

Abstract
Rapid, specific, and sensitive detection of bacterial pathogens is essential toward clinical management of infectious diseases. Traditional approaches for pathogen detection, however, often require time-intensive bacterial culture and amplification procedures. Herein, a microparticle enhanced double-stranded DNA probe is demonstrated for rapid species-specific detection of bacterial 16S rRNA. In this molecular assay, the binding of the target sequence to the fluorophore conjugated probe thermodynamically displaces the quencher probe and allows the fluorophore to fluoresce. By incorporation of streptavidin-coated microparticles to localize the biotinylated probes, the sensitivity of the assay can be improved by 3 orders of magnitude. The limit of detection of the assay is as few as eight bacteria without target amplification and is highly specific against other common pathogens. Its applicability toward clinical diagnostics is demonstrated by directly identifying bacterial pathogens in urine samples from patients with urinary tract infections.
Infectious disease caused by bacterial pathogens is a major healthcare challenge worldwide. For instance, urinary tract infection (UTI), which is the most common bacterial infection of any organ system,1–3 accounts for seven million office visits and more than one million hospital admissions each year.4 For clinical management of infectious diseases, identification and quantification of bacterial pathogens in patient-derived samples are required to assess the severity of the infection. The standard method for identifying pathogenic agents in clinical samples, such as urine in UTI, requires transportation of the samples to a central microbiology laboratory. After overnight culture, the bacterial colonies are counted and identified based on morphology and other phenotypic parameters. A major shortcoming of the standard culture-based diagnostic approach is the significant delay of at least 2–3 days from sample collection to result reporting. The absence of definitive microbiological diagnosis has largely driven the over- and misuse of antibiotics. The development of specific and sensitive molecular biosensing techniques for rapid detection of bacterial pathogens would revolutionize the clinical practice of UTI and other infectious diseases by allowing evidence-based, rather than empiric, management of infectious diseases and effective treatment to the patients.
A variety of molecular biosensing techniques, such as Southern blot, polymerase chain reaction (PCR), DNA microarray, nucleic acid sequence-based amplification, mass spectroscopy, and immunoblot, have been adopted for the detection of bacterial pathogens.5–13 These genotypic and proteomic approaches have allowed highly sensitive and specific detection of bacterial pathogen. On the other hand, development of rapid bacterial detection strategies toward point-of-care applications has been receiving increasing attention due to the time and labor intensive protocols associated with most of the existing assays. In particular, assays that are free of separation, amplification, and bacterial culture are highly desirable.14–16 These assays could dramatically simplify the assay protocol and facilitate rapid diagnostics in resource-limited settings.
Double-stranded DNA (dsDNA) probe is a homogeneous assay for rapid detection of specific nucleic acid sequences and can potentially be applied for pathogen identification.17–29 In the dsDNA sensing scheme, a DNA sequence containing a fluorophore labeled on the 5′ end is designed to be complementary to the nucleotide sequence of interest. To allow homogeneous detection of the target, a complementary sequence is designed with respect to the fluorophore probe but with a shorter length and its 3′ end is labeled with a quencher. In the absence of the target, the fluorophore and quencher probes are in close proximity diminishing the fluorescence signal. With the target, the quencher probe is replaced due to the thermodynamically driven binding event between the fluorophore probe and the target. Therefore, the fluorophore is separated from the quencher and is able to fluoresce. Compared to other homogeneous assays for nucleic acids, such as a molecular beacon, advantages of dsDNA probes include the possibility of adjusting the quencher-to-fluorophore ratio for noise minimization and the flexibility of modifying the lengths of the quencher sequence and the sticky end for improving the specificity and kinetics of the assay. The dsDNA probes have been demonstrated in various biomedical applications, including detection of single nucleotide mismatches, quantification of PCR products, and quantification of DNA binding proteins.17–29 However, most dsDNA assays require target amplification, such as PCR, to improve the sensitivity, and the applicability of dsDNA probes for rapid detection of bacterial pathogens has not been demonstrated. This is partially due to the limited sensitivity of the assay, which is often a result of the strong background in the biological sample, as a washing step is not involved in the homogeneous assay. This is particularly challenging for detecting pathogen in clinical samples such as urine and serum, which is known to have a strong matrix effect.30,31 This represents a major technical hurdle for applying dsDNA probes and other homogeneous assays for quantifying bacterial pathogens without amplification or bacterial culture.
