

Abstract
The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels belong to the superfamily of voltage-gated potassium ion channels. They are, however, activated by hyperpolarizing potentials and are permeable to cations. Four HCN subunits have been cloned, of which HCN1 and HCN2 subunits are predominantly expressed in the cortex. These subunits are principally located in pyramidal cell dendrites, although they are also found at lower concentrations in the somata of pyramidal neurons as well as other neuron subtypes. HCN channels are actively trafficked to dendrites by binding to the chaperone protein TRIP8b. Somato-dendritic HCN channels in pyramidal neurons modulate spike firing and synaptic potential integration by influencing the membrane resistance and resting membrane potential. Intriguingly, HCN channels are present in certain cortical axons and synaptic terminals too. Here, they regulate synaptic transmission but the underlying mechanisms appear to vary considerably amongst different synaptic terminals. In conclusion, HCN channels are expressed in multiple neuronal subcellular compartments in the cortex, where they have a diverse and complex effect on neuronal excitability.
 |
Mala Shah completed her PhD at University College London (UCL, UK) under the supervision of Dr Dennis Haylett. She then obtained a Wellcome Prize Travel Research Fellowship to work in Professor Daniel Johnston's laboratory at Baylor College of Medicine (Houston, TX, USA) and Professor David Brown's laboratory at UCL (UK). She subsequently received a lectureship at UCL School of Pharmacy (UK) where she is currently a Reader in Neuroscience. Her current research interests include understanding how hippocampal and cortical axonal and dendritic function is regulated under physiological as well as epileptogenic conditions.
Voltage-gated ion channels play a fundamental role in regulating neuronal activity and synaptic transmission. The abundance and biophysical properties of voltage-gated ion channels varies within neuronal subcellular compartments: axons, dendrites and somata (Lai & Jan, 2006; Johnston & Narayanan, 2008; Nusser, 2009). This variation in localization has a significant impact on neuronal and neural network excitability and thus physiological processes such as learning and memory and patho-physiological conditions, such as epilepsy.
The hyperpolarization-activated cyclic nucleotide-gated (HCN) channels are voltage-gated ion channels that are permeable to Na+ and K+ ions and open at potentials more negative than −50 mV (Pape, 1996; Robinson & Siegelbaum, 2003; Biel et al. 2009; Shah et al. 2010). These channels are therefore active at the normal resting membrane potentials (RMPs) of most neurons and contribute to depolarizing the RMP. In addition, HCN channels regulate the membrane resistance. By affecting both the RMP and membrane resistance, HCN channels critically influence intrinsic neuronal excitability, synaptic potential integration and neurotransmitter release (Biel et al. 2009; Shah et al. 2010).
Four HCN subunits (HCN1–4) have so far been cloned (Ludwig et al. 1998; Santoro et al. 1998). All four are expressed in the central nervous system (CNS) but their patterns of expression vary. Of these, only HCN1 subunits are abundantly found in the cortex, hippocampus, cerebellum and brain stem (Moosmang et al. 1999; Notomi & Shigemoto, 2004). In contrast, HCN2 subunits are distributed ubiquitously through the CNS, with highest expression levels in the thalamus and brain stem nuclei. HCN3 subunits are expressed at a low level in the CNS and HCN4 subunits are found in selective brain regions such as the mitral cell layer of the olfactory bulb (Moosmang et al. 1999; Notomi & Shigemoto, 2004). These subunits can form homomeric or heteromeric channels when expressed in heterologous systems (S. Chen et al. 2001; Biel et al. 2009). The activation time constants and steady-state voltage dependence of the individual HCN currents differ considerably (Biel et al. 2009). Thus, HCN1 subunits have very fast activation kinetics whilst HCN4 subunits have the slowest kinetics. In addition, all HCN subunits are modulated by cyclic nucleotides, though the extent to which HCN1–4 currents are modified by these varies considerably. A number of other intracellular signalling molecules such as phosphoinositides and kinases, as well as auxiliary subunits such as tetratricopeptide repeat (TPR)-containing Rab8b interacting protein (TRIP8b), also affect the biophysical properties and expression of HCN subunits (Robinson & Siegelbaum, 2003; Biel et al. 2009; Wahl-Schott & Biel, 2009; Shah et al. 2010). This diversity in their expression, biophysical properties and modulation by intracellular molecules is, therefore, likely to differentially and dynamically regulate neuronal excitability (for in-depth reviews on HCN channel structure and biophysical properties, please see Robinson & Siegelbaum, 2003; Biel et al. 2009; Wahl-Schott & Biel, 2009).
In this review, I will focus on the role of HCN channels in determining cortical neuronal excitability. I will also discuss how HCN subunits are trafficked to selective subcellular compartments (axons and dendrites) and how their activity and plasticity affects pyramidal cell dendritic excitability and presynaptic function.
Somato-dendritic HCN channel function
Electrophysiological studies have revealed that the HCN current, Ih, is greatest in the distal dendrites of cortical and hippocampal pyramidal neurons (Magee, 1998; Stuart & Spruston, 1998; Williams & Stuart, 2000; Berger et al. 2001; Shah et al. 2004; Huang et al. 2009; Fig. 1). These findings have been corroborated with immunohistochemical studies showing the abundance of HCN1 and HCN2 subunits in pyramidal cell distal dendrites (Lorincz et al. 2002; Notomi & Shigemoto, 2004). HCN channels, though, are also located in the somata of some pyramidal neurons (albeit at a lower density than that in distal dendrites), interneuron subtypes and stellate cells (Robinson & Siegelbaum, 2003; Biel et al. 2009). Here, treatment with HCN channel inhibitors augments the input resistance. Since the RMP is also lowered substantially, there is either a reduction or little change in the number of spikes generated in response to depolarizing stimuli (Maccaferri et al. 1993; Maccaferri & McBain, 1996; Gasparini & DiFrancesco, 1997; Magee, 1998; Robinson & Siegelbaum, 2003; Fransén et al. 2004; Shah et al. 2004; Aponte et al. 2006; van Welie et al. 2006; Nolan et al. 2007; Thuault et al. 2013). In some neurons, such as the entorhinal cortical stellate cells, HCN channels are activated during the afterhyperpolarization following spikes and thereby affect spike firing patterns (Nolan et al. 2007). In contrast, in distal dendrites, where the expression of HCN channels is the greatest, pharmacological blockade of Ih or genetic ablation of HCN results in augmented dendritic excitability despite a hyperpolarized RMP (Magee, 1998, 1999; Berger et al. 2001; Poolos et al. 2002; Shah et al. 2004; Huang et al. 2009; Fig. 1). This is because the increase in membrane resistance following the inhibition of HCN channels in larger in distal dendrites than at the soma such that the change in voltage induced by depolarizing stimuli in the absence of Ih results in spikes despite the RMP being hyperpolarized (Magee, 1998; Poolos et al. 2002; Shah et al. 2004; Huang et al. 2009; Fig. 1).
