoking the guest-responsive structural dynamism feature to allow the selective inclusion of the p-isomer over the other two sterically bulkier congeners. This kind of guest-selective structural changes occurring in the crystalline state of porous materials can be exploited in future for the targeted application-oriented development of novel functional materials which might result in important industrial applications for chemical separation. The present material might prove to be an excellent candidate to further investigate its sorption kinetics, which will be requisite to evaluate its suitability for practical separations based on this observed ambient temperature PX-selective sorption phenomena.
Methods
Materials
All the reagents and solvents were commercially available and used as received, without further purification.
Physical Measurements
Powder X-ray diffraction (PXRD) patterns were measured on Bruker D8 Advanced X-Ray diffractometer at room temperature using Cu-Kα radiation (λ = 1.5406 Å) with a scan speed of 0.5° min–1 and a step size of 0.01° in 2 theta. Thermogravimetric analysis results were obtained in the temperature range of 30–800°C on Perkin-Elmer STA 6000 analyzer under N2 atmosphere, at a heating rate of 10°C min−1, The Fourier transform (FT-IR) infra-red spectra were recorded on NICOLET 6700 FT-IR Spectrophotometer using KBr Pellets.
X-ray Structural Studies
Single-crystal X-ray data of compound 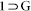 and 1 were collected at 200 K on a Bruker KAPPA APEX II CCD Duo diffractometer (operated at 1500 W power: 50 kV, 30 mA) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å), mounting on nylon CryoLoops (Hampton Research) with Paraton-N (Hampton Research) oil. Single-crystal X-Ray data of compound
and 1 were collected at 200 K on a Bruker KAPPA APEX II CCD Duo diffractometer (operated at 1500 W power: 50 kV, 30 mA) using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å), mounting on nylon CryoLoops (Hampton Research) with Paraton-N (Hampton Research) oil. Single-crystal X-Ray data of compound 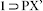 crystals was collected at 100 K on a Bruker KAPPA APEX II CCD Duo diffractometer (operated at 1500 W power: 50 kV, 1 mA) using graphite-monochromated Cu Kα radiation (λ = 1.5418Å), mounting on nylon CryoLoop (Hampton Research) with Paraton-N (Hampton Research) oil. The data integration and reduction were processed with SAINT[S2] software. A multi-scan absorption correction was applied to the collected reflections. The structures were solved by the direct method using SHELXTL[S3] and were refined on F2 by full-matrix least-squares technique using the SHELXL-97[S4] program package within the WINGX[S5] programme. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located in successive difference Fourier maps and they were treated as riding atoms using SHELXL default parameters. The structures were examined using the Adsym subroutine of PLATON[S6] to assure that no additional symmetry could be applied to the models. Tables S1, S2 and S3 contain crystallographic data for the compounds
crystals was collected at 100 K on a Bruker KAPPA APEX II CCD Duo diffractometer (operated at 1500 W power: 50 kV, 1 mA) using graphite-monochromated Cu Kα radiation (λ = 1.5418Å), mounting on nylon CryoLoop (Hampton Research) with Paraton-N (Hampton Research) oil. The data integration and reduction were processed with SAINT[S2] software. A multi-scan absorption correction was applied to the collected reflections. The structures were solved by the direct method using SHELXTL[S3] and were refined on F2 by full-matrix least-squares technique using the SHELXL-97[S4] program package within the WINGX[S5] programme. All non-hydrogen atoms were refined anisotropically. All hydrogen atoms were located in successive difference Fourier maps and they were treated as riding atoms using SHELXL default parameters. The structures were examined using the Adsym subroutine of PLATON[S6] to assure that no additional symmetry could be applied to the models. Tables S1, S2 and S3 contain crystallographic data for the compounds 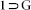 , 1 and
, 1 and 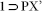 respectively.
respectively.
