

SUMMARY
Receptor engagement by HIV-1 during host cell entry activates signaling pathways that can reprogram the cell for optimal viral replication. To obtain a global view of the signaling events induced during HIV-1 entry, we conducted a quantitative phosphoproteomics screen of primary human CD4+ T cell after infection with an HIV-1 strain that engages the receptors CD4 and CXCR4. We quantified 1,757 phosphorylation sites with high stringency. The abundance of 239 phosphorylation sites from 175 genes, including several proteins in pathways known to be impacted by HIV-receptor binding, changed significantly within a minute after HIV-1 exposure. Several previously uncharacterized HIV-1 host factors were also identified and confirmed through RNAi depletion studies. Surprisingly, 5 serine/arginine-rich (SR)-proteins involved in mRNA splicing, including the splicing factor SRm300 (SRRM2) were differentially phosophorylated. Mechanistic studies with SRRM2 suggest that HIV-1 modulates host cell alternative splicing machinery during entry in order to facilitate virus replication and release.
INTRODUCTION
Viruses not only rely on host molecules to successfully infect and replicate in new cells, but often directly manipulate cellular processes to facilitate more efficient virus replication. Vaccinia virus (Mercer and Helenius, 2008), herpes viruses (Soroceanu et al., 2008), and enteroviruses (Coyne and Bergelson, 2006) induce signaling cascades resulting from viral engagement of cell surface receptors that can induce the internalization of viral particles and facilitate viral replication. While many studies have contributed to the growing list of cellular host factors that are required for viral replication, relatively little is known of the full extent to which viruses can facilitate their entry and replication by manipulating host signal transduction pathways.
Human immunodeficiency virus-1 (HIV-1) primarily infects CD4+ T cells in vivo. The gp120 subunit of the viral envelope protein first engages CD4 then binds to one of two chemokine receptors, CCR5 and CXCR4 (Wilen et al., 2012). These interactions enable the viral envelope protein to undergo the conformational changes needed to elicit fusion between the viral and host cell membranes and also result in signal transduction via CD4 and the CCR5 or CXCR4 coreceptors that can enhance T cell activation and virus replication (Briant et al., 1996; Davis et al., 1997). More specifically, HIV-regulated phosphorylation of cofilin (Yoder et al., 2008), moesin (Barrero-Villar et al., 2009), filamin (Jimenez-Baranda et al., 2007) and LARG (Hodges et al., 2007) during entry can modulate various early steps of viral replication. Whether these or other virus-induced signaling events enhance later steps of the HIV-1 life cycle is not well understood.
Given the growing list of virus-induced cellular signaling events that can influence entry and replication, we hypothesized that uncovering signaling events resulting from HIV-host cell interactions would reveal additional HIV-1 host factors. Using mass spectrometry (MS)-based phosphoproteomics in combination with stable isotope labeling by amino acids in cell culture (SILAC), we quantified 1,757 phosphorylation sites with high stringency in primary human CD4+ T cells during entry of an HIV-1 strain that engages CD4 and CXCR4. We found that 239 phosphorylation sites from 175 genes significantly changed in abundance within 1 min of exposure to HIV-1, including several proteins in signaling pathways that are known to be impacted by HIV-receptor binding. Unexpectedly, we found that five serine/arginine-rich (SR)-proteins involved in mRNA splicing were differentially phosophorylated in T cells upon HIV-receptor engagement. These included nine phosphorylation sites in the splicing factor SRm300 (SRRM2), a cellular protein without previously described functional links to HIV-1 replication. We found that suppression of SRRM2 with RNAi in Jurkat and MAGI cell lines enhanced HIV-1 gene expression only when virions containing Env proteins that engage CXCR4 were used. In addition, SRRM2 modulated alternative splicing of HIV-1 transcripts and was required for efficient virion release. These findings show that HIV-1 induced signaling events are far more extensive than previously suspected, and that by inducing signaling pathways, HIV-1 can reprogram cells to not only enhance entry, but to make later stages of the virus replication cycle, such as splicing, more efficient as well.
RESULTS
SILAC-based quantitative phosphoproteomics in nondividing primary CD4+ T cells
To identify signaling events induced soon after HIV-1 engagement of cell surface receptors, we titrated highly purified HIV-1 MN - a virus strain that utilizes CD4 and CXCR4 - on unstimulated primary human CD4+ T cells. A mock virus prep, produced from the same cells and in the same manner as the virus, was used as a negative control. We reasoned that the relatively quiescent state of these cells make it possible to monitor virus-induced signaling events that might otherwise be masked following potent in vitro stimulation regimes that are commonly used to maximize virus infection. To synchronize virus binding, we added a high concentration of virus at 37°C in order to obtain a sufficient number of binding events within a very short time-frame. Using cells from multiple donors, we found that addition of 20 μg/mL of viral p24 (~15nM gp120) to unstimulated CD4+ T cells for one minute gave optimal phosphorylation of MAPK p38, a known HIV-responsive phosphorylation site (data not shown and Figure 1B) (Furler and Uittenbogaart, 2010). We used these conditions to study early signaling events induced by HIV-1 upon binding to the cell surface.
Figure 1. Phosphoproteomics workflow.
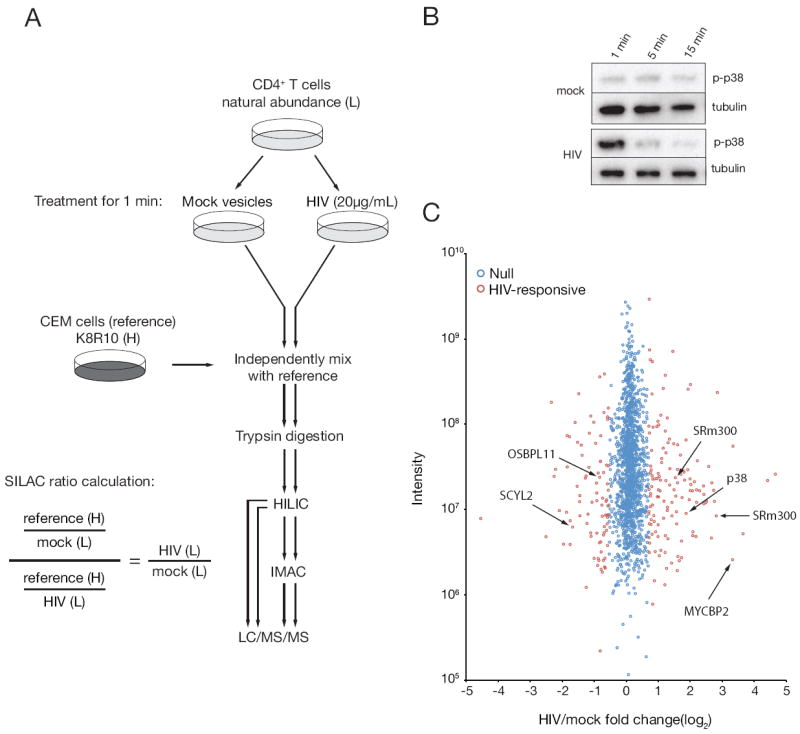
(A) Phosphoproteomics workflow for T cell stimulations, processing, and fractionation. Virus and mock-treated samples were independently processed following mixing with a common reference lysate. HIV/mock phosphorylation site fold-changes were calculated by dividing the ratio of reference/mock by reference/HIV phosphorylation site fold-changes. Abbreviations: light (L), heavy (H), hydrophilicity interaction liquid chromatography (HILIC), immobilized metal affinity chromatography (IMAC), liquid chromatography tandem mass spectrometer (LC/MS/MS). (B) Primary CD4+T cells were incubated with purified HIV-1 or a mock vesicle preparation for various lengths of time at 37°C and probed for phosphorylated p38 (pT180/pY182) by immunoblot to determine optimal kinetics for large scale phosphoproteomics stimulations. (C) Distribution of phosphorylation site fold-changes. HIV/mock phosphorylation site fold-changes are shown according to the mixture model designation of HIV-responsive (red) or HIV-nonresponsive, i.e. null (blue). For clarity, only a subset of SRm300-responsive phosphorylation sites is indicated. See also Figure S1.