Figure 1. Effects of HCN1 channels on cortical dendritic excitability.
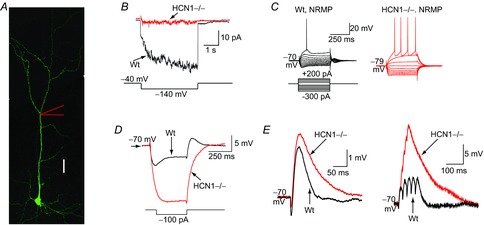
A, morphology of an entorhinal cortical (EC) layer III pyramidal neuron. Scale bar, 50 μm. B, cell-attached recordings of the HCN channel current (Ih) from HCN1 null (HCN1–/–) and wild-type (Wt) EC layer III dendrites. The current was generated by applying a hyperpolarizing step from −40 mV to −140 mV as shown below the traces. C, representative current-clamp recordings from wild-type and HCN1–/– EC layer III dendrites at their normal resting membrane potential (NRMP) when a series of 400 ms hyperpolarizing and depolarizing steps were applied as shown schematically. The scale bar shown applies to both recordings. D, typical traces obtained when a 100 pA hyperpolarizing pulse was applied to EC layer III dendrites from a fixed potential of −70 mV, demonstrating that the input resistance of HCN1–/– dendrites is much greater than that of wild-type. E, single and trains of simulated EPSPs recorded from HCN1–/– and wild-type dendrites at the common potential of −70 mV in response to alpha waveform injections. Adapted from Huang et al. (2009).
Changes in membrane resistance will also affect synaptic potential shapes and integration. Indeed, when HCN channel activity is reduced, somato-dendritic excitatory postsynaptic potential (EPSP) amplitudes are greater and their decay slower, leading to enhanced summation of a train of synaptic potentials (Fig. 1; Magee, 1998, 1999, 2000; Williams & Stuart, 2000; Berger et al. 2001; Poolos et al. 2002; Nolan et al. 2004; Shah et al. 2004; Huang et al. 2009; Sheets et al. 2011). Thus, trains of EPSPs are more likely to generate action potentials in axons, boosting neuronal excitability (Shah et al. 2004). The increased EPSP summation caused by somato-dendritic HCN channel inhibition is, at least in part, due to the enhanced membrane resistance. In distal dendrites, relief of inactivation of T- and N-type Ca2+ channels by RMP hyperpolarization also contributes to enhanced EPSP summation following pharmacological block of HCN channels (Tsay et al. 2007). Certainly, Ca2+ entry via these Ca2+ channels during dendritic Ca2+ spikes is greater in the presence of HCN channel inhibitors (Tsay et al. 2007). It should be noted, though, that certain subthreshold potassium channels such as KV7/M-channels are likely to have a larger impact on EPSP summation in the absence of Ih if expressed in the same subcellular domain within a particular neuron subtype (George et al. 2009). Hence, the overall effect of HCN channels on EPSP summation is likely to depend to a certain extent on which other ion channels are present locally.
Somato-dendritic HCN channels affect the integration of inhibitory synaptic inputs (IPSPs) too (Williams & Stuart, 2003; Atherton et al. 2010; Pavlov et al. 2011). HCN channels are activated by hyperpolarization and their activation during trains of IPSPs serves to limit synaptic hyperpolarization (Williams & Stuart, 2003; Atherton et al. 2010; Pavlov et al. 2011). In cortical pyramidal cell neurons, distal dendritic Ih enhances dendro-somatic IPSP attenuation and constrains axo-somatic depolarization (Williams & Stuart, 2003). Moreover, in certain neurons, the activation of HCN channels during synaptic inhibition restricts de-inactivation of T-type Ca2+ channels and rebound action potential firing (Atherton et al. 2010). Therefore, enhanced HCN channel activity during trains of synaptic inhibitory potentials can profoundly alter the state of neurons and their response to subsequent stimuli.
Somato-dendritic HCN channels have also been implicated in regulating neuronal oscillations and subthreshold resonance properties of neurons (Fransén et al. 2004; van Welie et al. 2006; Haas et al. 2007; Narayanan & Johnston, 2007, 2008; Nolan et al. 2007; Hu et al. 2009; Marcelin et al. 2009; Vaidya & Johnston, 2013). In distal dendrites, HCN channels are thought to act as inverse leaky voltage-dependent inductors and thereby act as a bandpass filter to allow synaptic inputs of a certain frequency to be preferentially transferred to the soma (Narayanan & Johnston, 2008; Vaidya & Johnston, 2013). Indeed, recent evidence suggests that HCN channels influence signal processing in distal hippocampal CA1 pyramidal dendrites such that slower, theta frequency synaptic inputs are preferentially transferred to the soma even when dendrites receive synaptic inputs at gamma frequency (Vaidya & Johnston, 2013). In addition, the inductance property of HCN channels provides an efficient means of synchronization of inputs so that voltage waveforms at the soma are not very sensitive to the location of the synaptic inputs in the dendritic tree (Vaidya & Johnston, 2013). Thus, HCN channels modulate somato-dendritic information processing in a variety of ways, all of which impact neuronal synaptic integration and activity.