To tackle the above-mentioned challenges, we present a rapid molecular approach for detecting bacterial pathogens using micro-particle conjugated dsDNA probes. Figure 1 shows the overall concept of the detection scheme. To achieve specific pathogen identification, the dsDNA probe is designed to be complementary to the species-specific 16S ribosomal RNA (rRNA) of the target pathogens with 20 000 copies in each bacterium.32,33 Detecting the bacterial 16S ribosomal RNA molecule allows not only specific pathogen identification but also high sensitivity due to the high copy number of 16S rRNA in each bacterium. To further improve the sensitivity of the assay without target amplification, streptavidin coated microparticles are applied to enhance the performance of the assay. In particular, the 3′ end of the fluorophore probe is labeled with biotin so that multiple probes can be captured by streptavidin coated microparticles. The resulting assembly brings a large number of fluorophore probes into a small region, which significantly increases the intensity, and facilitates further manipulation.
Figure 1.
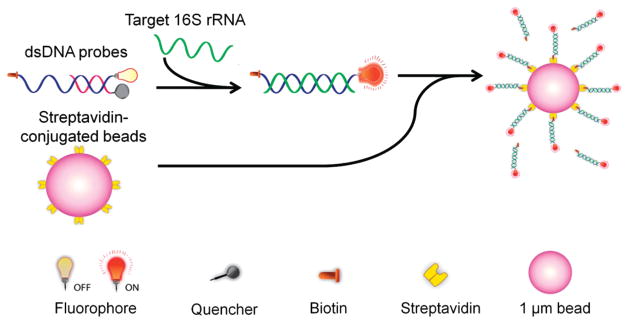
Schematic of a microparticle conjugated dsDNA probe for detecting specific nucleic acid sequences. In the existence of target, the fluorophore probe is thermodynamically driven to hybridize the target, which replaces the quencher probe. The probes are further captured by streptavidin coated microparticles.
In this study, a dsDNA probe that specifically detects Escherichia coli (E. coli), which accounts for over 80% of uncomplicated UTI, is designed to explore the microparticle enhanced molecular assay for pathogen detection. The signal-to-noise ratio of the assay is first optimized by adjusting the quencher-to-fluorophore ratio. Then, the analytical sensitivities and dynamic ranges of the dsDNA probes with and without microparticles are determined using synthetic targets and uropathogenic E. coli clinical isolates. The analytical specificity of the assay is tested against common pathogens including Staphylococcus saprophyticus (S. saprophyticus), Enterococcus species (spp.), and Proteus mirabilis (P. mirabilis). In addition, the applicability of the molecular assay to clinical testing is evaluated by using urine samples from UTI patients as a proof of concept.
MATERIALS AND METHODS
Bacteria and Clinical Urine Samples
Uropathogenic clinical isolates, including E. coli, S. saprophyticus, Enterococcus spp., P. mirabilis, and E. coli strain DH5α were applied in the experiment. The uropathogenic clinical isolates were obtained from four different patients with documented urinary tract infections. For E. coli, the bacteria were inoculated with Luria broth (LB) in a shaker at 37 °C and grown to 107–108 cfu/mL. The bacteria were then mixed with 25% glycerol (BD, MD) and stored at −80 °C. S. saprophyticus, Enterococcus spp., and P. mirabilis were cultured in LB overnight at 37 °C and diluted to 107 cfu/mL in the experiments. Two de-identified infected clinical urine samples were tested to evaluate the applicability of the assay for clinical diagnostics. Both patients (age 67 and 58) are male and has a history of spinal cord injury. Each sample contained a single species of uropathogen. One of them (sample no. 120) had 108 cfu/mL of E. coli and the other (sample no. 334) had 107 cfu/mL P. mirabilis. The samples were aliquoted upon sample receipt. Samples were pelleted by centrifugation for 5 min at 14 000g (microfuge), supernatant removed, pellets snap frozen on dry ice, and stored at −80 °C. The clinical isolates and urine samples were collected with approval from the Stanford University and VA Palo Alto Health Care System (VAPAHCS) Institutional Review Board. Identification of microorganisms was performed in the VAPAHCS clinical microbiology laboratory.