To quantify relative fold-changes of phosphorylation sites with MS, we used stable isotope labeling of amino acids in cell culture (SILAC). Since unstimulated T cells do not have sufficient metabolic activity for complete isotope labeling, we labeled the human CD4+ T cell line CEM with media containing 13C and 15N arginine and lysine so as to construct a ‘heavy’ isotope reference proteome (Geiger et al., 2011). Lysates from mock or virus-treated T cells were then independently mixed with this common reference lysate, making it possible to measure the fold-change in abundance of any phosphopeptide that was identified in the mock, virus-treated, and reference proteomes (Figure 1A). Importantly, the distribution of SILAC ratios for each reference mixture was unimodal and over 90% of ratios were 7.2-fold or less, suggesting that the reference phosphoproteome sufficiently represented the primary cell phosphoproteome (Figure S1). Following several filtering steps, 1,757 phosphorylation sites (from 799 genes) were quantified with high stringency in the mock-reference and HIV-reference samples (Figure S1, Table S1-3). At a 1% false discovery rate threshold, a total of 239 phosphorylation sites (from 175 genes) were deemed HIV-responsive, with 144 increasing and 95 decreasing in abundance upon HIV-1 treatment (Figure 1C, Table S3-4).
HIV-induced phosphorylation of the transcription factor ETS
We took several approaches to validate the SILAC phosphorylation site ratios, with the most direct being to use phosphospecific antibodies. Unfortunately, only two antibodies against the identified HIV-responsive phosphorylation sites, p38 (pT180/pY182) and ETS-1 (pS282/pS285), worked sufficiently well by immunoblot to make this analysis possible. We used each to assess changes in their respective phoshosites by immunoblot following addition of purified HIV-1 to CD4+ T cells. Enhanced p38 phophorylation (pT180/pY182) upon addition of HIV-1 for one minute was confirmed (Figure 2A), as was HIV-induced phosphorylation of ETS-1 (pS282/pS285) (Figure 2A). We also confirmed that the kinase ERK1/2 was not phosphorylated in an HIV-dependent manner, though it was responsive to CXCL12.
Figure 2. Immunoblot and bioinformatic validation of HIV-responsive phosphorylation site ratios.
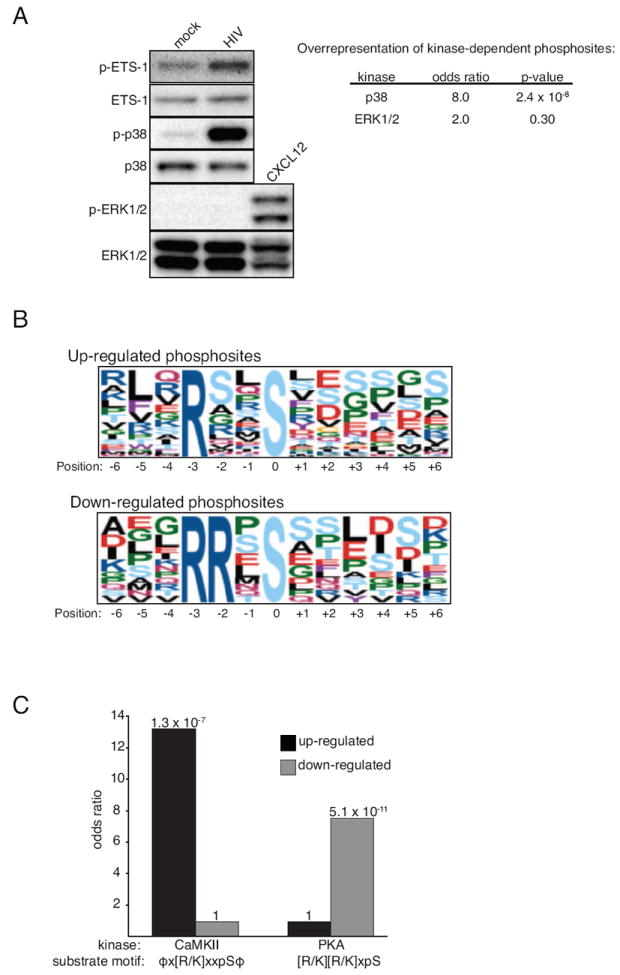
Primary CD4+ T cells were incubated with purified HIV-1 or a mock vesicle preparation and probed for phosphorylated p38 (pT180/pY182), ETS-1 (pS282/pS285), and ERK1/2 (pT202/pY204) by immunoblot. The chemokine CXCL12 (SDF-1) served as a positive control for pERK1/2. Odds ratio and p-values calculated with Fisher’s Exact with Benjamini and Hochberg corrections. (B) Putative kinase substrate motifs were searched for within up-, or down-regulated HIV-responsive phosphorylation. The amino acid position weighted matrix, centered on phosphorylated serine (position 0), is shown for each top-scoring motif. (C) Overrepresentation of the CaMKII and PKA motif within HIV up- and down-regulated phosphorylation sites was calculated with the Fisher’s Exact test with each p-value indicated above each bar. ϕ = L/V/I/M/Y/F, pS = phosphoserine. See also Table S4.
Phosphoproteome-wide validation of HIV-induced MAPK activity
To further validate the HIV-responsive phosphorylation site ratios, we took advantage of the fact that differential phosphorylation is in part regulated by changes in the catalytic activity of upstream kinases. Since we observed robust phosphorylation of p38 (pT180/pY182) but not of ERK1/2 (pT202/pY204) upon addition of HIV-1 to CD4+ T cells (Figure 2A), we hypothesized that HIV-responsive phosphorylation sites should contain an overrepresentation of p38 but not ERK1/2-dependent phosphorylation sites. To examine this, we took advantage of p38 and ERK1/2-responsive phosphorylation sites as defined by Pan et al (Pan et al., 2009). We then measured the overrepresentation of these p38 or ERK1/2-dependent phosphorylation sites within HIV-responsive phosphorylation sites with the Fisher’s exact test. Only p38-dependent phosphorylation sites were significantly overrepresented among HIV-responsive phosphorylation sites (OR: 6.7, p = 2.0 × 10-6 vs. OR: 1.5, p = 0.52) (Figure 2A, Table S4), supporting the fact that both by immunoblot and SILAC analysis the addition of HIV-1 to CD4+ T cells leads to rapid phosphorylation of p38 phosphorylation sites that are known to enhance its kinase activity.
Identification of overrepresented kinase substrate motifs
To determine if HIV-1 activated additional kinases, we took advantage of the fact that the phosphorylation of many kinase substrates is highly influenced by the amino acids that immediately surround the phosphorylation site. Therefore, we surveyed the amino acid residues surrounding each HIV-responsive phosphorylation site using the Group-based Prediction System (GPS) (Xue et al., 2011) and motif-x tool (Schwartz and Gygi, 2005). The GPS uses a hierarchical algorithm to rank the likelihood that a particular kinase or kinase family phosphorylates a given phosphorylation site while motif-x measures the overrepresentation of amino acid sequence patterns, providing an unbiased list of potential kinase substrate motifs. With the GPS tool, substrates for MAPKAPK were significantly overrepresented according to the Fisher’s Exact test (OR: 7.3 p = 6.1 × 10-6) (Table S4). Members of the MAPKAPK family are known substrates for p38 (Cargnello and Roux, 2011), which is rapidly activated upon HIV-1 binding to CD4+ T cells (Figure 1B).
The motif-x tool revealed that the [R/K]xxS motif, which displayed a bias towards hydrophobic amino acids at the P-5 and P+1 positions (ϕx[R/K]xxSϕ), was significantly overrepresented among upregulated HIV-responsive phosphorylation sites (OR: 13.2, 1.1 × 10-7) (Figure 2B-C, Table S4). This motif is one of many that are targets for calmodulin dependent kinase II (CaMKII) (Schwartz and Gygi, 2005). CaMKII activity is regulated by fluxes in free Ca2+ levels and HIV-1 gp120 induces a transient Ca2+ flux in primary CD4+ T cells (Weissman et al., 1997). These data, combined with the fact that the HIV-responsive phosphorylation site ETS-1 (pS282/pS285) is phosphorylated by CaMKII (Fisher et al., 1994) supports an increase in CaMKII activity during HIV-1 entry. For the down-regulated HIV-responsive phosphorylation site motifs, RRxS matches the consensus motif for protein kinase A (PKA), [R/K][R/K]xS (OR: 7.5, p = 5.1 × 10-11) (Figure 2B-C, Table S4). PKA kinase activity is regulated by intracellular levels of the second messenger cyclic AMP (cAMP) (Mosenden and Tasken, 2011). The observed decrease in phosphorylated PKA substrates is supported by the observation that gp120 treatment of primary CD4+ T cells decreases cAMP levels within minutes and that cAMP is a negative regulator of HIV-1 replication in primary cells (Navarro et al., 1998).