Role of presynaptic HCN channels in synaptic release
In addition to their location in dendrites, immunohistochemical evidence has suggested that HCN1 subunits are expressed in cortical and hippocampal axons and synaptic terminals of inhibitory and excitatory neurons (Notomi & Shigemoto, 2004; Lujan et al. 2005; Bender et al. 2007; Boyes et al. 2007; Huang et al. 2011, 2012; Fig. 2). In the rodent hippocampus, HCN channels are present in basket cell axons and terminals, where they inhibit synaptic release by an as yet undefined mechanism (Aponte et al. 2006). HCN1 subunits are also present in immature perforant path axons, which synapse onto dentate gyrus granule cells (Bender et al. 2007). Here, interestingly, pharmacological inhibitors of HCN channels moderately reduced short-term depression of long trains of EPSPs at 20 Hz but not at lower frequencies. These findings suggested that HCN1 subunits in immature perforant path axons form functional channels, which modulate action potential-dependent release (Bender et al. 2007).
Figure 2. Presynaptic HCN channels and synaptic release in the adult entorhinal cortex (EC).
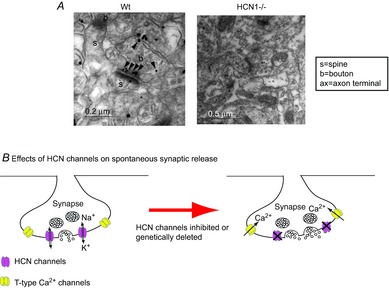
A, immunogold particles for HCN1 in asymmetric synaptic terminals (presumably excitatory glutamatergic synapses) in EC layer III in wild-type (Wt). Labelling was absent in HCN1–/– tissue. Adapted from Huang et al. (2011). B, schematic diagram showing that presynaptic HCN channels and T-type Ca2+ channels are present on the same terminals. Pharmacological inhibition or genetic ablation of HCN1 channels leads to membrane hyperpolarization and enhanced Ca2+ influx through T-type Ca2+ channels, boosting spontaneous synaptic release.
In adult neurons, the influence of HCN channels on synaptic transmission has so far only been found in the entorhinal cortex (EC). Here, immunogold labelling showed that HCN1 subunits were preferentially localized to the active zone in asymmetric (presumably glutamatergic) synaptic terminals (Huang et al. 2011, 2012; Fig. 2). These terminals also contained the T-type Ca2+ channel subunits CaV3.2 as evident from co-labelling for HCN1 and CaV3.2 subunits (Huang et al. 2011). Pharmacological inhibition or genetic ablation of HCN1 channels resulted in enhanced frequency of miniature excitatory postsynaptic currents (mEPSCs) selectively onto EC layer III pyramidal neurons, suggesting that presynaptic HCN channels regulate basal glutamatergic synaptic release (Huang et al. 2011; Fig. 2). The significantly increased mEPSC frequency was a consequence of relief of inactivation of CaV3.2 channels caused by membrane hyperpolarization when HCN channel function was reduced (Huang et al. 2011). Interestingly, the paired pulse ratio of EPSCs evoked by stimulating distal dendrites of EC layer III pyramidal neurons was significantly lowered in the presence of HCN channel blockers, suggesting that action potential-driven release onto EC layer III pyramidal neurons is also regulated by HCN channels (Huang et al. 2011). Hence, HCN channels affect multiple modes of synaptic transmission onto EC layer III pyramidal neurons.
Dendritic and presynaptic HCN1 subunit trafficking
HCN channels are actively trafficked to dendrites by binding to chaperone proteins known as TPR-containing Rab8b interacting protein (TRIP8b) (Santoro et al. 2004, 2009; Lewis et al. 2009; Zolles et al. 2009; Shah et al. 2010). Moreover, TRIP8b is essential for membrane expression of HCN channels in hippocampal and cortical dendrites (Santoro et al. 2004, 2009; Lewis et al. 2009). In addition, the presence of TRIP8b shifts the activation curve of Ih to the left (Lewis et al. 2009; Santoro et al. 2009; Zolles et al. 2009). Further, the presence of TRIP8b alters the sensitivity of HCN channels to cyclic nucleotides (Zolles et al. 2009; Han et al. 2011; Hu et al. 2013). Thus, the presence of TRIP8b is important for the expression and voltage dependence of dendritic HCN channels.
Nine isoforms of TRIP8b have so far been identified, most of which enhance the expression of dendritic HCN subunits (Lewis et al. 2009; Santoro et al. 2009). Some TRIP8b isoforms, however, suppress HCN subunit expression (Zolles et al. 2009; Lewis & Chetkovich, 2011; Santoro et al. 2011). Interestingly, TRIP8b isoforms that hinder HCN subunit expression have been suggested to predominantly exist in adult principal cell (including pyramidal neuron) axons (Piskorowski et al. 2011). It has thus been proposed that TRIP8b prevents mislocalization of HCN subunits to adult axons (Piskorowski et al. 2011). In agreement with this notion, HCN1 subunits are localized to immature perforant path axons when TRIP8b expression is low, suggesting that upregulation of TRIP8b expression during development leads to reduced axonal HCN subunit expression (Wilkars et al. 2012). Moreover, the absence of all TRIP8b isoforms results in HCN1 expression in adult perforant path axons (Wilkars et al. 2012).
HCN1 subunits, however, are expressed in certain adult axons and synaptic terminals in the entorhinal cortex (EC) (Huang et al. 2011, 2012). Are TRIP8b isoforms, therefore, not localized to these? There was no difference in HCN1 subunit expression in these axons and synaptic terminals when wild-type and TRIP8b tissue was compared (Huang et al. 2012). Further, functional presynaptic HCN1 channels regulated synaptic release onto EC layer III pyramidal neurons to a similar extent in both wild-type and TRIP8b null mice, suggesting that TRIP8b is not involved in the targeting of HCN1 subunits to these axons (Huang et al. 2012). In heterologous systems as well as neurons, other proteins such as filamin A have been shown to regulate HCN subunit expression (Biel et al. 2009). It remains to be determined if these proteins might be involved in the targeting and stabilization of HCN subunits in adult axons.