Probe Design
One set of dsDNA probe was designed to evaluate the assay in this study. Oligonucleotide probes targeting the 16S rRNA were designed using Primrose and Ribosomal Database Project Release 8. Probe selectivity was verified by alignment against 16S rDNA sequences from NCBI using Geneious and confirmed with bacterial clinical isolates from VAPAHCS. The fluorophore probe was designed to bind specifically to the loop region of the 16S rRNA, and the quencher probe is complementary to the fluorophore probe. The fluorophore and quencher sequences of the E. coli probes were 24 and 12 bases in length, respectively. As previously shown, these lengths are optimized for both improvement of thermodynamically driven replacement between the target and quencher and reduction of nonspecific binding.27 The fluorophore probe was labeled with biotin and fluorophore, TAMRA(NHS Ester), in the 3′ and 5′ ends, respectively. The fluorophore can be excited at 559 nm and has an emission peak at 583 nm. The quencher probes were labeled with Iowa Black RQ at the 3′ end, which was chosen based on its high quenching efficiency for TAMRA. In addition to the E. coli probe, a set of random probe was designed as a control. In the case of microparticle enhanced dsDNA assay, streptavidin-coated 1.0 μm particles (4 μL of 0.001% w/v) with 2.7 nmol/mg binding sites capacity (which can bind up to 1.16 × 1015 molecules per particle) were added to the supernatant (Figure S1 in the Supporting Information). Additional details of the experimental protocol can be found in the Supporting Information. Table S1 in the Supporting Information summarizes the probe designs in this study.
RESULTS AND DISCUSSION
Background Minimization
The disassociation of the free fluorophore probe from the quencher probe is a major source of the background noise and represents a major factor that limits the overall sensitivity of the dsDNA assay. In order to minimize the background level, the concentration of the quencher probe relative to the concentration of the fluorophore probe (quencher-to-fluorophore ratio) was adjusted systematically to minimize the concentration of free fluorophore probe in the solution (Figure S2 in the Supporting Information). The background fluorescence generally diminishes with the quencher-to-fluorophore ratio. The fluorescence intensity is minimized when the quencher-to-fluorophore ratio reaches 3-to-1 ratio and a further increase in the ratio does not show significant reduction of the fluorescence intensity. A higher quencher-to-fluorophore ratio can affect the probe sensitivity and shifts the dynamic range to the higher target concentration. This is consistent with our previous theoretical and experimental studies that a 3-to-1 ratio allows a high signal-to-noise ratio. Furthermore, two different fluorophores, 6-FAM (excitation 495 nm/emission 520 nm) and TAMIN (excitation 559 nm/emission 583 nm), were examined for reducing the background noise. In our experiment, TAMIN was shown to have a better signal-to-noise ratio (data not shown). It is likely a result of the strong autofluorescence from cellular components as well as the bacterial growth media at the shorter wavelength. As a result, TAMIN is used in all the other experiments.
dsDNA Probes for Detecting Bacterial 16S rRNA
The performance of the dsDNA probe without the microparticle was first characterized to evaluate the use of the assay for rapid molecular analysis without target amplification. Figure 2 shows the titration curves of the dsDNA probe for detecting synthetic targets. The fluorescence intensity generally increases with the concentration of the target and have a large dynamic range spanning over several orders of magnitude in concentration. This is consistent with previous analyses of the dsDNA probe.26,27 With a probe concentration of 3.4 nM, the synthetic target can be detected from the nanomolar to picomolar range. The LOD (95%) of the assay is estimated to be 4.8 pM (Table S2 in the Supporting Information). To evaluate its ability for bacterial detection, we have conducted experiments with the dsDNA probe to detect 16S rRNA from E. coli. The result is shown in Figure 3. The titration curve for detecting bacterial 16S rRNA displays a similar trend compared to the titration curve for detecting synthetic target. The dynamic range of the assay is 107 to 104 cfu with a LOD (95%) of 10 520 cfu in a 100 μL volume sample (Table S2 in the Supporting Information). This is equivalent to approximately 105 cfu/mL or 3.5 pM by assuming that there are 20 000 copies of 16S rRNA in each bacterium.34 This result is in good agreement with the LOD determined using synthetic targets and suggests that the dsDNA probe can effectively detect bacterial 16S rRNA.