Cellular pathways
To gauge which cellular pathways HIV-1 may activate during entry, we tested whether HIV-responsive phosphoproteins contained an overrepresentation of gene ontology or cellular pathway terms from manually curated databases. The most notable ontologies and pathways involved various aspects of GTPase biology, actin and T cell activation (Table S5).
Strengthening the association with T cell activation, upregulated T cell receptor (TCR)-responsive phosphorylation sites from both Jurkat (Mayya et al., 2009) (OR: 19.2, p = 0.039) and primary CD4+ T cells (Ruperez et al., 2012) (OR: 4.3, p = 8.7 × 10-5) were overrepresented among HIV-responsive phosphorylation sites (Table S4). We also generated an interaction network using the STRING database (Figure 3A) and found a striking segregation between cytoplasmic and nuclear localized genes and discreet functional modules of genes that regulate microtubules, actin and T cell activation – all processes that have been clearly linked to HIV-1 biology (Figure 3A) (Liu et al., 2009; Stevenson et al., 1990). The fact that the majority of genes that regulate T cell activation also regulate actin further strengthens the association between these processes and HIV-induced signaling (Liu et al., 2009).
Figure 3. Bioinformatic analysis of genes from HIV-responsive phosphorylation sites.
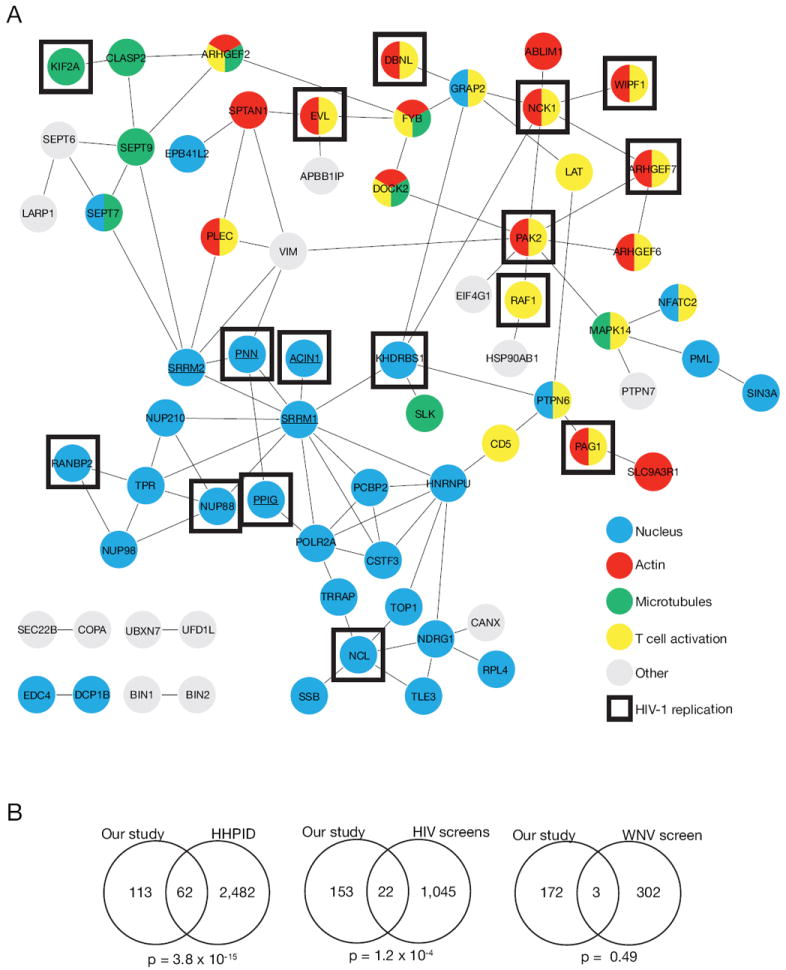
(A) Interaction network of HIV-responsive phosphoproteins. Functional or physical interactions (see methods) between genes from HIV-responsive phosphopsites were extracted from the STRING database (version 9.0). Underlined genes indicate SR-proteins. Genes indicated as ‘HIV-1 replication’ were taken from Table 1. (B) HIV-1 host factors are overrepresented within HIV-responsive phosphoproteins. Overlaps of genes from HIV-responsive phosphoproteins and genes from the HIV-1 human protein interaction database (HHPID), HIV-1 RNAi screens, and West Nile Virus (WNV) siRNA screen were calculated with the hypergeometric test using the human genome as background (n=20,402). See also Table S6.
Since our hypothesis is that HIV-responsive phosphoproteins are important for HIV-1 infection and replication, we measured whether there was an overrepresentation of previously defined HIV-1 host factors within our dataset. The HIV-1 human protein interaction database (HHPID) is a manually curated list of both physical and functional interactions between HIV-1 and human proteins (Fu et al., 2009). The overlap with the HHPID dataset was highly significant (hypergeometric test, p = 3.8 × 10-15) (Figure 3B, Table S6). In addition, the overlap with genes from the four published HIV-1 RNAi screens (Brass et al., 2008; Konig et al., 2008; Krishnan et al., 2008; Yeung et al., 2009) was also significant (hypergeometric test, p = 1.3 × 10-4), but not an siRNA screen conducted with West Nile Virus (Krishnan et al., 2008) (p = 0.49) (Figure 3B, Table S6). These overlaps, combined with literature mining, resulted in a total of 83/175 genes from HIV-responsive phosphorylation sites having been previously described as HIV-1 host factors (Table S6), supporting our hypothesis that HIV-responsive phosphoproteins are involved in HIV-1 infection.
Role of HIV-responsive phosphoproteins during HIV-1 infection
To examine whether HIV-responsive phosphoproteins regulate HIV-1 infection, we depleted 69 of the 175 genes from HIV-responsive phosphosites using siRNAs (four per gene) in MAGI cells and measured infection levels of three full-length infectious molecular clones (IMCs): NL4.3, 89.6, and SF162 (Table S7-8). We deemed a gene important for HIV-1 infection if at least three out of four siRNAs decreased MAGI cell β-galactosidase reporter activity by 2-fold (compared to a non-targeting siRNA) in at least one IMC, 37 genes of which satisfied this criteria (Table 1). While roughly half of these genes have been previously linked to HIV-1 biology in some way, 27 have not yet been functionally linked to HIV-1 to our knowledge (Table 1, Table S8). Cellular pathways such as actin, microtubules, T cell activation, and various nuclear processes such as splicing were heavily represented, further linking known aspects of HIV-1 signaling and our phosphoproteomics dataset (Figure 3A, Table 1).
Table 1. HIV-responsive phosphoproteins that impact HIV-1 replication in MAGI cells.
A subset (n=69) of HIV-responsive phosphoproteins were depleted in MAGI cells then infected with replication-competent HIV-1. Genes whose depletion led to at least a 2-fold decrease in infection with three out of four siRNAs tested are shown.
| Gene symbol | Cellular pathway | Previously linked to HIV? |
|---|---|---|
| ACIN1 | Apoptosis | Yes |
| AHNAK | T cell activation | |
| ANXA2 | Actin | Yes (+) |
| ARHGEF18 | Actin | |
| ARHGEF7 | Actin | Yes |
| BUD13 | Splicing | |
| CRTC3 | Cyclic AMP | Yes (+) |
| DBN1 | Actin | Yes (+) |
| DBNL | Actin | |
| DIDO1 | Apoptosis | |
| DNM1L | Apoptosis, T cell activation | Yes |
| DOCK11 | Actin | |
| EVL | Actin, T cell activation | Yes |
| FKBP15 | Actin | |
| GORASP2 | Vesicle trafficking | Yes (+) |
| KHDRBS1 | Splicing, NF-kB | Yes (+) |
| KIAA1967 | Chromatin | Yes |
| KIF2A | Microtubules | |
| LSM14A | Splicing | |
| MAP4 | Microtubules | Yes (+) |
| MTDH | NF-kB | Yes |
| NCK1 | Actin, T cell activation | Yes |
| NCL | Ribosome synthesis | Yes (+) |
| NUP88 | Nuclear transport | Yes |
| PAG1 | T cell activation | |
| PAK2 | Actin | Yes |
| PNN | Splicing | Yes |
| PPIG | Transcription | |
| RAF1 | NF-kB | Yes (+) |
| RANBP2 | HIV integration site selection | Yes (+) |
| RAPGEF6 | NF-kB | Yes (+) |
| REPS1 | Vesicle trafficking | |
| RNF31 | Transcription | |
| SH3BP1 | Actin | |
| TBC1D10B | Vesicle trafficking | |
| TBC1D5 | Vesicle trafficking | |
| WIPF1 | Actin |
(+) indicates reported functional link between HIV-1 replication and gene. See also Table S8.