Dendritic and presynaptic HCN channel plasticity under physiological and patho-physiological states
HCN channel activity in hippocampal CA1 pyramidal neurons is modified by activity-dependent mechanisms involving changes in intracellular Ca2+ concentrations (van Welie et al. 2004). Hebbian plasticity, including NMDA receptor-dependent long-term potentiation (LTP), also alters dendritic HCN subunit expression and channel function in hippocampal CA1 pyramidal neurons (Shah et al. 2010). In CA1 pyramidal neurons, induction of NMDA receptor-dependent LTP via a theta burst protocol enhances HCN channel expression by activating calcium–calmodulin-dependent protein kinase II (CaMKII) (Fan et al. 2005; Campanac et al. 2008). LTP induced by high frequency stimulation, though, reduces dendritic HCN channel function and causes greater synaptic potential summation and EPSP-spike coupling (Campanac et al. 2008). Further, activation of the α2 adrenoreceptors in prefrontal cortical dendritic spines led to enhanced LTP and working memory via a decrease in spine cAMP and HCN1 channel activity (Wang et al. 2007). Alterations in modifications in Ih induced by stimulation of Schaffer collateral inputs to CA1 pyramidal neurons were absent in TRIP8b null neurons, indicating that changes in TRIP8b expression or function might underlie plasticity-induced changes in Ih (Brager et al. 2013). Interestingly, though, pairing of Schaffer collateral and perforant path inputs produced LTP in TRIP8b null CA1 pyramidal neurons (Brager et al. 2013). LTP induced by stimulation of the distal perforant path was also greater in HCN1 null CA1 pyramidal neurons (Nolan et al. 2004). Consistent with this, HCN1 null mice have increased hippocampal-dependent learning and memory (Nolan et al. 2004). In addition to LTP, induction of long-term depression (LTD) modulates Ih. Thus, metabotropic glutamate receptor-dependent LTD lowered dendritic HCN channel activity due to Ca2+ release from internal stores and activation of protein kinase C (Brager & Johnston, 2007). Therefore, whilst activity and Hebbian forms of plasticity modulate dendritic HCN channel function and expression, the consequent change in HCN channel activity (up- or downregulation) is likely to be dependent upon the locus where plasticity is induced and the intracellular signalling cascades triggered by plasticity-inducing mechanisms.
Abnormal neuronal activity is a common feature of many neurological disorders too. Indeed, modifications in HCN channel expression and function have been associated with disorders such as neuropathic pain and epilepsy (Biel et al. 2009; Noam et al. 2011). Considerable evidence for alterations in HCN channel function has been reported in animal models of epilepsy, particularly those mimicking temporal lobe epilepsy (TLE). TLE is the most common, drug-resistant form of acquired epilepsy (Engel, 1996). It can be mimicked in animals by administering chemoconvulsants such as kainic acid or pilocarpine (White, 2002). Intriguingly, cortical dendritic HCN1 and HCN2 subunit expression and activity is reduced within hours of chemoconvulsant-induced status epilepticus and remains persistently downregulated for weeks (Shah et al. 2004; Jung et al. 2007, 2011; Shin et al. 2008; Marcelin et al. 2009). There is also a long-term decrease in cortical presynaptic HCN channel activity following kainic acid-induced status epilepticus (Huang et al. 2012). Further, a decrease in HCN1 mRNA and current function has also been found in cortical and hippocampal tissue obtained from TLE patients (Bender et al. 2003; Wierschke et al. 2010). HCN2 mRNA levels, though, appear slightly enhanced in epileptic human hippocampus (Bender et al. 2003). Interestingly, though, HCN2 null mice have absence epilepsy (Ludwig et al. 2003) and loss of function mutations in HCN2 subunits have been associated with idiopathic generalized epilepsies in patients (DiFrancesco et al. 2011). HCN1 null mice, however, are not spontaneously epileptic but are more susceptible to chemoconvulsant-induced status epilepticus or kindling (Huang et al. 2009; Santoro et al. 2010). These results suggest that the loss of HCN subunits following status epilepticus is likely to contribute to the process of epileptogenesis. The sustained reduction in HCN subunits has been attributed to multiple mechanisms including repressed transcription of HCN1 by upregulation of neuron-restrictive silencer factor (NRSF) (McClelland et al. 2011) and altered activity of the phosphatase calcineurin and the kinase p38 MAPK, resulting in a leftward shift in the HCN current activation curve and fewer HCN channels available at rest (Jung et al. 2010). Indeed, transiently restoring HCN channel expression by disrupting the interaction between the NRSF and HCN1 delays the onset of spontaneous seizure activity following termination of status epilepticus (McClelland et al. 2011).
Intriguingly, HCN2 channel expression and function is increased in hippocampal pyramidal neurons following febrile seizures (K. Chen et al. 2001; Brewster et al. 2002; Dyhrfjeld-Johnsen et al. 2008). A heterozygous missense mutation in HCN2 (S126L) has also been correlated with increased incidence of febrile seizures (Nakamura et al. 2013). Expressed HCN2 channels containing this mutation displayed faster kinetics with higher temperatures, resulting in enhanced availability of the current under these conditions (Nakamura et al. 2013). Moreover, dendritic HCN current is enhanced in hippocampal pyramidal neurons in mice with targeted deletions in the Fragile X FMR1 gene (Brager et al. 2012). Fmr1 knockout mice do not exhibit spontaneous seizures but are more susceptible to audiogenic seizures (Musumeci et al. 2000). Similarly, only about a third of the rodents subjected to febrile seizures develop chronic epilepsy (Walker & Kullmann, 1999; Dube et al. 2007). Hence, whether HCN channel upregulation under these conditions is a homeostatic change or a cause of the epileptogenic activity remains to be further investigated.
Concluding remarks
In summary, in the cortex, HCN channels are predominantly located in pyramidal neuron distal dendrites (Johnston & Narayanan, 2008; Nusser, 2009; Shah et al. 2010). They are actively trafficked here by binding to TRIP8b (Shah et al. 2010). Dendritic HCN channels lower the membrane resistance and depolarize the resting membrane potential, limiting calcium channel activation. Consequently, inhibition of these channels enhances dendritic excitability and synaptic potential summation despite the membrane potential being hyperpolarized (Robinson & Siegelbaum, 2003; Shah et al. 2010). Further, dendritic HCN channels contribute to the synchronization of synaptic potentials such that the voltage waveform at the soma is not influenced by the location of the synaptic inputs (Vaidya & Johnston, 2013). In addition to TRIP8b, dendritic HCN channels are modulated by a number of different intracellular molecules including cyclic nucleotides, kinases and phosphatases (Robinson & Siegelbaum, 2003; Biel et al. 2009; Shah et al. 2010). Thus, Hebbian and homeostatic plasticity-inducing mechanisms, as well as pathological conditions that result in alterations of the activity of various intracellular substances, modulate HCN channel function and neuronal excitability.
