

Abstract
Viral infections frequently alter mitochondrial function with suppression or induction of apoptosis and enhanced generation of reactive oxygen species. The mechanisms of these effects are varied and mitochondria are affected by both direct interactions with viral proteins as well as by secondary effects of viral activated signaling cascades. This chapter describes methods used in our laboratory to assess the effects of the Hepatitis C virus core protein on mitochondrial ROS production, electron transport and Ca2+ uptake. These include measurements of the effects of in vitro incubation of liver mitochondria with purified core protein as well as assessment of the function of mitochondria in cells and tissues expressing core and other viral proteins. These methods are generally applicable to the study of viral-mitochondrial interactions.
1. Introduction
In addition to their roles in respiration and ATP production, mitochondria are key regulators of apoptosis and cell survival. It is therefore not surprising that many viruses regulate mitochondrial function for suppression of apoptosis, enhancement of viral replication or modulation of the host environment (Boya et al., 2004). Several pathologically relevant examples include HIV, human T-cell leukemia virus, hepatitis B and C viruses, and Herpes viruses (Galluzzi et al., 2008; D'Agostino et al., 2005).
In some cases, viral proteins are known to bind directly to mitochondrial outer membrane components (Rahmani et al., 2000; Hickish et al., 1994) but in others the mechanisms of the viral mitochondrial effects appear to be secondary to other cellular events. Viral infection often leads to changes in mitochondrial function including inhibition of mitochondrial electron transport and ATP production, increased mitochondrial superoxide production, suppression or enhancement of apoptosis, alterations in mitochondrial structure, and alterations in mitochondrially based signaling processes (Loo et al., 2006).
In order to determine the mechanisms of virally mediated mitochondrial changes and assess their relevance to viral life cycle and host pathogenesis, it is necessary to develop methods to analyze viral effects on mitochondria. Since viral effects may be direct results of interactions of viral proteins with mitochondrial targets, or indirect effects of viral changes in signaling events, different cellular and subcellular systems are required. In this chapter we describe methods employed in our laboratories to assess the direct and indirect effects of HCV core protein on mitochondrial electron transport, calcium uptake, and redox status. These methods should be generally applicable to the broader study of virus-mitochondrial interactions as well.
Hepatitis C virus is an hepatotropic positive strand RNA virus which causes chronic hepatitis, cirrhosis and hepatocellular carcinoma (Thomson and Finch, 2005). Following infection the viral proteins form an ER-based replication complex associated with mitochondria and lipid droplets (Moradpour et al., 2007). The HCV core protein is synthesized as a 23kD protein (core 1–191) and then cleaved to a mature 21kD form (core 1–179) lacking the signal peptide. Core protein contains both basic and hydrophobic regions and, in addition to serving as the viral nucleocapsid protein, it has been shown to alter transcription, cell cycle control, mitochondrial electron transport and ER stress pathways (Irshad and Dhar, 2006). Transgenic mice that express core protein have mitochondrial electron transport defects and develop hepatocellular carcinoma. Core protein's effects on the mitochondria are both direct and indirect. While work from our laboratory has shown that core directly stimulates the mitochondrial Ca2+ uptake uniporter (Li et al., 2007), others have shown mitochondrial effects secondary to ER stress (Benali-Furet et al., 2005). Both pathways appear important. Even though the specific consequences of these HCV-mitochondria interactions are still under investigation it is clear that virally induced effects on the mitochondria can have profound effects in the host cell intracellular environment.
To study the effects of core protein on mitochondria we have used multiple model systems both in-vitro and in-vivo. All of the model systems described here have their respective advantages and disadvantages but the use of alternate approaches allows us to confirm and validate different results. This article describes methods that we have employed to examine the effects of core protein on mitochondrial function.
2. Materials and Model systems
2.1 Materials
Instruments
Branson Sonifier 450 (VWR, West Chester, PA, USA); Respirometry system with respirometer cell (MT200A) and Clark-type Oxygen electrode (model 1302) and meter (model 782) (Strathkelvin, North Lanarkshire, Scotland); FLUOstar OPTIMA plate reader(BMG LABTECH, Durham, NC, USA) (Ex/Em 544/590 nm); Becton-Dickinson FACScan Flow Cytometer (BDbioscience, San Jose, CA USA); Confocal (fluorescence) microscope or fluorescence microscope such as TE200-IUC Quantitative Fluorescence Live-Cell Imaging System (Nikon, Melville, NY, USA).