To address whether HIV-responsive phosphoproteins are important for HIV-1 infection, we selected four HIV-responsive phosphoproteins, MYCPB2, OSBPL11, SCYL2, and SRRM2 on the basis that an identical phosphorylation site was both HIV- and CXCL12-reponsive (Wojcechowskyj et al., 2011). CXCL12 is the chemokine ligand for CXCR4, which is the receptor used by the HIV-1 strain in our study. We depleted each protein with four individual siRNAs per gene in MAGI cells (Figure 4B) and with pools of siRNAs in Jurkat cells (Figure 4C). We then infected depleted cells with a single cycle HIV-1 reporter virus bearing either an X4 -or R5-tropic Env or the glycoprotein of vesicular stomatitis virus (VSV-G). As expected, treatment with CD4-specific siRNAs inhibited infection by HIV-1 but not VSV, the siRNAs to CXCR4 only inhibited infection by the X4 virus strain and the siRNAs to IGSF3 (an irrelevant gene for HIV-1 infection) had no effect on any virus (Figure 4A). Among the experimental samples, only suppression of SRRM2 consistently enhanced HIV-1 reporter gene expression in both MAGI and Jurkat cells (Figure 4A, D). We also measured the fraction of virus infected MAGI cells by intracellular p24 staining of NL4.3 and 89.6 IMC or GFP expression and found that suppression of SRRM2 had no consistent effect on the permissivity of cells to HIV-1 infection (Figure 4E). Therefore, we conclude that suppression of SRRM2 results in higher levels of HIV-1 gene expression per infected cell and thus focused on the possible role of SRRM2 in HIV-1 infection in subsequent experiments.
Figure 4. RNAi-mediated knockdown of HIV-responsive phosphoproteins.
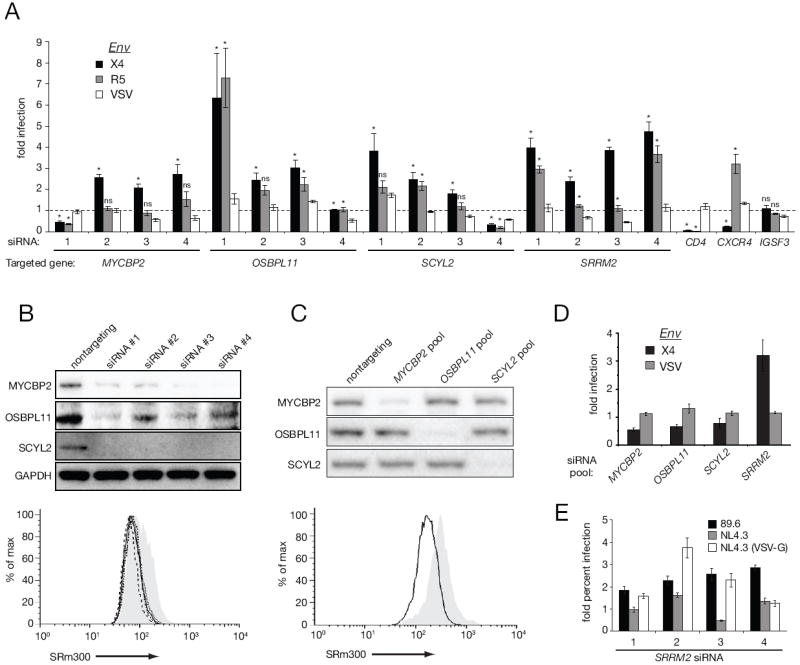
(A) MAGI cells were transfected with individual siRNAs then infected with NL4.3 pseudovirions with an X4-tropic Env, R5-tropic Env, or VSV-G. Infection values (luciferase activity) were normalized to a non-targeting control siRNA. (B) Lysates from depleted MAGI cells were analyzed by immunoblot (top panel). SRm300 protein levels were monitored by FACS (bottom panel): grey area (non-targeting control), solid line (siRNA#1), dotted line (siRNA#2), short dashed line (siRNA#3), long dashed line (siRNA#4). (C) Jurkat cells were nucleofected with indicated pools of siRNA and analyzed by immunoblot (top panel). SRm300 protein levels were monitored by FACS (bottom panel): grey area (non-targeting control), solid line (SRm300 pool). (D) Nucleofected jurkat cells were infected as in (A). Bars=SEM, n=3. (E) Relative percent infection of SRm300-depleted MAGI cells with replication competent 89.6, NL4.3, and GFP-expressing VSV-G pseudovirions. Bars=SEM, n=4, *p<0.05 Mann-Whitney, ns: not significant (p>0.05).
Regulation of HIV-1 alternative splicing and infection by SRRM2
SRRM2 encodes SRm300, an SR protein that contains two characteristic arginine-serine (RS) domains (Figure S2). SR proteins are central regulators of cellular splicing whose activity can be modulated by phosphorylation of RS-domains (Long and Caceres, 2009). Of the 37 unique phosphorylation site ratios in SRm300 we were able to quantify, nine were HIV-responsive (Figure S2, Table S3). During HIV-1 infection, dozens of fully and partially spliced viral mRNAs are generated through a complex interplay between viral and cellular elements, of which many of the latter are cellular SR proteins (Stoltzfus, 2009). To address whether SRRM2 regulates HIV-1 alternative splicing, we specifically amplified fully spliced transcripts from mRNA isolated from SRRM2-depleted MAGI cells that were infected with LAI or VSV-G pseudovirions or with the primary IMC 89.6 (Figure 5A). A representative gel is shown for 89.6 with the corresponding exon composition of each transcript. With all viruses, depletion of SRRM2 led to an approximately 2-fold increase in the ratio of tat-2/tat-1 and nef-3/nef-2 transcripts (Figure 5B). The ratio of tat-3/tat-1 and nef-4/nef-2 also increased with 89.6, yet we were not able to clearly resolve tat-3 and nef-4 with the pseudovirions. We conclude that SRRM2 regulates the inclusion of exons 2 and 3, perhaps by modulating the strength of the splice acceptors A1 and A2. Unexpectedly, LAI pseudovirions had on average 1.8 times less nef-3 relative to nef-2 than VSV-G pseudovirions, regardless of SRm300 levels (Figure 5B), suggesting that signals induced by HIV-1 Env may themselves influence alternative splicing.
Figure 5. SRRM2 regulates alternative splicing of HIV-1.