Whilst HCN channel function in cortical pyramidal cell dendrites has been studied the most, HCN channels are also located at the somata of some pyramidal neurons, interneurons and stellate cells (Robinson & Siegelbaum, 2003; Biel et al. 2009; Wahl-Schott & Biel, 2009) as well as in a subset of excitatory and inhibitory synaptic terminals (Notomi & Shigemoto, 2004; Lujan et al. 2005; Bender et al. 2007; Huang et al. 2011). At the somata, HCN channels contribute to maintaining intrinsic excitability too. However, in contrast to their effects in dendrites, pharmacological block or genetic deletion of HCN channels at pyramidal neuron, interneuron or stellate cell somata has little effect on spike firing (Maccaferri et al. 1993; Maccaferri & McBain, 1996; Gasparini & DiFrancesco, 1997; Magee, 1998; Robinson & Siegelbaum, 2003; Fransén et al. 2004; Shah et al. 2004; Aponte et al. 2006; van Welie et al. 2006; Nolan et al. 2007; Thuault et al. 2013). Synaptic potential integration at the soma is affected by HCN channels as well but the overall effect on somatic excitability is likely to depend on the complement of other ion channels expressed. Similarly, HCN channels in synaptic terminals regulate synaptic release but the overall effect on the change in neurotransmission appears to be dependent on which other ion channels might also be located in particular synaptic terminals (Aponte et al. 2006; Bender et al. 2007; Huang et al. 2011, 2012). In comparison to dendritic HCN channels, though, relatively little is known about the regulation, modulation and trafficking of presynaptic HCN channels. Hence, HCN channels are diversely expressed within the cerebral cortex, where they contribute to maintaining intrinsic neuronal activity and synaptic transmission, thereby serving to dynamically influence cortical network activity.
Glossary
- EC
entorhinal cortex
- HCN channel
hyperpolarization-activated cyclic nucleotide-gated channel
- LTP
long-term potentiation
- RMP
resting membrane potential
- TRIP8b
tetratricopeptide repeat (TRP)-containing Rab8b interacting protein
Additional information
Competing interests
None declared.
References
- Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol. 2006;574:229–243. doi: 10.1113/jphysiol.2005.104042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atherton JF, Kitano K, Baufreton J, Fan K, Wokosin D, Tkatch T, Shigemoto R, Surmeier DJ, Bevan MD. Selective participation of somatodendritic HCN channels in inhibitory but not excitatory synaptic integration in neurons of the subthalamic nucleus. J Neurosci. 2010;30:16025–16040. doi: 10.1523/JNEUROSCI.3898-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Kirschstein T, Kretz O, Brewster AL, Richichi C, Ruschenschmidt C, Shigemoto R, Beck H, Frotscher M, Baram TZ. Localization of HCN1 channels to presynaptic compartments: novel plasticity that may contribute to hippocampal maturation. J Neurosci. 2007;27:4697–4706. doi: 10.1523/JNEUROSCI.4699-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender RA, Soleymani SV, Brewster AL, Nguyen ST, Beck H, Mathern GW, Baram TZ. Enhanced expression of a specific hyperpolarization-activated cyclic nucleotide-gated cation channel (HCN) in surviving dentate gyrus granule cells of human and experimental epileptic hippocampus. J Neurosci. 2003;23:6826–6836. doi: 10.1523/JNEUROSCI.23-17-06826.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger T, Larkum ME, Lüscher HR. High Ih channel density in the distal apical dendrite of layer V pyramidal cells increases bidirectional attenuation of EPSPs. J Neurophysiol. 2001;85:855–868. doi: 10.1152/jn.2001.85.2.855. [DOI] [PubMed] [Google Scholar]
- Biel M, Wahl-Schott C, Michalakis S, Zong X. Hyperpolarization-activated cation channels: from genes to function. Physiol Rev. 2009;89:847–885. doi: 10.1152/physrev.00029.2008. [DOI] [PubMed] [Google Scholar]
- Boyes J, Bolam JP, Shigemoto R, Stanford IM. Functional presynaptic HCN channels in the rat globus pallidus. Eur J Neurosci. 2007;25:2081–2092. doi: 10.1111/j.1460-9568.2007.05463.x. [DOI] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, Johnston D. Impaired dendritic expression and plasticity of h-channels in the fmr1–/y mouse model of fragile X syndrome. Cell Rep. 2012;1:225–233. doi: 10.1016/j.celrep.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Johnston D. Plasticity of intrinsic excitability during long-term depression is mediated through mGluR-dependent changes in Ih in hippocampal CA1 pyramidal neurons. J Neurosci. 2007;27:13926–13937. doi: 10.1523/JNEUROSCI.3520-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brager DH, Lewis AS, Chetkovich DM, Johnston D. Short- and long-term plasticity in CA1 neurons from mice lacking h-channel auxiliary subunit TRIP8b. J Neurophysiol. 2013;110:2350–2357. doi: 10.1152/jn.00218.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewster A, Bender RA, Chen Y, Dube C, Eghbal-Ahmadi M, Baram TZ. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J Neurosci. 2002;22:4591–4599. doi: 10.1523/JNEUROSCI.22-11-04591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanac E, Daoudal G, Ankri N, Debanne D. Downregulation of dendritic Ih in CA1 pyramidal neurons after LTP. J Neurosci. 2008;28:8635–8643. doi: 10.1523/JNEUROSCI.1411-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Aradi I, Thon N, Eghbal-Ahmadi M, Baram TZ, Soltesz I. Persistently modified h-channels after complex febrile seizures convert the seizure-induced enhancement of inhibition to hyperexcitability. Nat Med. 2001;7:331–337. doi: 10.1038/85480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang J, Siegelbaum SA. Properties of hyperpolarization-activated pacemaker current defined by coassembly of HCN1 and HCN2 subunits and basal modulation by cyclic nucleotide. J Gen Physiol. 