Figure 2.

Titration curve of the dsDNA probe determined using synthetic DNA target. Insert shows the intensities at the lower concentration range (9 pM to 9 fM).
Figure 3.

Titration curve of the dsDNA probe determined using E. coli bacteria (DH5α). Insert shows the intensities at the lower concentration range (20 000 cfu to 20 cfu).
Microparticle Enhanced dsDNA Probe
To further improve the sensitivity of the assay, microparticles coated with streptavidin are applied. The microparticle-enhanced dsDNA probe aims to improve the sensitivity by localizing the target concentration into a small region (Figure S2 in the Supporting Information). Figure 4 shows the intensities of the microparticle enhanced dsDNA probes with different concentrations of synthetic target. In general, the microparticle-enhanced dsDNA probe achieves a higher signal than merely the dsDNA probe at every single target concentration tested. Similarly, the assay has a dynamic range across several orders of magnitude in concentration. With utilization of the microparticles, the minimum target concentration that is distinguishable from the background noise is on the order of femtomolar. Statistical analysis shows the LOD of the assay is 4.5 fM, which is 1000-fold better than that achieved by the dsDNA probe itself (Table S2 in the Supporting Information). This result suggests that the microparticle provides a simple and effective approach to enhance the sensitivity of the dsDNA probe assay without target amplification. The microparticle enhanced assay was also evaluated for its capability to detect E. coli. The result is shown in Figure 5. Similar to the synthetic target experiment, we observed a significant improvement in the sensitivity of the assay compared to the dsDNA probe alone. The LOD of the assay is estimated to be 8 cfu in a 100 μL sample (i.e., 80 cfu/mL), which is equivalent to 2.6 fM of 16S rRNA. These data show that the incorporation of the microparticles can significantly improve the sensitivity of the dsDNA probe and that the assay requires less than 10 bacteria for pathogen identification.
Figure 4.

Titration curve of the microparticle-enhanced dsDNA probe determined using synthetic DNA target. Insert shows the intensities at lower concentration range (9 pM to 9 fM).
Figure 5.

Titration curve of the microparticle-enhanced dsDNA probe determined using E. coli bacteria (DH5α). Insert shows the intensities at the lower concentration range (20 000 cfu to 20 cfu).
We further investigated the specificity of the microparticle enhanced dsDNA probe. The specificity of the assay was evaluated against three other common uropathogenic bacteria including S. saprophyticus, Enterococcus spp., and P. mirabilis. The result is shown in Figure 6. In the experiment, the signals for these pathogens cannot be statistically distinguished from the background. This shows the dsDNA probe has excellent specificity against other bacteria. In fact, the probe sequence has been previously demonstrated for its specificity for E. coli against other bacteria in an electrochemical format.3,35 Our results support that bacterial 16S rRNA can be specifically detected by dsDNA probes and that the incorporation of the microparticle does not compromise the specificity of the probe sequence.
Figure 6.

Specificity of the microparticle conjugated probe for the E. coli clinical isolate against other common uropathogens, including S. saprophyticus (SS), Enterococcus spp. (ES), and P. mirabilis (PM). Samples without bacteria were utilized as a negative control.
Clinical Urine Samples
A major challenge in molecular diagnostics using physiological samples is the matrix effect, which is effect of components in a sample other than the target analyte.30,31 Conventional assays for bacterial detection require time-consuming bacterial culture steps to isolate the target bacteria from the sample matrix. To avoid the culture step for rapid detection, a molecular assay for point-of-care diagnostics should be insensitivity to the matrix effect. To test the ability of the microparticle enhanced dsDNA assay to detect bacteria directly from physiological samples, two clinical urine samples from UTI patients with 108 cfu/mL E. coli and 107 cfu/mL P. mirabilis bacteria, respectively, were tested (Figure 7). The relative intensity of the clinical urine sample with E. coli is significantly higher than that with both P. mirabilis bacteria and negative control. The intensity of the sample with P. mirabilis cannot be distinguished from the signal in the negative control. These results indicate that the microparticle enhanced assay is capable of specific detection of the target bacteria in urine and is not affected by the matrix effects.