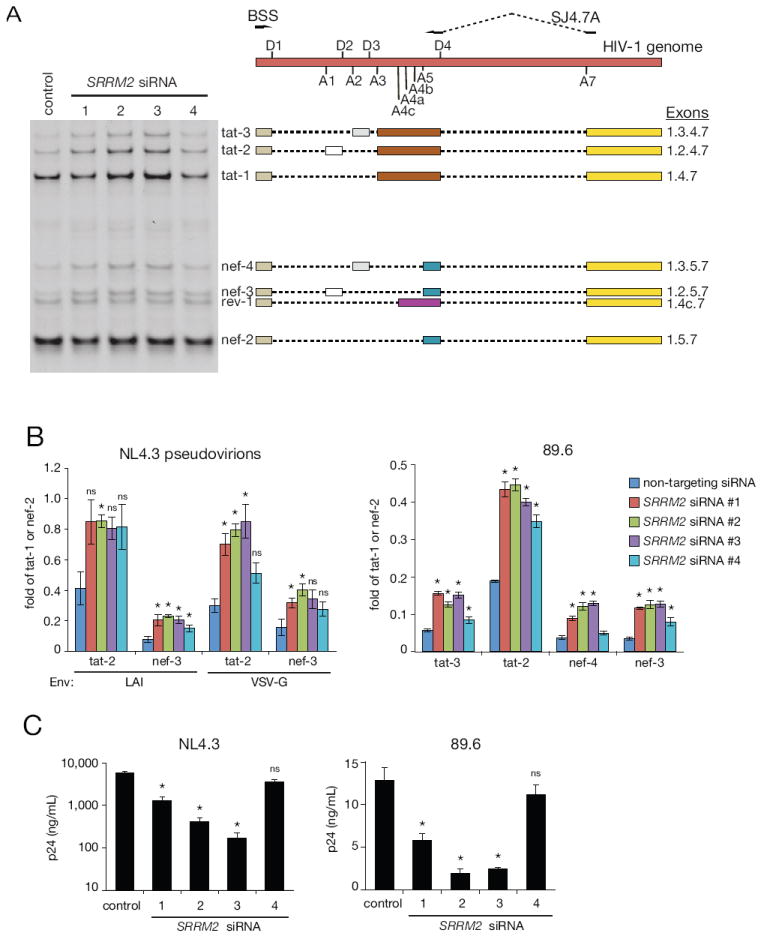
(A) RT-PCR analysis of HIV-1 transcripts. Exon composition of sequence-confirmed transcripts are indicated to the right of a representative 89.6 DNA gel, positioned with respect to the HIV-1 genome above. D1-4: 5’ splice sites. A1-7: 3’splice sites. (B) Relative quantification of HIV-1 transcripts. Band intensities of corresponding RT-PCR products of tat and nef alternatively spliced products were divided by tat-1 and nef-2, respectively. Comparisons were made between each SRRM2 siRNA and the non-targeting control siRNA. (C) HIV-1 release following depletion of SRRM2 as measured by p24 collected on day 5 post infection. Comparisons were made between each SRRM2 siRNA and the non-targeting control siRNA. Bars=SEM, n=4, *p<0.05 Mann-Whitney, ns: not significant (p>0.05). See also Figure S2.
The balance of HIV-1 splicing products is finely tuned and highly dependent on an overlapping, interconnected network of diverse of cis and trans factors which in turn can affect HIV-1 replication (Stoltzfus, 2009). Consistent with reports that increased splicing at A1 and A2 leads to decreased virus production (Jacquenet et al., 2005), we observed up to a 6- and 30-fold reduction in p24 release upon infection with 89.6 and NL4.3 respectively, in three out of four siRNAs targeting SRRM2 (Figure 5C). We also tested four additional siRNAs against SRRM2 and observed the same increase in tat and nef alternatively spliced transcripts and impact on virion release (Figure S4). When the fold change magnitudes of tat and nef transcripts were correlated with p24 release for all siRNAs tested (n=8), the correlation with nef-4 was statistically significant (p = 0.022, Spearman) (Table S9), suggesting that SRRM2-dependent dysregulation of nef-4 transcript abundance is most tightly linked to productive HIV-1 infection.
Regulation of HIV-1 alternative splicing and infection by HIV-responsive SR-proteins
Five other SR proteins, PNN, PPIG, TRA2A, ACIN1, and SRRM1, were also HIV-responsive phosphoproteins, leading us to hypothesize that they too are important for HIV-1 splicing. We depleted each with pools of siRNAs in MAGI cells and measured fully spliced HIV-1 transcripts (Figure 6A). Depletion of all SR proteins except PPIG led to a unique HIV-1 splicing pattern. Depletion of PNN, led to increases in all exon 2 and 3-containing tat transcripts, yet only decreased levels of nef-4, suggesting that PNN regulates the inclusion of exons 2 and 3 only in the context of tat transcription (Figure 6B). A similar pattern with tat transcripts was seen with TRA2A (Figure 6B). Depletion of ACIN1 led to a dramatic increase in tat-4 and tat-3 transcripts and a corresponding decrease in all nef transcripts, making quantification of nef transcripts difficult (Figure 6B). Depletion of SRRM1, a binding partner of SRRM2, led to increases in tat-2 and nef-3, but not tat-3 and nef-4 (Figure 6B). This suggests that SRRM1 regulates the activity of site A1, but not A2 in both tat and nef transcripts, and that SRRM1 and SRRM2 may modulate HIV-1 splicing in both overlapping and distinct protein complexes.
Figure 6. HIV-responsive SR-proteins regulate HIV-1 splicing and virion release.

(A) Lysates from depleted MAGI cells were analyzed by immunoblot with each antibody. (B) Relative quantification of HIV-1 transcripts. Band intensities of tat and nef alternatively spliced products (top panel) were divided by tat-1 and nef-2, respectively for quantification (bottom panel). n=6 (C) HIV-1 release following depletion of SR proteins as measured by p24 collected on day 5 post infection. Comparisons were made between each SRRM2 siRNA and the non-targeting control siRNA. Bars=SEM, n=4, *p<0.05, **p<0.01 Mann-Whitney, ns: not significant (p>0.05). See also Figure S3.
DISCUSSION
Viral infection is intimately tied to interactions with host proteins and pathways that facilitate virtually every step of the virus life cycle. By binding to and cross-linking cell surface molecules, viruses can induce intracellular signaling cascades that can enhance the permissiveness of the host cell to. The receptors used by HIV-1 to enter cells are well known signaling molecules and important immune modulators. While binding of Env to these receptors results in downstream phosphorylation events and the modulation of various intracellular second messengers, the breadth of HIV-induced signaling cascades, their temporal relationship to virus infection and the roles that these signaling events play in specific aspects of virus infection are far from well characterized.
We employed unstimulated CD4+ T cells as these most closely reflect the major cell type encountered by HIV-1 in vivo, and because a growing body of evidence indicates that virus-induced signaling events play a significant role in virus infection of primary T cells. We used an X4-tropic strain of HIV-1 in part due to the fact that virus engagement of CXCR4 induces signals that lead to cytoskeleton reorganization in primary T cells and more efficient virus infection (Balabanian et al., 2004; Barrero-Villar et al., 2009; Jimenez-Baranda et al., 2007; Yoder et al., 2008). Thus, at a minimum, we anticipated finding changes in the phosphorylation status of proteins linked to cytoskeletal rearrangements upon binding of HIV-1 to host cells. Our findings not only supported this supposition, but also showed that HIV-receptor interactions rapidly modulate phosphorylation of numerous host cell proteins with the potential to impact multiple steps of viral replication. Of 1,757 quantified phosphorylation sites, 14% proved to be HIV-responsive. While some fraction of these HIV-responsive phosphorylation sites may prove to be false-positives, our analysis only detected a small fraction of total cellular phosphorylation sites and we only examined a single, early time-point. Thus, it is likely that binding of HIV-1 to its cell surface receptors leads to altered phosphorylation of at least hundreds of host proteins.
Several complementary lines of evidence including western blots, analysis of experimentally and computationally derived kinase substrates, and bioinformatics of cellular gene ontologies support the validity of the MS-derived phosphorylation site ratios described here. Fluxes of intracellular calcium were one of the first and most consistently observed examples of HIV-induced cellular signaling (Kornfeld et al., 1988; Liu et al., 2000; Weissman et al., 1997). The calmodulin-dependent kinase (CaMK) family is activated by calcium and we detected a strong overrepresentation of CaMKII substrates among upregulated HIV-responsive phosphorylation sites along with phosphorylation of the transcription factor ETS-1 (pS282/pS285), a well characterized substrate of CaMKII that has been linked to HIV-1 gene expression (Sieweke et al., 1998; Soderling, 1999; Yang et al., 2009). In addition, overexpression of a splice variant of ETS-1 lacking exon VII reactivated latent HIV-1 (Yang et al., 2009). Interestingly, exon VII contains serines 282 and 285 and the ETS-1 isoform lacking this exon binds to DNA more strongly than WT ETS-1 independently of CaMKII activity (Fisher et al., 1994).
In a previous study, we characterized the CXCL12-responsive phosphoproteome in a T cell line (Wojcechowskyj et al., 2011) and the overlap of significantly changed phosphorylation sites between these datasets was statistically significant (OR: 3.1, p = 0.039) (Table S4). However, based on the unique nature of each study and the modest overlap of quantified phosphorylation sites (~50%), it is difficult to determine which of the HIV-responsive sites were dependent on CXCR4 signaling. To increase the likelihood of capturing relevant signaling events we focused on proteins that contained both HIV- and CXCL12-responsive phosphorylation sites. While suppression of only SRRM2 showed a consistent phenotype on virus replication in both MAGI and Jurkat cells, MYCBP2, OSBPL11 and SCYL2 did show effects with some siRNAs. The most consistent of these was with MYCBP2, a negative regulator of cAMP (Scholich et al., 2001). Interestingly, our data support the observations of others that the role of cAMP during HIV-1 replication is cell type dependent. In HeLa cells, components of PKA signaling enhance HIV-1 replication (Konig et al., 2008; Lemay et al., 2008; Zhou et al., 2008), while the opposite is true in Jurkat and primary T cells (Navarro et al., 1998). Finally, a recent study showed that SCYL2 limits the release of HIV-1 from cells – something that we did not measure in our assays (Miyakawa et al., 2012).