2001;117:491–504. doi: 10.1085/jgp.117.5.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco JC, Barbuti A, Milanesi R, Coco S, Bucchi A, Bottelli G, Ferrarese C, Franceschetti S, Terragni B, Baruscotti M, DiFrancesco D. Recessive loss-of-function mutation in the pacemaker HCN2 channel causing increased neuronal excitability in a patient with idiopathic generalized epilepsy. J Neurosci. 2011;31:17327–17337. doi: 10.1523/JNEUROSCI.3727-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube CM, Brewster AL, Richichi C, Zha Q, Baram TZ. Fever, febrile seizures and epilepsy. Trends Neurosci. 2007;30:490–496. doi: 10.1016/j.tins.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyhrfjeld-Johnsen J, Morgan RJ, Foldy C, Soltesz I. Upregulated H-current in hyperexcitable CA1 dendrites after febrile seizures. Front Cell Neurosci. 2008;2:2. doi: 10.3389/neuro.03.002.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel JJ. Introduction to temporal lobe epilepsy. Epilepsy Res. 1996;26:141–150. doi: 10.1016/s0920-1211(96)00043-5. [DOI] [PubMed] [Google Scholar]
- Fan Y, Fricker D, Brager DH, Chen X, Lu HC, Chitwood RA, Johnston D. Activity-dependent decrease of excitability in rat hippocampal neurons through increases in Ih. Nat Neurosci. 2005;8:1542–1551. doi: 10.1038/nn1568. [DOI] [PubMed] [Google Scholar]
- Fransén E, Alonso AA, Dickson CT, Magistretti J, Hasselmo ME. Ionic mechanisms in the generation of subthreshold oscillations and action potential clustering in entorhinal layer II stellate neurons. Hippocampus. 2004;14:368–384. doi: 10.1002/hipo.10198. [DOI] [PubMed] [Google Scholar]
- Gasparini S, DiFrancesco D. Action of the hyperpolarization-activated current (Ih) blocker ZD 7288 in hippocampal CA1 neurons. Pflugers Arch. 1997;435:99–106. doi: 10.1007/s004240050488. [DOI] [PubMed] [Google Scholar]
- George MS, Abbott LF, Siegelbaum SA. HCN hyperpolarization-activated cation channels inhibit EPSPs by interactions with M-type K+ channels. Nat Neurosci. 2009;12:577–584. doi: 10.1038/nn.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JS, Dorval AD, 2nd, White JA. Contributions of Ih to feature selectivity in layer II stellate cells of the entorhinal cortex. J Comp Neurosci. 2007;22:161–171. doi: 10.1007/s10827-006-0005-7. [DOI] [PubMed] [Google Scholar]
- Han Y, Noam Y, Lewis AS, Gallagher JJ, Wadman WJ, Baram TZ, Chetkovich DM. Trafficking and gating of hyperpolarization-activated cyclic nucleotide-gated channels are regulated by interaction with tetratricopeptide repeat-containing Rab8b-interacting protein (TRIP8b) and cyclic AMP at distinct sites. J Biol Chem. 2011;286:20823–20834. doi: 10.1074/jbc.M111.236125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Vervaeke K, Graham LJ, Storm JF. Complementary theta resonance filtering by two spatially segregated mechanisms in CA1 hippocampal pyramidal neurons. J Neurosci. 2009;29:14472–14483. doi: 10.1523/JNEUROSCI.0187-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu L, Santoro B, Saponaro A, Liu H, Moroni A, Siegelbaum S. Binding of the auxiliary subunit TRIP8b to HCN channels shifts the mode of action of cAMP. J Gen Physiol. 2013;142:599–612. doi: 10.1085/jgp.201311013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Kadurin I, Uebele VN, Renger JJ, Dolphin AC, Shah MM. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci. 2011;14:478–486. doi: 10.1038/nn.2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Lujan R, Martinez-Hernandez J, Lewis AS, Chetkovich DM, Shah MM. TRIP8b-independent trafficking and plasticity of adult cortical presynaptic HCN1 channels. J Neurosci. 2012;32:14835–14848. doi: 10.1523/JNEUROSCI.1544-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Walker MC, Shah MM. Loss of dendritic HCN1 subunits enhances cortical excitability and epileptogenesis. J Neurosci. 2009;29:10979–10988. doi: 10.1523/JNEUROSCI.1531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston D, Narayanan R. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 2008;31:309–316. doi: 10.1016/j.tins.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Jung S, Bullis JB, Lau IH, Jones TD, Warner LN, Poolos NP. Downregulation of dendritic HCN channel gating in epilepsy is mediated by altered phosphorylation signaling. J Neurosci. 2010;30:6678–6688. doi: 10.1523/JNEUROSCI.1290-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Jones TD, Lugo JN, Jr, Sheerin AH, Miller JW, D'Ambrosio R, Anderson AE, Poolos NP. Progressive dendritic HCN channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J Neurosci. 2007;27:13012–13021. doi: 10.1523/JNEUROSCI.3605-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Warner LN, Pitsch J, Becker AJ, Poolos NP. Rapid loss of dendritic HCN channel expression in hippocampal pyramidal neurons following status epilepticus. J Neurosci. 2011;31:14291–14295. doi: 10.1523/JNEUROSCI.1148-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai HC, Jan LY. The distribution and targeting of neuronal voltage-gated ion channels. Nat Rev Neurosci. 2006;7:548–562. doi: 10.1038/nrn1938. [DOI] [PubMed] [Google Scholar]