Low Pressure Gas Sorption Measurements
Low pressure gas and solvent sorption measurements were performed using BelSorpmax (Bel Japan). The sorption-recyclability experiments were recorded in BelAqua (Bel Japan). All the gases used were of 99.999% purity. As-synthesized crystals of compound 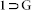 were heated at 180°C under vacuum for 24 h, to get guest-free crystals of compound 1. Prior to adsorption measurement, the guest free sample 1 was pretreated at 170°C under vacuum for 2 h, using BelPrepvacII, and purged with N2 on cooling.
were heated at 180°C under vacuum for 24 h, to get guest-free crystals of compound 1. Prior to adsorption measurement, the guest free sample 1 was pretreated at 170°C under vacuum for 2 h, using BelPrepvacII, and purged with N2 on cooling.
Solvent exposure study
Crystalline solid powder of compound 1 taken in smaller glass vials were kept open inside larger capped closed glass vials containing different guest solvents (benzene, toluene, cyclohexane, o-xylene, m-xylene and p-xylene respectively) over a period of 48 h to allow vapor-phase exposure of solvents and characterized by PXRD.
Synthesis of Intermediate L'
4-tert-butyl catechol (11 g, 0.0662 mole), 4-fluorobenzonitrile (16.029 g, 0.1324 mole) and potassium carbonate (~27.6 g, 0.2 mole) were refluxed at 180°C in a mixture of N, N-dimethylformamide (DMF) and p-xylene (110 mL/55 ml) for 2days. After cooling the reaction mixture to r.t., it was poured into ~100 mL ice-cold water, followed by acidification with dil. HCl until pH ~ 3. The reaction mixture was evaporated to dryness (maximum amount as possible) by rotary evaporation under reduced pressure, to get a reddish yellow product. This was extracted with Ethyl acetate (2 × 250 mL), washed with brine/water (twice each) and the resultant EtOAc layer was dried over anhydrous Na2SO4. Evaporation of this EtOAc layer, followed by drying under high vacuum yielded the intermediate compound L'. Yield: 23.28 g, 95.48%. 1H NMR (400 MHz, CDCl3) δ(ppm): 7.55–7.51 (m, 4 H), 7.33 (dd, J = 8.24 Hz, J = 2.28 Hz, 1 H), 7.22 (d, J = 2.28 Hz, 1 H), 7.14 (d, J = 8.28 Hz, 1 H), 6.82–6.77 (m, 4 H), 1.34 (s, 9 H); 13C NMR (100 MHz, CDCl3) δ(ppm): 161.1, 151.2, 145.2, 143.4, 134.2, 124.21, 122.9, 120.7, 119, 116.8, 106, 35, 31.5; HRMS (ESI) Calcd. for C24H20N2O2 369.160 [M + H]+, found 369.160 (Figs S2, S3 and S4).
Synthesis of Ligand LH2
A mixture of L' (10 g, 0.0271 mole) (Fig. S1) and potassium hydroxide (9.138 g, 0.163 mole) was refluxed in 200 mL water/ethanol (1:1) solvent mixture for 1day. After the reaction mixture was allowed to cool to r.t., it was acidified by dil. HCl keeping on an ice bath till pH ~ 1. The crude material that precipitated out was recrystallized from hot EtOH/water to get colorless crystalline product LH2. Yield: 9.56 g, 86.66%. 1H NMR (400 MHz, DMSO-d6) δ(ppm):7.87–7.84 (m, 4 H), 7.36 (dd, J = 8.7 Hz, J = 2.28 Hz, 1 H), 7.30 (d, J = 2.28 Hz, 1 H), 7.22 (d, J = 8.68 Hz, 1 H),6.87–6.83 (m, 4 H), 1.29 (s, 9 H); 13C NMR (100 MHz, DMSO-d6) δ(ppm): 167.1, 161.0, 150, 145.5, 144, 131.9, 125.3, 123.8, 122.6, 120.5, 116.4, 34.8, 31.4: HRMS (ESI) Calcd. for C24H22O6 429.131 [M + Na]+, found 429.130 (Figs S5, S6 and S7).