Chemicals and Kits
Sodium Glutamate was from Fisher (Pittsburgh, PA, USA); 5-sulfosalicylic acid, sodium malate, sodium succinate dibasic, adenosine 5'-diphosphate sodium salt (ADP), cyclosporin A, nicotinamide adenine dinucleotide phosphate (NADPH) and carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP) were all purchased from Sigma-Aldrich (St. Louis, MO, USA); Rhod-2 AM (Molecular probes, Eugene, OR, USA) was prepared freshly as a 1.0 mM stock solution in dry DMSO each time; tert-butyl hydroperoxide (Sigma-Aldrich) was diluted in water to 2 mM solution; Ru360 and Thapsigargin (TG) were from CalBiochem (Gibbstown, NJ, USA); JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolcarbocyanine iodide) (Molecular probes) was diluted in dry DMSO to 5 mg/ml; Glutathione reductase (2.2 units/ml), Glutathione peroxidase (200 U/ml) and Valinomycin (10 μM stock solution in DMSO) were from Sigma-Aldrich. MitoSOX™ Red (Invitrogen, Carlsbad, CA, USA), 2',7'-dichlorodihydrofluorescin diacetate (DCFDA) (Molecular Probes) was dissolved 5mM in dry DMSO.
Ratiometric pericam-mt cDNA expression plasmid was kindly provided by Dr. Atsushi Miyawaki (Wako, Saitama, Japan) (Nagai et al., 2001).
GSH/GSSG-412™ kit and GSH 400™ kit were from Oxis Research (Foster City, CA, USA).
2.2 Plasmid design and core protein isolation
To perform detailed and controlled in vitro experiments, it is necessary to express and purify the protein of interest in its native conformation. The following section describes a robust method to obtain HCV core protein that contains amino acids 1 to 179, inclusive (Kunkel and Watowich, 2004).
HCV core protein (HCVC179), residues 1 to 179, derived from the AG94 isolate of genotype 1a sequence, was amplified by polymerase chain reaction (PCR) using the sense primer 5'-GGGAAATCCATATGAGCACGAATCCTAAACCTCAAAGAAAA-3' and the antisense primer 5'-CCGGAATTCTCATTACAGAAGGAAGATAGAGAAAGAGCAACC. Sequence of the cloned PCR fragment was confirmed by DNA sequencing and the fragment was then subcloned into a pET30a expression vector (Novagen, Gillstown, NJ).
The above HCVC179/pET30a expression vector was transformed into Escherichia coli BL21 (DE3) cells and the bacterial cultures were maintained in 2xYT medium for core protein expression. When the optical density OD600nm of the culture reached 1.0, 1 mmol/L isopropyl-β-D-thiogalactopyranoside (IPTG) was added to induce HCVC179 expression. Bacterial cultures were maintained for an additional 4 hours at 25°C, and then centrifuged at 5,000 × g for 30 min to pellet the cells. For HCVC179 protein purification, bacterial pellet was sequentially resuspended in ice-cold Lysis buffer (20mM Tris-HCl, pH 7.0, 2mM DTT), Urea Lysis buffer (8M Urea, 20mM NaPhosphate (NaH2PO4), pH 7.0), and Urea/Salt Lysis buffer (8 M Urea, 500 mM NaCl, 20mM (NaH2PO4), pH 6.5). Each time the pellet was sonicated using ten 30 s cycles of sonication and cooling, followed by centrifugation at 50,000 × g for 20 min at 4°C in an SS-34 rotor. The supernatants remaining after each centrifugation were saved and stored at 4°C. Ten μl of each of the supernatants obtained above (Lysis supernatant, Urea Lysis supernatant, and Urea/Salt Lysis supernatant) and of the Urea/Salt pellet, were treated with sample buffer (75 mM Tris pH 6.8, 2% SDS, 5% glycerol, 0.003% bromophenol blue, 0.9% ß-mercaptoethanol) and incubated at 37°C for 30 min and run on16% SDS-PAGE. The sample containing the most enriched HCVC179 was determined by staining the gel with Gel Code Blue Stain (Rockford, IL, USA) for one hour to overnight. If the most HCVC179 enriched fraction occurred in the Lysis supernatants and/or the Urea Lysis supernatants, these fractions were pooled and loaded directly on the HPLC column; if the most HCVC179 enriched fraction occurred in the Urea/Salt