- Lewis AS, Chetkovich DM. HCN channels in behavior and neurological disease: too hyper or not active enough. Mol Cell Neurosci. 2011;46:357–367. doi: 10.1016/j.mcn.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis AS, Schwartz E, Chan CS, Noam Y, Shin M, Wadman WJ, Surmeier DJ, Baram TZ, Macdonald RL, Chetkovich DM. Alternatively spliced isoforms of TRIP8b differentially control h channel trafficking and function. J Neurosci. 2009;29:6250–6265. doi: 10.1523/JNEUROSCI.0856-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz A, Notomi T, Tamas G, Shigemoto R, Nusser Z. Polarized and compartment-dependent distribution of HCN1 in pyramidal cell dendrites. Nat Neurosci. 2002;5:1185–1193. doi: 10.1038/nn962. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Budde T, Stieber J, Moosmang S, Wahl C, Holthoff K, Langebartels A, Wotjak C, Munsch T, Zong X, Feil S, Feil R, Lancel M, Chien KR, Konnerth A, Pape HC, Biel M, Hofmann F. Absence epilepsy and sinus dysrhythmia in mice lacking the pacemaker channel HCN2. EMBO J. 2003;22:216–224. doi: 10.1093/emboj/cdg032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Lujan R, Albasanz JL, Shigemoto R, Juiz JM. Preferential localization of the hyperpolarization-activated cyclic nucleotide-gated cation channel subunit HCN1 in basket cell terminals of the rat cerebellum. Eur J Neurosci. 2005;21:2073–2082. doi: 10.1111/j.1460-9568.2005.04043.x. [DOI] [PubMed] [Google Scholar]
- Maccaferri G, McBain CJ. The hyperpolarization-activated current (Ih) and its contribution to pacemaker activity in rat CA1 hippocampal stratum oriens–alveus interneurones. J Physiol. 1996;497:119–130. doi: 10.1113/jphysiol.1996.sp021754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maccaferri G, Mangoni M, Lazzari A, DiFrancesco D. Properties of the hyperpolarization-activated current in rat hippocampal CA1 pyramidal cells. J Neurophysiol. 1993;69:2129–2136. doi: 10.1152/jn.1993.69.6.2129. [DOI] [PubMed] [Google Scholar]
- McClelland S, Flynn C, Dube C, Richichi C, Zha Q, Ghestem A, Esclapez M, Bernard C, Baram TZ. Neuron-restrictive silencer factor-mediated hyperpolarization-activated cyclic nucleotide gated channelopathy in experimental temporal lobe epilepsy. Ann Neurol. 2011;70:454–464. doi: 10.1002/ana.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. J Neurosci. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic Ih normalizes temporal summation in hippocampal CA1 neurons. Nat Neurosci. 1999;2:508–514. doi: 10.1038/12229. [DOI] [PubMed] [Google Scholar]
- Magee JC. Dendritic integration of excitatory synaptic input. Nat Rev Neurosci. 2000;1:181–190. doi: 10.1038/35044552. [DOI] [PubMed] [Google Scholar]
- Marcelin B, Chauvière L, Becker A, Migliore M, Esclapez M, Bernard C. h channel-dependent deficit of theta oscillation resonance and phase shift in temporal lobe epilepsy. Neurobiol Dis. 2009;33:436–447. doi: 10.1016/j.nbd.2008.11.019. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Biel M, Hofmann F, Ludwig A. Differential distribution of four hyperpolarization-activated cation channels in mouse brain. Biol Chem. 1999;380:975–980. doi: 10.1515/BC.1999.121. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Bosco P, Calabrese G, Bakker C, De Sarro GB, Elia M, Ferri R, Oostra BA. Audiogenic seizures susceptibility in transgenic mice with fragile X syndrome. Epilepsia. 2000;41:19–23. doi: 10.1111/j.1528-1157.2000.tb01499.x. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Shi X, Numata T, Mori Y, Inoue R, Lossin C, Baram TZ, Hirose S. Novel HCN2 mutation contributes to febrile seizures by shifting the channel's kinetics in a temperature-dependent manner. PLoS One. 2013;8:e80376. doi: 10.1371/journal.pone.0080376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. Long-term potentiation in rat hippocampal neurons is accompanied by spatially widespread changes in intrinsic oscillatory dynamics and excitability. Neuron. 2007;56:1061–1075. doi: 10.1016/j.neuron.2007.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan R, Johnston D. The h channel mediates location dependence and plasticity of intrinsic phase response in rat hippocampal neurons. J Neurosci. 2008;28:5846–5860. doi: 10.1523/JNEUROSCI.0835-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noam Y, Bernard C, Baram TZ. Towards an integrated view of HCN channel role in epilepsy. Curr Opin Neurobiol. 2011;21:873–879. doi: 10.1016/j.conb.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Dudman JT, Dodson PD, Santoro B. HCN1 channels control resting and active integrative properties of stellate cells from layer II of the entorhinal cortex. J Neurosci. 2007;27:12440–12451. doi: 10.1523/JNEUROSCI.2358-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan MF, Malleret G, Dudman JT, Buhl DL, Santoro B, Gibbs E, Vronskaya S, Buzsaki G, Siegelbaum SA, Kandel ER, Morozov A. A behavioral role for dendritic integration: HCN1 channels constrain spatial memory and plasticity at inputs to distal dendrites of CA1 pyramidal neurons. Cell. 2004;119:719–732. doi: 10.1016/j.cell.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Notomi T, Shigemoto R. Immunohistochemical localization of Ih channel subunits, HCN1–4, in the rat brain. J Comp Neurol. 2004;471:241–276. doi: 10.1002/cne.11039. [DOI] [PubMed] [Google Scholar]
- Nusser Z. Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 2009;32:267–274. doi: 10.1016/j.tins.2009.01.003. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annu Rev Physiol. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Pavlov I, Scimemi A, Savtchenko L, Kullmann DM, Walker MC. Ih-mediated depolarization enhances the temporal precision of neuronal integration. Nat Commun. 2011;2:199. doi: 10.1038/ncomms1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskorowski R, Santoro B, Siegelbaum SA. TRIP8b splice forms act in concert to regulate the localization and expression of HCN1 channels in CA1 pyramidal neurons. Neuron. 2011;70:495–509. doi: 10.1016/j.neuron.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Migliore M, Johnston D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat Neurosci. 2002;5:767–774. doi: 10.1038/nn891. [DOI] [PubMed] [Google Scholar]
- Robinson RB, Siegelbaum SA. Hyperpolarization-activated cation currents: from molecules to physiological function. Annu Rev Physiol. 2003;65:453–480. doi: 10.1146/annurev.physiol.65.092101.142734. [DOI] [PubMed] [Google Scholar]