Synthesis of {[Zn4O(L)3(DMF)2].xG}n (1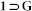 G)
G)
A mixture of H2L (40 mg, 0.1 mmol), Zn(NO3)2.6H2O (29.7 mg, 0.1 mmol), DMF (1 mL) and 1-butanol (1 mL) was placed in a teflon capped glass vial. This was heated at 90°C for 48 h and then cooled to room temperature for 1day. Growth of colorless block-shaped crystals was observed upon cooling to RT, the desired product 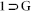 appeared in ~72% yield.
appeared in ~72% yield.
Dehydrated phase [Zn4O(L)3]n (1)
Shiny single crystals of 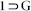 were heated at 160°C under reduced pressure for 8 h, to obtain the slightly pale colorless dehydrated crystals of compound 1.
were heated at 160°C under reduced pressure for 8 h, to obtain the slightly pale colorless dehydrated crystals of compound 1.
Resolvated phase {[Zn4O(L)3(DMF)2].(C8H10)}n (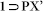 )
)
Colorless Single Crystals of 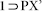 were obtained on exposing the crystals of 1 to the vapor of a solution of p-Xylene (2 mL) and DMF (1 mL) for 72 h, without allowing any disturbance of the system.
were obtained on exposing the crystals of 1 to the vapor of a solution of p-Xylene (2 mL) and DMF (1 mL) for 72 h, without allowing any disturbance of the system.
Author Contributions
S.M. carried out the project. B.J. solved the SC-XRD crystal structures. B.M., A.V.D. and A.K.C. helped in improving pictorial presentations. All of them helped throughout the project. S.K.G. supervised the project.
Supplementary Material
Framework-Flexibility Driven Selective Sorption of p-Xylene over Other Isomers by a Dynamic Metal-Organic Framework
Acknowledgments
B.J. and B.M. are thankful to CSIR for research fellowship. IISER Pune, DAE (Project No.2011/20/37C/06/BRNS) and DST (Project No.GAP/DST/CHE-12-0083) are acknowledged for the financial support. Sandeep Pimparkar is acknowledged for helping in GC analysis.
References
- Zhou H.-C., Long J. R. & Yaghi O. M. Introduction to Metal-Organic Frameworks. Chem. Rev. 112, 673–674 (2012). [DOI] [PubMed] [Google Scholar]
- Wang C., Zhang T. & Lin W. Rational Synthesis of Noncentrosymmetric Metal_Organic Frameworks for Second-Order Nonlinear Optics. Chem. Rev. 112, 1084–1104 (2012). [DOI] [PubMed] [Google Scholar]
- Mason J. A., Veenstra M. & Long J. R. Evaluating metal–organic frameworks for natural gas storage. Chem. Sci. 5, 32–51 (2014). [Google Scholar]
- Li J.-R., Kuppler R. J. & Zhou H.-C. Selective gas adsorption and separation in metal–organic frameworks. Chem. Soc. Rev. 38, 1477–1504 (2009). [DOI] [PubMed] [Google Scholar]
- Lee J. et al. Metal–organic framework materials as catalysts. Chem. Soc. Rev. 38, 1450–1459 (2009). [DOI] [PubMed] [Google Scholar]
- Corma A., García H. & Xamena F. X. L. Engineering Metal Organic Frameworks for Heterogeneous Catalysis. Chem. Rev. 110, 4606–4655 (2010). [DOI] [PubMed] [Google Scholar]
- Horcajada P. et al. Metal–Organic Frameworks as Efficient Materials for Drug Delivery. Angew. Chem. Int. Ed. 45, 5974–5978 (2006). [DOI] [PubMed] [Google Scholar]
- Nagarkar S. S., Joarder B., Chaudhari A. K., Mukherjee S. & Ghosh S. K. Highly Selective Detection of Nitro Explosives by a Luminescent Metal–Organic Framework. Angew. Chem., Int. Ed. 52, 2881–2885 (2013). [DOI] [PubMed] [Google Scholar]