HIV-1 can disseminate among T cells either through cell-free or cell-cell routes and this data set may help elucidate components of cell-cell transmission of HIV-1. The contact of infected and uninfected CD4 T cells form structures that are termed ‘virologicalsynapses’ (VS) that resemble immunological synapses (IS) (Haller and Fackler, 2008). We found several HIV-responsive phosphoproteins that are known to participate in either the VS or IS such as LAT, FYB (ADAP), GRAB2 (Gads), NCK1, PTPN6 (SHIP-1). The HIV-responsive phosphoproteins APBB1IP (RIAM), RAPGEF6 (rap1 GEF) and SIPA1 (rap1 GAP) are also components of integrin ‘inside-out’ signaling, a feature of both the VS and IS (Hioe et al., 2011).
Our application of quantitative MS-based phosphoproteomics made it possible to explore a heretofore unexplored possibility: that HIV-1 may induce cellular signals that not only enhance virus entry, but that render the cell more permissive to later steps in the viral replication cycle as well. Six SR proteins and five other genes that can regulate cellular splicing were among the 175 phosphoproteins that were HIV-responsive, raising the possibility that HIV-induced signaling during entry may influence the splicing of incoming viral transcripts. Indeed, signaling events can regulate the localization, intrinsic splicing activity, protein-protein, or protein-RNA interactions of various splicing factors. Tools to firmly establish this link are crude, yet this hypothesis is supported by the fact that five SR-proteins and three others were important for HIV-1 infection and that altered ratios of nef transcripts correlated with p24 release levels. It is also possible that HIV-dependent regulation of these SR proteins is indirect, i.e. that they may affect the alternative splicing of additional HIV-1 host factors. In fact, it is well known that cellular alternative splicing is altered during T cell activation (Martinez et al., 2012). Further complicating matters, SR-proteins are heavily phosphorylated – over 600 unique phosphorylation sites have been catalogued for SRm300 alone making the task of assigning roles for each phosphorylation site a daunting task.
At first glance, the kinetic and spatial disconnect between the plasma membrane and the nucleus in regulating downstream stages of the HIV-1 lifecycle may seem unlikely. However, phosphorylation of cellular proteins during HIV-1 entry can promote viral gene expression from the provirus (Gringhuis et al., 2010) and EGF induces the phosphorylation of 62 nuclear phosphorylation sites in HeLa cells with one minute (Olsen et al., 2006). In addition, the transcription factors NF-kB (Briant et al., 1996), NFAT (Cicala et al., 2006) and AP-1 (Briant et al., 1996; Chirmule et al., 1995), all of which bind to the HIV-1 long terminal repeat and facilitate transcription, localize into the nucleus upon HIV-1 binding to the cell surface.
The screening approach taken here suggests that the interplay between HIV-1 and its receptors results in a much broader array of signaling events than has been suspected, and that by these interactions the virus can modulate the host cell environment in ways that impact virus replication at multiple steps, including splicing of viral transcripts. Examining the status of SRm300 phosphorylation as well as that of other splicing factors and phosphoproteins identified in this study over the course of virus infection in the presence or absence of agents that inhibit virus-receptor signaling should more fully reveal how the virus can reprogram cells so as to make them optimal hosts.
EXPERIMENTAL PROCEDURES
Cell lines and viruses
Jurkat E6-1 cells (ATCC) were propagated in RPMI supplemented with 10% (v/v) FBS. MAGI cells, which are a clone of HeLa cells stably expressing CD4, CCR5, and β-galactosidase under the control of the HIV-1 LTR, were grown in DMEM supplemented with 10% (v/v) FBS. CEM cells were grown in DMEM medium (AthenaES) with 10% dialyzed fetal calf serum and lysine and arginine were replaced by [13C6, 15N2]-lysine and [13C6, 15N4]-arginine. Protein isolates from these cells served as the stable isotope labeled reference material for the SILAC quantitative analyses. HIV-1 strain MN was used for primary cell stimulations and was grown in SupT1 cells, purified and concentrated with sucrose gradients, and inactivated with aldrithiol-2 (AT-2) (Chertova et al., 2003; Rossio et al., 1998). A mock prep was produced from uninfected cells using the same procedures. Replication incompetent HIV-1 pseudovirions (pNL43-ΔEnv-vpr+-luc+) were generated as previously described (Parrish et al., 2012). HIV-1 Env strains used for pseudovirion generation were the lab-adapted X4 strain LAI and the Clade B transmitted/founded R5 strain REJO.D12. Replication competent 89.6 and NL4.3 strains of HIV-1 were grown in CEMx174 cells and SupT1 cells, respectively.
Primary human CD4+ T cell stimulations
Primary human CD4+ T cells from healthy donors were obtained from the Center for AIDS Research Human Immunology Core at the University of Pennsylvania and purified by negative selection against HLA-DR, CD21, CD16, CD11b, CD14 and CD8. Cells rested overnight in 10% FBS containing RPMI at 37°C and were serum-starved for 90 min before stimulation. AT-2-treated HIV-1 preparations or mock vesicles (volume matched) were added to 4.5 × 108 cells (5 × 106 cells/mL) to achieve a final concentration of 20 μg/mL p24. AT-2-treated HIV-1 This concentration of p24 corresponds to approximately 15nM gp120 and an MOI of 100, a concentration that is commonly used for biochemical measurements of virus-induced signaling events. After 1 min incubation at 37°C, two volumes of ice cold PBS containing phosphatase inhibitors (Sigma) were added to each culture of cells then spun at 450 × g at 4°C. Supernatant was aspirated and cell pellets frozen in liquid nitrogen.
Mass Spectrometry and data processing
Details of sample preparation and enrichment, mass spectrometry, sequence database searching, mixture model calculations and statistical analyses are included as Supplementary Methods.
Immunoblot analysis
Cell pellets were lysed in 1% Triton X-100 with phosphatase (Sigma P5726, P0044) and protease inhibitors (Roche 1836170) for 5 min on ice, then centrifuged at 20,000 × g for 10 min. Samples were denatured and reduced, incubated at 70°C for 10 min then run on 10% Bis-Tris gels. Gels were transferred to PVDF membranes and blocked for 30 min in evaporated milk. Blots were incubated at 4°C overnight with a 1:1,000 dilution of primary antibodies. Antibodies against ERK1/2 (pT202/pY204), ERK1/2, α-tubulin, p38 (pT180/pY182), p38, and PPIG were obtained from Cell Signaling Technology, ETS-1 (pS282/pS285) and ETS-1 from Invitrogen, MYCPB2, SCYL2, PNN, and ACIN1 from Abcam, OSBPL11 and TRA2A from Sigma, GAPDH from Calbiochem, SRRM1 from Proteintech, and SRm300 (B4A11) was a generous gift from Jeff Nickerson.
RNA interference
Jurkat cells were transfected with an Amaxa nucleofector (Lonza) as follows: 2 × 106 cells were resuspended in 100 μL solution V containing 100 μM siRNA, electroporated with program X-001, then incubated in 4 mL of complete RPMI for 48 hr before harvesting for immunoblot or infection with HIV-1. MAGI cells were transfected with Lipofectamine RNAiMax reagent (Invitrogen). 2.5 × 103 cells in a 96-well plate were reverse transfected with 50 nM (final) siRNA according to the manufacturer’s instructions. Cells were then harvested for immunoblot or infected with HIV-1 72 hr later. siRNA sequences are listed in Table S7 and S9.
HIV-1 infections
Approximately 5 × 105 nucleofected Jurkat cells were spinoculated with pseudovirions at 1,200 × g for 2 hr at RT. Cells were harvested for firefly luciferase expression 48 hours post infection (hpi). For infection of MAGI cells for RT-PCR splicing analysis, 100 ng p24 of either NL4.3 or 89.6 was added per 1 × 106 cells, rinsed following a 2 hr incubation at 37°C, then harvested for FACS or RNA 48 hpi. For β-galactosidase-based infections of MAGI cells, siRNA-treated cells in 96 wells were infected with 2.5 ng p24/well of replication competent IMC of NL4.3, 89.6 and SF162 and β -galactosidase activity measured 48h later. For measurement of percent infected cells, infected MAGI cells were stained for intracellular p24 and analyzed by FACS. For p24 release, siRNA treated MAGI cells were infected with NL4.3 and 89.6 as above, and media replaced and collected 5 days later. Supernatant p24 was measured using alphaLISA (Perkin Elmer) in 384 well plates.