Figure 7.

Specificity of the microparticle conjugated probe for detecting bacteria in clinical urine samples. Three samples were tested including urine 120 (with 108 cfu/mL E. coli), urine 334 (with 107 cfu/mL P. mirabilis), and negative control.
Evolution of biosensor systems to develop practical and cost-effective approaches with procedures that are easy to implement and have high sensitivity are of great importance for disease diagnostic applications. Within this context, we have demonstrated a homogeneous dsDNA probe to rapidly quantify bacteria. Bacterial 16S rRNA was chosen as the target for molecular analysis due to its high copy numbers in bacteria. Moreover, 16S rRNA gene sequences are well characterized with regions of interspecies diversity that are useful for probe design to differentiate different bacterial species.36 As a proof of concept, we designed the dsDNA probe for E. coli and tested it with clinical isolates and urine samples from UTI patients. As demonstrated in this study, the probe has good specificity against other common bacteria and sample matrix from clinical samples. To the best of our knowledge, this is the first demonstration of the applicability of dsDNA probes for specific detection of bacterial 16S rRNA. Compared to other hybridization assays that detect the species-specific bacterial 16S rRNA,3,24,35,37,38 the dsDNA probe dramatically simplifies the assay protocol and is particularly suitable in resource limited settings, such as rural clinics and temporary clinics at a disaster zone.
We have also demonstrated enhancement of the sensitivity of dsDNA probes by utilizing microparticles to localize the probes. Our results show that the combination of microparticles and dsDNA probes leads to a highly sensitive and specific optical biosensor for bacteria identification and quantification. Because of the small sample volume in this assay, the detection limit is reported in terms of absolute number of bacteria instead of bacterial concentrations. Without microparticles, the assay requires over 104 bacteria for pathogen identification. When the molecular probes are localized with microparticles, the sensitivity of the probe can be improved for over 1000-fold. Similar concepts were applied in a molecular beacon-labeled micro-sphere assay for detecting synthetic nucleic acid sequences that mimic SARS coronavirus.39 On the other hand, we demonstrate that microparticles can be combined with the dsDNA probe for improving its sensitivity for over 3 orders of magnitude and less than 10 bacteria are required for pathogen identification. The improvement in the sensitivity allows direct detection of bacteria without overnight culture or target amplification procedures.
Other advantages of microparticle enhanced dsDNA assay include short assay time and simplicity of the assay protocol. These characteristics are essential considerations for point-of-care applications. The total assay time from sample-to-result is less than 40 min including sample centrifugation for 5 min, bacterial lysis for 10 min, probe hybridization for 10 min, and target localization by microparticles for 5 min. This is a significant reduction compared to days in standard culture based approaches and can potentially be further reduced if automated using microfluidic techniques.40,41 Implementing the microparticle enhanced dsDNA assay using microfluidics will also facilitate multiplexed detection, sample preconcentration, and antimicrobial susceptibility testing.42,43 Finally, only a small volume of sample is required for the assay. This is particularly useful when a large amount of sample is not available, such as diagnostics for the pediatric population.
CONCLUSIONS
In this study, we have developed and demonstrated a microparticle enhanced dsDNA assay for quantitative detection of bacteria with high sensitivity and specificity. Our results suggest the assay can be directly applied for clinical diagnostic applications, although a larger scale clinical validation study with greater number of clinical samples will be required to determine the diagnostic sensitivity and specificity. With its rapidity, simplicity, and small volume requirement, the assay can potentially be applied to various biomedical applications, such as pathogen identification for urinary tract infection and sepsis diagnostics in neonatal intensive care units.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Director’s New Innovator Award (Grant 1DP2OD007161-01), NIAID (Grants 1U01AI082457-01 and R43AI088756-01), NICHD (Grant R43HD065303-01), and the National Science Foundation (Grants 0930900 and 0900899).