- Lan A. J. et al. Luminescent Microporous Metal–Organic Framework for the Fast and Reversible Detection of High Explosives. Angew. Chem. Int. Ed. 48, 2334–2338 (2009). [DOI] [PubMed] [Google Scholar]
- Kreno L. E. et al. Metal–Organic Framework Materials as Chemical Sensors. Chem. Rev. 112, 1105–1125 (2012). [DOI] [PubMed] [Google Scholar]
- Cui Y., Yue Y., Qian G. & Chen B. Luminescent Functional Metal Organic Frameworks. Chem. Rev. 112, 1126–1162 (2012). [DOI] [PubMed] [Google Scholar]
- Tanabe K. K., Allen C. & Cohen S. M. Photochemical Activation of a Metal–Organic Framework to Reveal Functionality. Angew. Chem. Intl. Ed. 49, 9730–9733 (2010). [DOI] [PubMed] [Google Scholar]
- Shimizu G. K. H., Taylor J. M. & Kim S. Proton Conduction with Metal-Organic Frameworks. Science 341, 354–355 (2013). [DOI] [PubMed] [Google Scholar]
- Yamada T., Otsubo K., Makiura R. & Kitagawa H. Designer coordination polymers: dimensional crossover architectures and proton conduction. Chem. Soc. Rev. 42, 6655–6669 (2013). [DOI] [PubMed] [Google Scholar]
- Nagarathinam M. & Vittal J. J. Anisotropic Movements of Coordination Polymers upon Desolvation: Solid-State Transformation of a Linear 1D Coordination Polymer to a Ladderlike Structure. Angew. Chem. Int. Ed. 45, 4337–4341 (2006). [DOI] [PubMed] [Google Scholar]
- Denayer J. F. M., Vos D. D. & Leflaive P. Separation of Xylene Isomers. In: Metal-Organic Frameworks: Applications from Catalysis to Gas Storage Farrusseng D., Ed.; Wiley-VCH: Weinheim, Germany, 173–189 (2011). [Google Scholar]
- Jin Z. et al. A novel microporous MOF with the capability of selective adsorption of xylenes. Chem. Commun. 46, 8612–8614 (2010). [DOI] [PubMed] [Google Scholar]
- Alaerts L. et al. Selective Adsorption and Separation of Xylene Isomers and Ethylbenzene with the Microporous Vanadium(IV) Terephthalate MIL-47. Angew. Chem. Int. Ed. 46, 4293–4297 (2007). [DOI] [PubMed] [Google Scholar]
- Finsy V. et al. Pore-Filling-Dependent Selectivity Effects in the Vapor-Phase Separation of Xylene Isomers on the Metal-Organic Framework MIL-47. J. Am. Chem. Soc. 130, 7110–7118 (2008). [DOI] [PubMed] [Google Scholar]
- Vermoortele F. et al. p-Xylene-Selective Metal Organic Frameworks: A Case of Topology-Directed Selectivity. J. Am. Chem. Soc. 133, 18526–18529 (2011). [DOI] [PubMed] [Google Scholar]
- Peralta D. et al. Comparison of the Behavior of Metal-Organic Frameworks and Zeolites for Hydrocarbon Separations. J. Am. Chem. Soc. 134, 8115–8126 (2012). [DOI] [PubMed] [Google Scholar]
- Stylianou K. C. et al. Dimensionality Transformation through Paddlewheel Reconfiguration in a Flexible and Porous Zn-Based Metal-Organic Framework. J. Am. Chem. Soc. 134, 20466–20478 (2012). [DOI] [PubMed] [Google Scholar]
- Lusi M. & Barbour L. J. Solid–Vapor Sorption of Xylenes: Prioritized Selectivity as a Means of Separating All Three Isomers Using a Single Substrate. Angew. Chem. Int. Ed. 51, 3928–3931 (2012). [DOI] [PubMed] [Google Scholar]
- Peralta D. et al. The separation of xylene isomers by ZIF-8: A demonstration of the extraordinary flexibility of the ZIF-8 framework. Microporous Mesoporous Mater. 173, 1–5 (2013). [Google Scholar]