Supplementary Material
HIGHLIGHTS.
A quantitative phosphoproteomics screen of HIV-1 infected primary human CD4+ T cells
Abundance of 239 phosphorylation sites from 175 genes changed after HIV-1 exposure
Previously uncharacterized HIV-1 host factors, including SR-proteins, confirmed by RNAi
SRm300 and other SR proteins regulate alternative splicing of HIV-1 transcripts
Acknowledgments
We especially thank Jim Riley and the Penn Immunology Core for human primary CD4+ T cells, Lynn Spruce and the Children’s Hospital of Philadelphia Research Institute Protein and Proteomics Core for expert technical assistance, Julian Bess and Jeffrey Lifson for AT-2 treated HIV-1 virions, and Jeffrey Nickerson for the anti-SRm300 antibody. We’d also like to thank Matt Pace, Rebecca Custers-Allen, Karen Ocwieja, Gary Koretzky, Vijaya Ramesh, Vesa Olkkonen, Bruce Freedman, Alexander Babich, Janis Burkhardt, James Murray, Robert Siliciano, Erika Holzbaur, Jim Alwine, Farida Shaheen, and Steven Bryan for various reagents and technical assistance and Logan Everett and Craig Wilen for helpful discussions.
These experiments utilized regents provided by the AIDS and Cancer Virus Program, SAIC Frederick, Inc./National Cancer Institute, Frederick, under contract HHSN261200800001E and by the Penn Center for AIDS Research (P30 AI 045008). JAW was supported by T32-AI007632 and and R01 AI40880
Conceived and designed the experiments: JAW SHS RWD. Performed the experiments: JAW CAD JYL NFP. Analyzed the data: JAW STJ SHS RWD. Contributed reagents/analysis tools: NFP RS BHH FDB STJ SHS. Wrote the paper: JAW STJ SHS RWD
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balabanian K, Harriague J, Decrion C, Lagane B, Shorte S, Baleux F, Virelizier JL, Arenzana-Seisdedos F, Chakrabarti LA. CXCR4-tropic HIV-1 envelope glycoprotein functions as a viral chemokine in unstimulated primary CD4+ T lymphocytes. J Immunol. 2004;173:7150–7160. doi: 10.4049/jimmunol.173.12.7150. [DOI] [PubMed] [Google Scholar]
- Barrero-Villar M, Cabrero JR, Gordon-Alonso M, Barroso-Gonzalez J, Alvarez-Losada S, Munoz-Fernandez MA, Sanchez-Madrid F, Valenzuela-Fernandez A. Moesin is required for HIV-1-induced CD4-CXCR4 interaction, F-actin redistribution, membrane fusion and viral infection in lymphocytes. J Cell Sci. 2009;122:103–113. doi: 10.1242/jcs.035873. [DOI] [PubMed] [Google Scholar]
- Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- Briant L, Coudronniere N, Robert-Hebmann V, Benkirane M, Devaux C. Binding of HIV-1 virions or gp120-anti-gp120 immune complexes to HIV-1-infected quiescent peripheral blood mononuclear cells reveals latent infection. J Immunol. 1996;156:3994–4004. [PubMed] [Google Scholar]
- Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chertova E, Crise BJ, Morcock DR, Bess JW, Jr, Henderson LE, Lifson JD. Sites, mechanism of action and lack of reversibility of primate lentivirus inactivation by preferential covalent modification of virion internal proteins. Curr Mol Med. 2003;3:265–272. doi: 10.2174/1566524033479889. [DOI] [PubMed] [Google Scholar]
- Chirmule N, Goonewardena H, Pahwa S, Pasieka R, Kalyanaraman VS. HIV-1 envelope glycoproteins induce activation of activated protein-1 in CD4+ T cells. J Biol Chem. 1995;270:19364–19369. doi: 10.1074/jbc.270.33.19364. [DOI] [PubMed] [Google Scholar]
- Cicala C, Arthos J, Censoplano N, Cruz C, Chung E, Martinelli E, Lempicki RA, Natarajan V, VanRyk D, Daucher M, et al. HIV-1 gp120 induces NFAT nuclear translocation in resting CD4+ T-cells. Virology. 2006;345:105–114. doi: 10.1016/j.virol.2005.09.052. [DOI] [PubMed] [Google Scholar]
- Coyne CB, Bergelson JM. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell. 2006;124:119–131. doi: 10.1016/j.cell.2005.10.035. [DOI] [PubMed] [Google Scholar]
- Davis CB, Dikic I, Unutmaz D, Hill CM, Arthos J, Siani MA, Thompson DA, Schlessinger J, Littman DR. Signal transduction due to HIV-1 envelope interactions with chemokine receptors CXCR4 or CCR5. J Exp Med. 1997;186:1793–1798. doi: 10.1084/jem.186.10.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RJ, Fivash M, Casas-Finet J, Erickson JW, Kondoh A, Bladen SV, Fisher C, Watson DK, Papas T. Real-time DNA binding measurements of the ETS1 recombinant oncoproteins reveal significant kinetic differences between the p42 and p51 isoforms. Protein Sci. 1994;3:257–266. doi: 10.1002/pro.5560030210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu W, Sanders-Beer BE, Katz KS, Maglott DR, Pruitt KD, Ptak RG. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009;37:D417–422. doi: 10.1093/nar/gkn708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furler RL, Uittenbogaart CH. Signaling through the P38 and ERK pathways: a common link between HIV replication and the immune response. Immunol Res. 2010;48:99–109. doi: 10.1007/s12026-010-8170-1. [DOI] [PubMed] [Google Scholar]
- Geiger T, Wisniewski JR, Cox J, Zanivan S, Kruger M, Ishihama Y, Mann M. Use of stable isotope labeling by amino acids in cell culture as a spike-in standard in quantitative proteomics. Nat Protoc. 2011;6:147–157. doi: 10.1038/nprot.2010.192. [DOI] [PubMed] [Google Scholar]
- Gringhuis SI, van der Vlist M, van den Berg LM, den Dunnen J, Litjens M, Geijtenbeek TB. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat Immunol. 2010;11:419–426. doi: 10.1038/ni.1858. [DOI] [PubMed] [Google Scholar]
- Hioe CE, Tuen M, Vasiliver-Shamis G, Alvarez Y, Prins KC, Banerjee S, Nadas A, Cho MW, Dustin ML, Kachlany SC. HIV envelope gp120 activates LFA-1 on CD4 T-lymphocytes and increases cell susceptibility to LFA-1-targeting leukotoxin (LtxA) PLoS One. 2011;6:e23202. doi: 10.1371/journal.pone.0023202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodges A, Sharrocks K, Edelmann M, Baban D, Moris A, Schwartz O, Drakesmith H, Davies K, Kessler B, McMichael A, et al. Activation of the lectin DC-SIGN induces an immature dendritic cell phenotype triggering Rho-GTPase activity required for HIV-1 replication. Nat Immunol. 2007;8:569–577. doi: 10.1038/ni1470. [DOI] [PubMed] [Google Scholar]
- Jacquenet S, Decimo D, Muriaux D, Darlix JL. Dual effect of the SR proteins ASF/SF2, SC35 and 9G8 on HIV-1 RNA splicing and virion production. Retrovirology. 2005;2:33. doi: 10.1186/1742-4690-2-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez-Baranda S, Gomez-Mouton C, Rojas A, Martinez-Prats L, Mira E, Ana Lacalle R, Valencia A, Dimitrov DS, Viola A, Delgado R, et al. Filamin-A regulates actin-dependent clustering of HIV receptors. Nat Cell Biol. 2007;9:838–846. doi: 10.1038/ncb1610. [DOI] [PubMed] [Google Scholar]
- Konig R, Zhou Y, Elleder D, Diamond TL, Bonamy GM, Irelan JT, Chiang CY, Tu BP, De Jesus PD, Lilley CE, et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell. 2008;135:49–60. doi: 10.1016/j.cell.2008.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan MN, Ng A, Sukumaran B, Gilfoy FD, Uchil PD, Sultana H, Brass AL, Adametz R, Tsui M, Qian F, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemay J, Maidou-Peindara P, Cancio R, Ennifar E, Coadou G, Maga G, Rain JC, Benarous R, Liu LX. AKAP149 binds to HIV-1 reverse transcriptase and is involved in the reverse transcription. J Mol Biol. 2008;383:783–796. doi: 10.1016/j.jmb.2008.08.055. [DOI] [PubMed] [Google Scholar]