- Santoro B, Hu L, Liu H, Saponaro A, Pian P, Piskorowski RA, Moroni A, Siegelbaum SA. TRIP8b regulates HCN1 channel trafficking and gating through two distinct C-terminal interaction sites. J Neurosci. 2011;31:4074–4086. doi: 10.1523/JNEUROSCI.5707-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Lee JY, Englot DJ, Gildersleeve S, Piskorowski RA, Siegelbaum SA, Winawer MR, Blumenfeld H. Increased seizure severity and seizure-related death in mice lacking HCN1 channels. Epilepsia. 2010;51:1624–1627. doi: 10.1111/j.1528-1167.2010.02554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Santoro B, Piskorowski RA, Pian P, Hu L, Liu H, Siegelbaum SA. TRIP8b splice variants form a family of auxiliary subunits that regulate gating and trafficking of HCN channels in the brain. Neuron. 2009;62:802–813. doi: 10.1016/j.neuron.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro B, Wainger BJ, Siegelbaum SA. Regulation of HCN channel surface expression by a novel C-terminal protein-protein interaction. J Neurosci. 2004;24:10750–10762. doi: 10.1523/JNEUROSCI.3300-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Anderson AE, Leung V, Lin X, Johnston D. Seizure-induced plasticity of h channels in entorhinal cortical layer III pyramidal neurons. Neuron. 2004;44:495–508. doi: 10.1016/j.neuron.2004.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Hammond RS, Hoffman DA. Dendritic ion channel trafficking and plasticity. Trends Neurosci. 2010;33:307–316. doi: 10.1016/j.tins.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets PL, Suter BA, Kiritani T, Chan CS, Surmeier DJ, Shepherd GM. Corticospinal-specific HCN expression in mouse motor cortex: Ih-dependent synaptic integration as a candidate microcircuit mechanism involved in motor control. J Neurophysiol. 2011;106:2216–2231. doi: 10.1152/jn.00232.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin M, Brager D, Jaramillo TC, Johnston D, Chetkovich DM. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol Dis. 2008;32:26–36. doi: 10.1016/j.nbd.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N. Determinants of voltage attenuation in neocortical pyramidal neuron dendrites. J Neurosci. 1998;18:3501–3510. doi: 10.1523/JNEUROSCI.18-10-03501.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thuault SJ, Malleret G, Constantinople CM, Nicholls R, Chen I, Zhu J, Panteleyev A, Vronskaya S, Nolan MF, Bruno R, Siegelbaum SA, Kandel ER. Prefrontal cortex HCN1 channels enable intrinsic persistent neural firing and executive memory function. J Neurosci. 2013;33:13583–13599. doi: 10.1523/JNEUROSCI.2427-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsay D, Dudman JT, Siegelbaum SA. HCN1 channels constrain synaptically evoked Ca2+ spikes in distal dendrites of CA1 pyramidal neurons. Neuron. 2007;56:1076–1089. doi: 10.1016/j.neuron.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya SP, Johnston D. Temporal synchrony and gamma-to-theta power conversion in the dendrites of CA1 pyramidal neurons. Nat Neurosci. 2013;16:1812–1820. doi: 10.1038/nn.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Welie I, Remme MW, van Hooft JA, Wadman WJ. Different levels of Ih determine distinct temporal integration in bursting and regular-spiking neurons in rat subiculum. J Physiol. 2006;576:203–214. doi: 10.1113/jphysiol.2006.113944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Welie I, van Hooft JA, Wadman WJ. Homeostatic scaling of neuronal excitability by synaptic modulation of somatic hyperpolarization-activated Ih channels. Proc Natl Acad Sci U S A. 2004;101:5123–5128. doi: 10.1073/pnas.0307711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl-Schott C, Biel M. HCN channels: structure, cellular regulation and physiological function. Cell Mol Life Sci. 2009;66:470–494. doi: 10.1007/s00018-008-8525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker MC, Kullmann DM. Febrile convulsions: a ‘benign’ condition. Nat Med. 1999;5:871–872. doi: 10.1038/11308. [DOI] [PubMed] [Google Scholar]
- Wang M, Ramos BP, Paspalas CD, Shu Y, Simen A, Duque A, Vijayraghavan S, Brennan A, Dudley A, Nou E, Mazer JA, McCormick DA, Arnsten AF. α2A-adrenoceptors strengthen working memory networks by inhibiting cAMP-HCN channel signaling in prefrontal cortex. Cell. 2007;129:397–410. doi: 10.1016/j.cell.2007.03.015. [DOI] [PubMed] [Google Scholar]
- White HS. Animal models of epileptogenesis. Neurology. 2002;59(9 Suppl. 5):S7–S14. doi: 10.1212/wnl.59.9_suppl_5.s7. [DOI] [PubMed] [Google Scholar]
- Wierschke S, Lehmann TN, Dehnicke C, Horn P, Nitsch R, Deisz RA. Hyperpolarization-activated cation currents in human epileptogenic neocortex. Epilepsia. 2010;51:404–414. doi: 10.1111/j.1528-1167.2009.02275.x. [DOI] [PubMed] [Google Scholar]
- Wilkars W, Liu Z, Lewis AS, Stoub TR, Ramos EM, Brandt N, Nicholson DA, Chetkovich DM, Bender RA. Regulation of axonal HCN1 trafficking in perforant path involves expression of specific TRIP8b isoforms. PLoS One. 2012;7:e32181. doi: 10.1371/journal.pone.0032181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Site independence of EPSP time course is mediated by dendritic Ih in neocortical pyramidal neurons. J Neurophysiol. 2000;83:3177–3182. doi: 10.1152/jn.2000.83.5.3177. [DOI] [PubMed] [Google Scholar]
- Williams SR, Stuart GJ. Voltage- and site-dependent control of the somatic impact of dendritic IPSPs. J Neurosci. 2003;23:7358–7367. doi: 10.1523/JNEUROSCI.23-19-07358.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolles G, Wenzel D, Bildl W, Schulte U, Hofmann A, Muller CS, Thumfart JO, Vlachos A, Deller T, Pfeifer A, Fleischmann BK, Roeper J, Fakler B, Klocker N. Association with the auxiliary subunit PEX5R/Trip8b controls responsiveness of HCN channels to cAMP and adrenergic stimulation. Neuron. 2009;62:814–825. doi: 10.1016/j.neuron.2009.05.008. [DOI] [PubMed] [Google Scholar]