Footnotes
Supporting Information. Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Nicolle LE. Infect Med. 2001;18:153–162. [Google Scholar]
- 2.Foxman B, Barlow R, D’Arcy H, Gillespie B, Sobel JD. Ann Epidemiol. 2000;10:509–515. doi: 10.1016/s1047-2797(00)00072-7. [DOI] [PubMed] [Google Scholar]
- 3.Liao JC, Mastali M, Gau V, Suchard MA, Moller AK, Bruckner DA, Babbitt JT, Li Y, Gornbein J, Landaw EM, McCabe ERB, Churchill BM, Haake DA. J Clin Microbiol. 2006;44:561–570. doi: 10.1128/JCM.44.2.561-570.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schappert SM, Burt CW. Vital Health Stat. 2006;13:1–66. [PubMed] [Google Scholar]
- 5.Zhao Y, Park S, Kreiswirth BN, Ginocchio CC, Veyret R, Laayoun A, Troesch A, Perlin DS. J Clin Microbiol. 2009;47:2067–2078. doi: 10.1128/JCM.02230-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jordan JA, Durso MB. J Mol Diagn. 2005;7:575–581. doi: 10.1016/S1525-1578(10)60590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang S, Lin S, Kelen GD, Quinn TC, Dick JD, Gaydos CA, Rothman RE. J Clin Microbiol. 2002;40:3449–3454. doi: 10.1128/JCM.40.9.3449-3454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joung HA, Lee NR, Lee SK, Ahn J, Shin YB, Choi HS, Lee CS, Kim S, Kim MG. Anal Chim Acta. 2008;630:168–173. doi: 10.1016/j.aca.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 9.Frye JG, Jesse T, Long F, Rondeau G, Porwollik S, McClelland M, Jackson CR, Englen M, Fedorka-Cray PJ. Int J Antimicrob Agents. 2006;27:138–151. doi: 10.1016/j.ijantimicag.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 10.Allen KJ, Poppe C. Can J Vet Res. 2002;66:137–144. [PMC free article] [PubMed] [Google Scholar]
- 11.Nogales B, Timmis KN, Nedwell DB, Osborn AM. Appl Environ Microb. 2002;68:5017–5025. doi: 10.1128/AEM.68.10.5017-5025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gikunju CM, Lev SM, Birenzvige A, Schaefer DM. Talanta. 2004;62:741–744. doi: 10.1016/j.talanta.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 13.Wang ZP, Russon L, Li L, Roser DC, Long SR. Rapid Commun Mass Spectrom. 1998;12:456–464. doi: 10.1002/(SICI)1097-0231(19980430)12:8<456::AID-RCM177>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 14.Tyagi S, Kramer FR. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 15.Bao G, Tsourkas A, Santangelo P. Abstr Pap Am Chem Soc. 2003;225:U982–U982. [Google Scholar]
- 16.Li N, Wong PK. Bioanalysis. 2010;2:1689–1699. doi: 10.4155/bio.10.116. [DOI] [PubMed] [Google Scholar]
- 17.Fuller NJ, Marie D, Partensky F, Vaulot D, Post AF, Scanlan DJ. Appl Environ Microbiol. 2003;69:2430–2443. doi: 10.1128/AEM.69.5.2430-2443.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manero A, Blanch AR. J Microbiol Methods. 2002;50:115–121. doi: 10.1016/s0167-7012(02)00027-1. [DOI] [PubMed] [Google Scholar]
- 19.Hsieh CC, Huang SB, Wu PC, Shieh DB, Lee GB. Biomed Microdevices. 2009;11:903–913. doi: 10.1007/s10544-009-9307-7. [DOI] [PubMed] [Google Scholar]
- 20.Blair RH, Rosenblum ES, Dawson ED, Kuchta RD, Kuck LR, Rowlen KL. Anal Biochem. 2007;362:213–220. doi: 10.1016/j.ab.2006.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crockett AO, Wittwer CT. Anal Biochem. 2001;290:89–97. doi: 10.1006/abio.2000.4957. [DOI] [PubMed] [Google Scholar]