- Nicolau M. P. M. et al. Single- and Multicomponent Vapor-Phase Adsorption of Xylene Isomers and Ethylbenzene in a Microporous Metal-Organic Framework. J. Phys. Chem. C. 113, 13173–13179 (2009). [Google Scholar]
- He Y. et al. A microporous lanthanide-tricarboxylate framework with the potential for purification of natural gas. Chem. Commun. 48, 10856–10858 (2012). [DOI] [PubMed] [Google Scholar]
- Li B., Wang H. & Chen B. Microporous Metal–Organic Frameworks for Gas Separation. Chem. Asian J. 9, 1474–1498 (2014). [DOI] [PubMed] [Google Scholar]
- Finsy V. et al. Framework Breathing in the Vapour-Phase Adsorption and Separation of Xylene Isomers with the Metal–Organic Framework MIL-53. Chem. Eur. J. 15, 7724–7731 (2009). [DOI] [PubMed] [Google Scholar]
- Bradshaw D., Warren J. E. & Rosseinsky M. J. Reversible Concerted Ligand Substitution at Alternating Metal Sites in an Extended Solid. Science 315, 977–980 (2007). [DOI] [PubMed] [Google Scholar]
- Zhang J.-P. & Chen X.-M. Exceptional Framework Flexibility and Sorption Behavior of a Multifunctional Porous Cuprous Triazolate Framework. J. Am. Chem. Soc. 130, 6010–6017 (2008). [DOI] [PubMed] [Google Scholar]
- Chen C. -L. & Beatty A. M. Guest Inclusion and Structural Dynamics in 2-D Hydrogen-Bonded Metal-Organic Frameworks. J. Am. Chem. Soc. 130, 17222–17223 (2008). [DOI] [PubMed] [Google Scholar]
- Ghosh S. K., Bureekaew S. & Kitagawa S. A Dynamic, Isocyanurate-Functionalized Porous Coordination Polymer. Angew. Chem. Int. Ed. 47, 3403–3406 (2008). [DOI] [PubMed] [Google Scholar]
- Nagarkar S. S., Desai A. V. & Ghosh S. K. Stimulus responsive metal-organic frameworks. Chem. Asian J. 10.1002/asia.201402004 (2014). [DOI] [PubMed] [Google Scholar]
- Horike S., Shimomura S. & Kitagawa S. Soft porous crystals. Nat. Chem. 1, 695–704 (2009). [DOI] [PubMed] [Google Scholar]
- Guillou N., Millangea F. & Walton R. I. Rapid and reversible formation of a crystalline hydrate of a metal–organic framework containing a tube of hydrogen-bonded water. Chem. Commun. 47, 713–715 (2011). [DOI] [PubMed] [Google Scholar]
- Joarder B., Chaudhari A. K., Nagarkar S. S., Manna B. & Ghosh S. K. Amino Acid Based Dynamic Metal–Biomolecule Frameworks. Chem. Eur. J. 19, 11178–11183 (2013). [DOI] [PubMed] [Google Scholar]
- Ferey G. et al. Why hybrid porous solids capture greenhouse gases?. Chem. Soc. Rev. 40, 550–562 (2011). [DOI] [PubMed] [Google Scholar]
- Hong D. H. & Suh M. P. Selective CO2 adsorption in a metal–organic framework constructed from an organic ligand with flexible joints. Chem. Commun. 48, 9168–9170 (2012). [DOI] [PubMed] [Google Scholar]
- Nagarkar S. S., Chaudhari A. K. & Ghosh S. K. Selective CO2 Adsorption in a Robust and Water-Stable Porous Coordination Polymer with New Network Topology. Inorg. Chem. 51, 572–576 (2012) [DOI] [PubMed] [Google Scholar]
- Webster C. E., Drago R. S. & Zerner M. C. Molecular Dimensions for Adsorptives. J. Am. Chem. Soc. 120, 5509–5516 (1998). [Google Scholar]
- Spencer S. F. Rapid Separation of Xylenes and Ethylbenzene by Gas Chromatography Using Packed Columns. Anal. Chem. 35, 592–592 (1963). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Framework-Flexibility Driven Selective Sorption of p-Xylene over Other Isomers by a Dynamic Metal-Organic Framework