- Liu QH, Williams DA, McManus C, Baribaud F, Doms RW, Schols D, De Clercq E, Kotlikoff MI, Collman RG, Freedman BD. HIV-1 gp120 and chemokines activate ion channels in primary macrophages through CCR5 and CXCR4 stimulation. Proc Natl Acad Sci U S A. 2000;97:4832–4837. doi: 10.1073/pnas.090521697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Belkina NV, Shaw S. HIV infection of T cells: actin-in and actin-out. Sci Signal. 2009;2:pe23. doi: 10.1126/scisignal.266pe23. [DOI] [PubMed] [Google Scholar]
- Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, Zhu W, Ajith S, Blencowe BJ, Lynch KW. Alternative splicing networks regulated by signaling in human T cells. RNA. 2012;18:1029–1040. doi: 10.1261/rna.032243.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayya V, Lundgren DH, Hwang SI, Rezaul K, Wu L, Eng JK, Rodionov V, Han DK. Quantitative phosphoproteomic analysis of T cell receptor signaling reveals systemwide modulation of protein-protein interactions. Sci Signal. 2009;2:ra46. doi: 10.1126/scisignal.2000007. [DOI] [PubMed] [Google Scholar]
- Mercer J, Helenius A. Vaccinia virus uses macropinocytosis and apoptotic mimicry to enter host cells. Science. 2008;320:531–535. doi: 10.1126/science.1155164. [DOI] [PubMed] [Google Scholar]
- Miyakawa K, Sawasaki T, Matsunaga S, Tokarev A, Quinn G, Kimura H, Nomaguchi M, Adachi A, Yamamoto N, Guatelli J, Ryo A. Interferon-induced SCYL2 limits release of HIV-1 by triggering PP2A-mediated dephosphorylation of the viral protein Vpu. Sci Signal. 2012;5:ra73. doi: 10.1126/scisignal.2003212. [DOI] [PubMed] [Google Scholar]
- Mosenden R, Tasken K. Cyclic AMP-mediated immune regulation--overview of mechanisms of action in T cells. Cell Signal. 2011;23:1009–1016. doi: 10.1016/j.cellsig.2010.11.018. [DOI] [PubMed] [Google Scholar]
- Navarro J, Punzon C, Jimenez JL, Fernandez-Cruz E, Pizarro A, Fresno M, Munoz-Fernandez MA. Inhibition of phosphodiesterase type IV suppresses human immunodeficiency virus type 1 replication and cytokine production in primary T cells: involvement of NF-kappaB and NFAT. J Virol. 1998;72:4712–4720. doi: 10.1128/jvi.72.6.4712-4720.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- Pan C, Olsen JV, Daub H, Mann M. Global effects of kinase inhibitors on signaling networks revealed by quantitative phosphoproteomics. Mol Cell Proteomics. 2009;8:2796–2808. doi: 10.1074/mcp.M900285-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrish NF, Wilen CB, Banks LB, Iyer SS, Pfaff JM, Salazar-Gonzalez JF, Salazar MG, Decker JM, Parrish EH, Berg A, et al. Transmitted/Founder and Chronic Subtype C HIV-1 Use CD4 and CCR5 Receptors with Equal Efficiency and Are Not Inhibited by Blocking the Integrin alpha4beta7. PLoS Pathog. 2012;8:e1002686. doi: 10.1371/journal.ppat.1002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossio JL, Esser MT, Suryanarayana K, Schneider DK, Bess JW, Jr, Vasquez GM, Wiltrout TA, Chertova E, Grimes MK, Sattentau Q, et al. Inactivation of human immunodeficiency virus type 1 infectivity with preservation of conformational and functional integrity of virion surface proteins. J Virol. 1998;72:7992–8001. doi: 10.1128/jvi.72.10.7992-8001.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruperez P, Gago-Martinez A, Burlingame AL, Oses-Prieto JA. Quantitative phosphoproteomic analysis reveals a role for serine and threonine kinases in the cytoskeletal reorganization in early T cell receptor activation in human primary T cells. Mol Cell Proteomics. 2012;11:171–186. doi: 10.1074/mcp.M112.017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholich K, Pierre S, Patel TB. Protein associated with Myc (PAM) is a potent inhibitor of adenylyl cyclases. J Biol Chem. 2001;276:47583–47589. doi: 10.1074/jbc.M107816200. [DOI] [PubMed] [Google Scholar]
- Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005;23:1391–1398. doi: 10.1038/nbt1146. [DOI] [PubMed] [Google Scholar]
- Sieweke MH, Tekotte H, Jarosch U, Graf T. Cooperative interaction of ets-1 with USF-1 required for HIV-1 enhancer activity in T cells. EMBO J. 1998;17:1728–1739. doi: 10.1093/emboj/17.6.1728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderling TR. The Ca-calmodulin-dependent protein kinase cascade. Trends Biochem Sci. 1999;24:232–236. doi: 10.1016/s0968-0004(99)01383-3. [DOI] [PubMed] [Google Scholar]
- Soroceanu L, Akhavan A, Cobbs CS. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature. 2008;455:391–395. doi: 10.1038/nature07209. [DOI] [PubMed] [Google Scholar]
- Stevenson M, Stanwick TL, Dempsey MP, Lamonica CA. HIV-1 replication is controlled at the level of T cell activation and proviral integration. EMBO J. 1990;9:1551–1560. doi: 10.1002/j.1460-2075.1990.tb08274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoltzfus CM. Chapter 1. Regulation of HIV-1 alternative RNA splicing and its role in virus replication. Adv Virus Res. 2009;74:1–40. doi: 10.1016/S0065-3527(09)74001-1. [DOI] [PubMed] [Google Scholar]
- Weissman D, Rabin RL, Arthos J, Rubbert A, Dybul M, Swofford R, Venkatesan S, Farber JM, Fauci AS. Macrophage-tropic HIV and SIV envelope proteins induce a signal through the CCR5 chemokine receptor. Nature. 1997;389:981–985. doi: 10.1038/40173. [DOI] [PubMed] [Google Scholar]
- Wilen CB, Tilton JC, Doms RW. HIV: Cell Binding and Entry. Cold Spring Harb Perspect Med. 2012;2 doi: 10.1101/cshperspect.a006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcechowskyj JA, Lee JY, Seeholzer SH, Doms RW. Quantitative phosphoproteomics of CXCL12 (SDF-1) signaling. PLoS One. 2011;6:e24918. doi: 10.1371/journal.pone.0024918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Liu Z, Cao J, Ma Q, Gao X, Wang Q, Jin C, Zhou Y, Wen L, Ren J. GPS 2.1: enhanced prediction of kinase-specific phosphorylation sites with an algorithm of motif length selection. Protein Eng Des Sel. 2011;24:255–260. doi: 10.1093/protein/gzq094. [DOI] [PubMed] [Google Scholar]
- Yang HC, Shen L, Siliciano RF, Pomerantz JL. Isolation of a cellular factor that can reactivate latent HIV-1 without T cell activation. Proc Natl Acad Sci U S A. 2009;106:6321–6326. doi: 10.1073/pnas.0809536106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung ML, Houzet L, Yedavalli VS, Jeang KT. A genome-wide short hairpin RNA screening of jurkat T-cells for human proteins contributing to productive HIV-1 replication. J Biol Chem. 2009;284:19463–19473. doi: 10.1074/jbc.M109.010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoder A, Yu D, Dong L, Iyer SR, Xu X, Kelly J, Liu J, Wang W, Vorster PJ, Agulto L, et al. HIV envelope-CXCR4 signaling activates cofilin to overcome cortical actin restriction in resting CD4 T cells. Cell. 2008;134:782–792. doi: 10.1016/j.cell.2008.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Xu M, Huang Q, Gates AT, Zhang XD, Castle JC, Stec E, Ferrer M, Strulovici B, Hazuda DJ, et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe. 2008;4:495–504. doi: 10.1016/j.chom.2008.10.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.