supernatant, the sample needs to be diluted with equal volume of 8M Urea (pretreated with Dowex MR-3 resin, 1 g resin/ 100ml urea) containing Dithiothreitol (DTT) to a final concentration of 50mM (stock 1.0M) and incubated overnight at 4°C. The HCVC179 containing supernatant was then loaded on a cation-exchange HPLC column (e.g. Poros 20 CM) pre-equilibrated with cation column equilibration buffer (50mM HEPES, pH 7.0, 8M urea, 150mM NaCl). Fractions were eluted from the column with a linear gradient of NaCl (from 150mM to 750mM) and identified by 16% SDS-PAGE for HCVC179 expression. Fractions containing HCVC179 were pooled and concentrated by centrifugation using Centriplus 10KD at 3,000 × g 4°C (typically concentrated by 10–15 fold). A reverse phase HPLC column (e.g., YMC-ODS) was pre-equilibrated with reverse phase column equilibration buffer (20 mM NaH2PO4, pH 3.0, 1% MeOH). Two ml of the concentrated samples (12.5μl concentrated H3PO4 per every ml was added before loading) were loaded on the reverse phase HPLC column and eluted with a linear gradient of 20mM sodium-phosphate-methanol, pH 3.0 (1% MeOH to 96% MeOH). HCVC179 elutes as a single peak around 90% MeOH. Fractions containing HCVC179 were pooled and dialyzed overnight at 4°C against refolding buffer (20 mM Tris, pH 7.0, 100 mM NaCl plus protease inhibitor cocktail [leupeptin 0.05mM,pepstatin A 0.001mM, PMSF 0.5mM]) and dialyzed with fresh refolding buffer plus protease inhibitor cocktail for an additional 6 hours. The dialyzed samples were concentrated by using Centriplus 10KD, 3,000 × g at 4°C (typically concentrated by 10 fold). The homogeneity and identity of purified HCVC179 was confirmed by SDS-PAGE (typically 98%) and immunoblot using a mouse monoclonal antibody to HCV core protein (Affinity BioReagents).
This method allows efficient and rapid purification of full-length folded HCV core protein, and typically produces ~2 mg of purified protein (>95% homogeneity as visualized by SDS-PAGE and Gel Code blue staining) per Liter of bacterial culture. Elution conditions will need to be optimized if different cation exchange and reverse phase columns are used. An example of purity of the core obtained by this method is shown in Fig. 1.
Figure 1. HCV core 179 expression and purification.
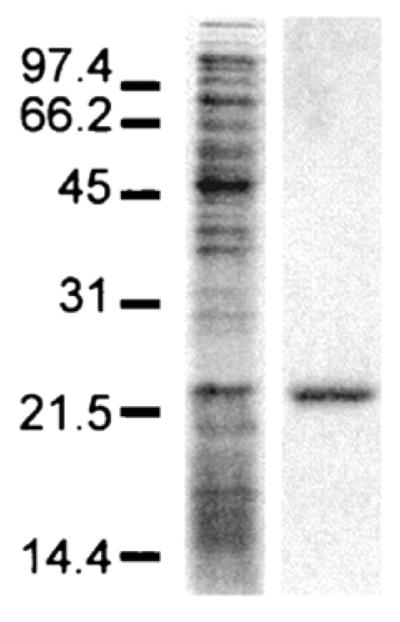
Samples were resolved on an SDS-16% polyacrylamide gel under reducing conditions and visualized by Coomassie blue stain. Lanes: A, expression of HCVC179 in bacterial cell pellets; B, HCVC179 following purification by cation exchange and reverse phase HPLC. Sizes are indicated in kilodaltons.
2.3 Mitochondrial isolation and cellular model systems
2.3.1 Mitochondrial isolation
Four hundred mg of mouse liver tissue was rinsed in ice cold 1XPBS (137 mM NaCl, 2.7 mM KCl, 4.3 mM Na2HPO4, 1.47 mM KH2PO4, pH 7.4) immediately after animal sacrifice and minced on ice in 4 ml of mitochondria isolation buffer (250mM sucrose, 10mM HEPES, 0.5mM EGTA, 0.1% BSA, pH 7.4). Minced tissue was gently homogenized (3–4 strokes) using a Dounce homogenizer with a loose fitting pestle and the homogenate was centrifuged at 500×g for 5 min at 4°C. The supernatant was collected and kept on ice and the pellet was resuspended again in mitochondria isolation buffer and centrifuged at 500×g for 5 min at 4°C. The supernatants obtained above were combined and centrifuged at 7800×g for 10 min at 4°C to obtain crude mitochondrial pellets. To wash the mitochondria, crude mitochondrial pellets were resuspended in mitochondria wash buffer (250mM sucrose, 10mM HEPES, pH 7.4) and centrifugation at 7800×g for 10 min at 4°C was repeated. The washed mitochondria were resuspended in suitable mitochondria resuspension buffer and an aliquot of suspension was taken to measure protein concentration.