- 22.Nutiu R, Li YF. J Am Chem Soc. 2003;125:4771–4778. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- 23.Nutiu R, Li YF. Angew Chem, Int Ed. 2005;44:1061–1065. doi: 10.1002/anie.200461848. [DOI] [PubMed] [Google Scholar]
- 24.Li N, Ho CM. J Am Chem Soc. 2008;130:2380–2381. doi: 10.1021/ja076787b. [DOI] [PubMed] [Google Scholar]
- 25.Li QQ, Luan GY, Guo QP, Liang JX. Nucleic Acids Res. 2002;30:e5. doi: 10.1093/nar/30.2.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gidwani V, Riahi R, Zhang DD, Wong PK. Analyst. 2009;134:1675–1681. doi: 10.1039/b906077d. [DOI] [PubMed] [Google Scholar]
- 27.Meserve D, Wang Z, Zhang DD, Wong PK. Analyst. 2008;133:1013–1019. doi: 10.1039/b804853c. [DOI] [PubMed] [Google Scholar]
- 28.Wang Z, Gidwani V, Sun Z, Zhang DD, Wong PK. J Assoc Lab Autom. 2008;13:243–248. [Google Scholar]
- 29.Wang Z, Gidwani V, Zhang DD, Wong PK. Analyst. 2008;133:998–1000. doi: 10.1039/b809113g. [DOI] [PubMed] [Google Scholar]
- 30.Chiu ML, Lawi W, Snyder ST, Wong PK, Liao JC, Gau V. J Assoc Lab Autom. 2010;15:233–242. [Google Scholar]
- 31.Mariella R. Biomed Microdevices. 2008;10:777–784. doi: 10.1007/s10544-008-9190-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delong EF, Wickham GS, Pace NR. Science. 1989;243:1360–1363. doi: 10.1126/science.2466341. [DOI] [PubMed] [Google Scholar]
- 33.Fuchs BM, Wallner G, Beisker W, Schwippl I, Ludwig W, Amann R. Appl Environ Microb. 1998;64:4973–4982. doi: 10.1128/aem.64.12.4973-4982.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neidhardt F, Umbarger H. Escherichia coli and Salmonella typhimurium. 2. I. ASM Press; Washington, D.C: 1996. Chemical composition of Escherichia coli; pp. 13–16. [Google Scholar]
- 35.Sun CP, Liao JC, Zhang YH, Gau V, Mastali M, Babbitt JT, Grundfest WS, Churchill BM, McCabe ERB, Haake DA. Mol Genet Metab. 2005;84:90–99. doi: 10.1016/j.ymgme.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 36.Relman DA. Science. 1999;284:1308–1310. doi: 10.1126/science.284.5418.1308. [DOI] [PubMed] [Google Scholar]
- 37.Lagally ET, Lee SH, Soh HT. Lab Chip. 2005;5:1053–1058. doi: 10.1039/b505915a. [DOI] [PubMed] [Google Scholar]
- 38.Gau JJ, Lan EH, Dunn B, Ho CM, Woo JCS. Biosens Bioelectron. 2001;16:745–755. doi: 10.1016/s0956-5663(01)00216-0. [DOI] [PubMed] [Google Scholar]
- 39.Horejsh D, Martini F, Poccia F, Ippolito G, Di Caro A, Capobianchi MR. Nucleic Acids Res. 2005;33:e13. doi: 10.1093/nar/gni015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ali MF, Kirby R, Goodey AP, Rodriguez MD, Ellington AD, Neikirk DP, McDevitt JT. Anal Chem. 2003;75:4732–4739. doi: 10.1021/ac034106z. [DOI] [PubMed] [Google Scholar]
- 41.Wang TH, Wong PK. J Assoc Lab Autom. 2010;15:A15–A16. [Google Scholar]
- 42.Gao J, Sin MLY, Liu T, Gau V, Liao JC, Wong PK. Lab Chip. 2011;11:1770–1775. doi: 10.1039/c1lc20054b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CH, Lu Y, Sin MLY, Mach KE, Zhang DD, Gau V, Liao JC, Wong PK. Anal Chem. 2010;82:1012–1019. doi: 10.1021/ac9022764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.