2.3.2 Cellular model systems
In studying mitochondrial function we have taken advantage of several model systems. These model systems are in general use in the field and have been described in detail elsewhere. Cellular model systems for studying HCV include simple expression systems for viral proteins (Ray et al., 2000), tetracycline repressor based inducible expression systems (Li et al., 2002;Otani et al., 2005), replicon cell lines autonomously replicating viral RNA (Blight et al., 2000;Ikeda et al., 2002), cell culture infectious virus (Wakita et al., 2005; Lindenbach et al., 2005;Zhong et al., 2005) and transgenic mice expressing one or more viral proteins in either constitutive or inducible fashion (Honda et al., 1999;Moriya et al., 1998;Lerat et al., 2002).
3. HCV core protein effects on mitochondria in vitro
The simplest way to demonstrate a direct mitochondrial effect of a viral protein is to demonstrate that in vivo observations can be reproduced in vitro through direct interaction of purified viral proteins with isolated mitochondria. The following methods are described for the effect of HCV core on mouse liver mitochondria.
3.1 Effects of core protein on mitochondrial respiration
Freshly isolated mitochondria were resuspended in respiration buffer (130 mM sucrose, 50 mM KCl, 5 mM MgCl2, 5 mM KH2PO4, 0.05 mM EDTA, 5 mM HEPES, pH 7.4) and incubated in the presence or absence of core protein (1–10 ng/mg mitochondrial protein) for 5 min at 4°C. Immediately following incubation, 1–1.5 mg of mitochondria were transferred to a respiration chamber pre-filled with 1ml of respiration buffer, mixed well using magnetic stirring and allowed to equilibrate at 25°C. Complex I-supported state 4 respiration was initiated by adding 5 mM glutamate and 5 mM malate to the sample chamber. Complex I-supported state 3 respiration was initiated by adding 100 nmol of ADP. Maximum oxygen consumption (uncoupled respiration) was measured after returning to state 4 respiration with the addition of 5 μM FCCP. Complex II-supported state 3 and 4 respiration was measured in a similar manner by adding 5mM succinate instead of glutamate and malate. P:O ratios and FCCP-induced consumption rates were calculated as described by Estabrook (Estabrook, 1967).
The advantage of this method is that it allows direct effects of core on mitochondria to be observed in the absence of more complex cellular signaling networks. As illustrated in Fig. 2, brief incubations with core protein in the range of 1–10 ng/mg mitochondrial protein produce a selective inhibition of complex I associated electron transport reflected as reduced P:O ratio and uncoupled O2 consumption from glutamate-malate substrate (Korenaga et al., 2005). Further details about core protein effects on mitochondrial respiratory complexes can also be obtained by incubating mitochondria with the viral protein as described above and then measuring respiratory complex activity in submitochondrial particles as previously shown (Korenaga et al., 2005).
Figure 2. Effects of core protein on mitochondrial respiration.

Mouse liver mitochondria were incubated with core protein as described. Maximum respiratory rate after addition of FCCP is presented. Note that core protein inhibits respiration with glutamate/malate but not succinate.
3.2 Direct effect of core on mitochondrial GSH/GSSG
The measurement of glutathione in tissue extracts can be performed by multiple assays including colorimetric assays of thiol reaction products that measure reduced glutathione (GSH), a “recycling” assay in which the rate of glutathione dependent reduction of 5,5'-dithiobis(2-nitrobenzoic acid) or DTNB is proportional to total glutathione (GSH + GSSG) concentration (Anderson, 1989), and several HPLC assays. In mouse liver mitochondria we have found the DTNB recycling assay to accurately measure total glutathione but attempts to first derivatize GSH in order to remove it for the measurement of the oxidized fraction were not successful possibly due to poor penetration of the derivatizing reagent into intact mitochondria. When used for mitochondria, the reactive thiol assays all suffer from some degree of reactivity with non-glutathione thiols and therefore tend to overestimate the total GSH content. We solved this problem by using a commercially available thiol assay for GSH and measuring GSH under three conditions: (1) untreated, (2) after quantitative reduction of the glutathione pool with glutathione reductase and NADPH, and (3) after quantitative oxidation of the glutathione pool with glutathione peroxidase (GPx) and tertiary butyl hydroperoxide (tBOOH). We found this to be the most reliable biochemical assay of GSH in the context of intact mouse liver mitochondria.
After preparing crude mitochondria as described above, mitochondrial pellets were resuspended in 1.5 ml of PBS alone or in 1.5 ml of PBS containing HCV core protein (50–500 ng/mg of mitochondrial protein) and incubated for 5 min at 4°C. The contents of each tube were then divided into three equal aliquots of 500 μl each. One aliquot was used to directly measure both GSH and GSH + GSSG. The second aliquot was used to measure GSH after reduction with glutathione reductase and NADPH. To do so, the sample was first freeze-thawed twice in a dry ice/ethanol bath to release GSH from mitochondria and then treated with 5 μl of glutathione reductase and 50 μl of NADPH, mixed by vortex and incubated for 10 min at 25°C. The third aliquot was used to measure reduced thiols after oxidation with GPx and tBOOH. To do so, the sample was first freeze-thawed as described above and then treated with 5 μl of GPx and 50 μl 2mM tBOOH, mixed by vortex and incubated for 10 min at 25°C. Following their respective treatments, the aliquots were placed on ice and acidified with 10% SSA, sonicated for 15 s and centrifuged at 10,000×g for 10 min at 4°C. Glutathione measurements were performed on the supernatants obtained following the centrifugation. We used the DTNB recycling assay for total glutathione (GSH + GSSG), and GSH-400™ thioester method for reduced glutathione (GSH). The recycling assay was performed only on the first aliquot. The thioester method was performed on all three samples. We used the above referenced commercial kits but the assays can be done by directly obtaining the individual reagents as well.
For calculation of total glutathione (GSH + GSSG) the DTNB recycling assay result is accurate. For GSH, the thioester result overestimates GSH due to contribution of non-GSH thiols. The magnitude of this nonspecific background is determined after GPx/tBOOH oxidation and the validity of the thiol method is confirmed by comparing the result from the reduced sample, after background subtraction, to the value obtained with the recycling assay. In our hands these results agree to within 5–10%. An example of the use of this method for the determination of the effect of in vitro core protein on mitochondrial glutathione is presented in Fig. 3.
Figure 3. Effects of in vitro core protein incubation on mitochondrial glutathione.

Total glutathione was measured by the DTNB recycling assay. GSH thiol was measured as described either before (dark grey bar) or after (black bar) reduction with glutathione reductase and NADPH. The non-GSH component of the thiol reaction was measured by oxidation of the sample with tBOOH and glutathione peroxidase (oxidation control).
3.3 Measurement of core effects on calcium uptake in isolated mitochondria
Mitochondria were resuspended in mitochondrial isolation buffer and incubated on ice in the presence or absence of HCV core protein for 30 min. The concentration of core protein used in these experiments ranged from 100–600 ng core protein/mg mitochondrial protein. After incubation with core protein, mitochondria were loaded with the Ca2+ indicator Rhod-2 AM at a final concentration of 4 μM and incubated for an additional 1 hour at 4°C. Following this, mitochondria were pelleted by centrifugation at 7,800×g for 10 min at 4°C and washed twice in wash buffer. Finally, washed mitochondria were resuspended in respiration buffer (final protein concentration 0.33 mg/ml). Depending on the protocol to be tested, mitochondria could be further treated with various inhibitors (e.g. Ca2+ transport inhibitor Ru-360 (125 nM) and/or permeability transition inhibitor cyclosporin A (1 μM)). We recommend carrying out these incubations at 4°C for 30 min. Once this was done, the mitochondrial suspension was aliquoted into 96-well plates with at least triplicates for each condition. The plate was incubated at room temperature for 30 min in the dark. After the incubation, 25 μl of Ca2+ containing respiration buffer (250 μM Ca2+ final concentration) was added to each well using a multi-channel pipette and the plate was immediately placed on the plate reader. Rhod-2 fluorescence (Ex/Em 544/590 nm) was recorded every 30 s for 60 min.
This method first depletes mitochondrial Ca2+ by prolonged incubation with EGTA and then suddenly exposes the mitochondria to an inwardly directed Ca2+ gradient. As shown in Fig. 4 the combination of the Ca2+ uniport inhibitor Ru-360 and the permeability transition pore inhibitor Cyclosporin A (CSA) effectively eliminated all Ca2+ uptake in this system. In vivo, the chemical Ca2+ gradient is outward and thus potential driven uniport is the only significant mechanism for Ca2+ entry. Opening of the permeability transition pore results in Ca2+ efflux from the matrix. However, in this in vitro system, the Ca2+ gradients are reversed and mitochondrial permeability transition becomes a second mechanism for Ca2+ entry. For this reason we found it necessary to conduct all experiments in the presence of CSA. The use of CSA allowed us to measure calcium uptake exclusively due to the calcium uniporter, known to be the only significant source of calcium entry into mitochondria in-vivo. Pre-incubation of mitochondria with 100ng of recombinant core protein increased the initial rate of calcium uptake as well as the plateau value for steady-state Ca2+ content (Fig. 4). As expected, the presence of Ru360 completely blocked the effect of core protein (Li et al., 2007).
Figure 4. Characteristics of Ca2+ uptake in isolated mitochondria. Left panel.

Confirmation of the intramitochondrial source of the Rhod-2 fluorescence signal. Liver mitochondria were loaded with Rhod-2 and incubated with FCCP (50 nM) or EGTA (10 mM) as indicated. Ca2+ (250 μM) was added 30 min prior to measurement of fluorescence. Note that the Ca2+-induced Rhod-2 signal could be completely prevented by mitochondrial depolarization with FCCP. This indicates that it results from mitochondrial uptake of Ca2+ and not from Rhod-2 leakage out of the mitochondria. Right panel: Time course of mitochondrial Ca2+ uptake. Note that the combination of Cyclosporin A and Ru360 was required to completely inhibit uptake under these conditions (see text). Reprinted with permission from FASEB Journal (Li et al., 2007).
4. Mitochondrial effects of viral proteins in cellular systems
Viral effects on mitochondria that result as a consequence of signaling events, ER stress, or replication complexes are not likely to occur in the reconstituted in vitro systems described in the previous section. It is thus important to measure viral effects on mitochondrial function in cellular systems and compare to direct effects. This section describes methods useful for measurement of mitochondrial function in cellular and animal model systems for Hepatitis C virus infection.
4.1 Measurement of mitochondrial depolarization by JC-1 flow cytometry
Several methods exist to measure mitochondrial membrane potential. JC-1 is a cationic fluorophore with a concentration dependent fluorescence spectral shift that allows it to be used in a ratiometric fashion to differentiate polarized from depolarized mitochondria. This has some advantages over single wavelength methods, such as rhodamine 123 in which depolarization is marked by a decrease in fluorescence intensity which may be less specific. Use of this method has been described previously (Otani et al., 2005).
For measurement of mitochondrial membrane potential, Huh-7 hepatoma cells with tetracycline-regulated expression of core protein (Li et al., 2002) were grown in 10mm dishes to 50–70% confluence (approximately 1×106 cells per dish.) As a positive control for depolarization one dish was first treated with valinomycin, final concentration 500nM for 30 min prior to cell harvesting. Medium was then removed, cells were washed once with PBS and trypsinized by adding 100 μl of trypsin solution for 3 min. Cells were then suspended in 5 ml culture medium and centrifuged at 500×g for 5min at room temperature. Cell pellets were resuspended in 5ml of 0.5% BSA in PBS containing 10 μl of 5mg/ml JC1 solution for 10min in the dark. JC-1 loaded cells were then centrifuged at 500×g for 5min, washed once with 0.25% BSA in PBS, and resuspended in 1ml of PBS prior to immediate analysis by flow cytometry.
This method has proven useful for demonstrating the effect of HCV core protein expression on oxidant and Ca2+-induced mitochondrial depolarization. As shown in Fig. 5, control cells cluster with high red fluorescence and submaximal green fluorescence. Upon depolarization there is first an increase of green fluorescence followed by a loss of red fluorescence. Suitable controls to validate the depolarization are essential. We have found that valinomycin works well for this purpose. We have used this method with core-expressing and non-core-expressing cells as well as HCV subgenomic and genome-length replicons. These studies demonstrate that core protein expression sensitizes mitochondria to oxidant-induced depolarization (Otani et al., 2005).
Figure 5. Core-induced mitochondrial depolarization measured with JC-1 flow cytometry.

Measurements of JC-1 red and green fluorescence were performed as described. A. Control Huh7 cells. B. Huh7 treated with valinomycin as a positive control for depolarization. C. L-14 cells with tet-off regulated core protein expression, in the presence of doxycycline (no core expression). D. L-14 cells in the presence of doxycycline (no core expression) after incubation with 100 μM tBOOH. E. L-14 cells in the absence of doxycycline (core expression induced). F. L-14 cells in the absence of doxycycline (core expression induced) after incubation with 100 μM tBOOH.
4.2 Measurement of core protein effects on live cell mitochondrial Ca2+
In many cases, viral proteins do not alter baseline mitochondrial properties but do alter response to stimuli. An important example of this is mitochondrial Ca2+ uptake in response to ER Ca2+ release. These processes can be measured using targeted protein reporter probes in live cells. The use of ratiometric pericam-mt to measure the effects of core protein on mitochondrial Ca2+ uptake in response to ER Ca2+ release is described. Ratiometric pericam-mt is a Ca2+ dependent fluorescent protein developed by Miyawaki and colleagues (Nagai et al., 2001) and targeted with a mitochondrial sequence encoding the N-terminal 12-aa presequence of subunit IV of cytochrome c oxidase. Dual fluorescence excitation allows Ca2+ measurement independently of reporter concentrations or cell geometry.
To measure mitochondrial matrix Ca2+, Huh-7 cells were seeded onto coverslips in 6 well plates and incubated overnight. Cells were then rinsed and transfected with the mitochondrial targeted Ca2+ indicator protein (ratiometric-pericam-mt) cDNA plasmid. After 48 hrs, coverslips were rinsed and mounted in Ca2+ free buffer in a fluorescence microscope system for quantitative imaging. Pericam-transfected cells were recognized by the presence of fluorescence in a cytoplasmic, perinuclear location typical for mitochondria (Fig. 6). Fluorescence emission images at 510 nm were acquired every 2 s after sequential excitation at 405 and 490 nm. Data analysis was performed using MetaMorph software. Fluorescence intensity at each wavelength was determined in the mitochondrial region and corrected by subtraction of background fluorescence. Mitochondrial Ca2+ was represented as the F495/405 nm ratio.
Figure 6. Thapsigargin-induced increases in mitochondrial calcium measured with mitochondirally-targeted pericam.
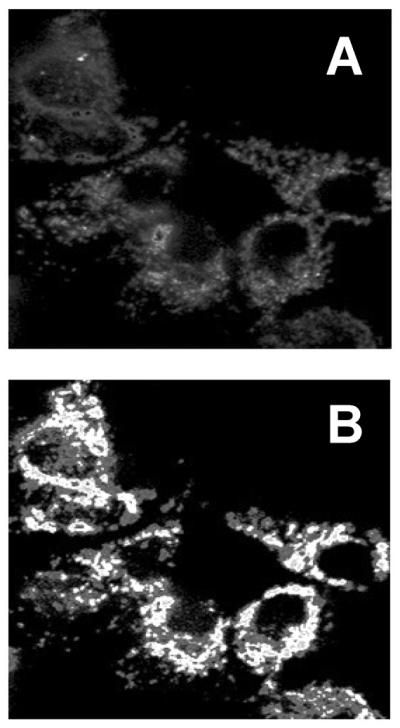
The images were obtained as described either before (A) or after (B) thapsigargin exposure. The images represent a ratio mapped image where pixel intensity corresponds to fluorescence ratio.
Efficiency of cell transfection can be monitored by the green fluorescence of ratiometric-pericam and is usually around 30–50% in Huh7 cells transfected with lipofectamine 2000. We have used this method to compare the response of core protein expressing cells and control cells to thapsigargin-induced ER Ca2+ release. Very distinct patterns were observed (Li et al., 2007). Similar to the results seen with isolated mitochondria, expression of core protein caused a rapid increase in mitochondrial Ca2+uptake, in this case following the release of Ca2+ from ER induced by thapsigargin.
4.3 Core protein effects on mitochondrial ROS production
Well-established fluorescence methodologies can be readily used in the HCV cellular model systems for measurement of mitochondrial ROS production. The commercially available fluorescent probes, mitoSOX™ Red and 2',7'-dichlorodihydrofluorescein diacetate (DCFDA) (Molecular Probes) are cell permeable and become highly fluorescent in the presence of ROS (mitoSOX™ being specifically sensitive to superoxide). Both of these probes were used in our cell system, to assess the specific effect of core protein on mitochondrial ROS production (Okuda et al., 2002; Otani et al., 2005; Li et al., 2007).
Live cell imaging was used to measure ROS production in response to ER Ca2+ release in HCV core expressing cells or control cells. Cells were grown on glass coverslips, culture medium was aspirated and cells incubated in phenol red-free medium containing 2.5 μM mitoSOX™ Red for 30 min at 37°C. Cells were then washed briefly with Ca2+ free Hanks Balanced Salt Solution, and mounted in a microscope chamber containing Ca2+ free buffer solution. Calcium release from the ER was induced by adding thapsigargin to a final concentration of 200 μM. Alternatively, cells can be exposed to DCFDA at final concentration of 500 nM immediately before imaging. Fluorescence images (Ex/Em 510/570nm for mitoSOX; Ex/Em 488/530 nM for DCFDA) were then obtained using a confocal or fluorescence microscope at various time points (generally 1 min intervals). Mitochondrial ROS production was quantified by analysis of cell fluorescence intensity in a perinuclear mitochondrial-rich region.
Similar assays were performed with a plate reader method obviating the need for imaging and cell selection. Cells were first seeded in 96-well plates and incubated for 12–24 hrs followed by incubation with 2.5 μM mitoSOX™ Red or 5 μM DCFDA for 30 min at 37° C in the dark. After washing with Ca2+ free buffer cells were exposed to 10 μM thapsigargin for 2 hrs. Fluorescence was measured in a plate reader (Ex/Em 510/570 nm for mitoSOX; Ex/Em 488/530 nM for DCFDA) and raw fluorescence intensity values were normalized to cell numbers plated on each well.
These methods describe the use of simple fluorescence probes for ROS determination in live cells. MitoSOX is an ethidium derivative that is specifically oxidized by superoxide to form a product that intercalates in DNA where it becomes fluorescent. Because of its DNA association it is retained well in cells and is suitable for flow cytometry measurements as well. DCFDA is somewhat more problematic. Although its fluorescent product is a result of oxidation, this reaction requires enzymatic conversion and the fluorescent product is somewhat membrane permeant and is only partially retained in the cells. Both oxidation and cell retention can be variable and these characteristics have made it difficult to use for flow cytometery. In our hands it is best suited for short-term imaging based measurements before the variable cell retention properties become relevant. Both methods have confirmed that core protein increases mitochondrial ROS production in a Ca2+-dependent fashion (Okuda et al., 2002; Li et al., 2007).
MitoSOX™ measurements showed that TG treatment on core-expressing cells resulted in greater mitochondrial superoxide production compared to that observed in non-core-expressing cells. This effect was specifically induced by Ca2+ uptake by the mitochondria; DCFDA measurements also showed results similar to those seen with mitoSOX™ demonstrating that core protein could sensitize cells to oxidative stimulus. The primary site of ROS production in response to the ROS donor was shown to be the mitochondria (Otani et al., 2005).
5. Conclusion
Since viruses have both direct and indirect effects on mitochondrial function, a series of methods which examine mitochondria in live cells as well as reconstitution systems are required. In the case of HCV core protein, similar effects on electron transport and ROS production have been observed in-vitro and in-vivo and this has allowed the identification of direct interaction of the viral protein with the Ca2+ uptake mechanism. These methods are generally applicable to the study of viral protein control of mitochondrial function.
Acknowledgements
The authors would like to thank M. Korenaga, K. Otani, M. Okuda, and D. Boehning for help in developing assays. We also thank S. Lemon, K. Li, M. Yi, J. Sun, and T. Chan, for helpful advice and providing the model systems for some of the work described here. This work was supported in part by NIH Grant AA12863 from the National Institute on Alcohol Abuse and Alcoholism.
Reference List
- Anderson ME. Enzymatic and chemical methods for the determination of glutathione. In: Dolphin D, Poulson R, Avramovic O, editors. Glutathione: chemical, biochemical and medical aspects. Part.A. John Wiley and Sons; New York: 1989. pp. 339–365. [Google Scholar]
- Benali-Furet NL, Chami M, Houel L, De GF, Vernejoul F, Lagorce D, Buscail L, Bartenschlager R, Ichas F, Rizzuto R, Paterlini-Brechot P. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene. 2005;24:4921–4933. doi: 10.1038/sj.onc.1208673. [DOI] [PubMed] [Google Scholar]
- Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCV RNA replication in cell culture. Science. 2000;290:1972–1974. doi: 10.1126/science.290.5498.1972. [DOI] [PubMed] [Google Scholar]
- Boya P, Pauleau AL, Poncet D, Gonzalez-Polo RA, Zamzami N, Kroemer G. Viral proteins targeting mitochondria: controlling cell death. Biochim Biophys Acta. 2004;1659:178–189. doi: 10.1016/j.bbabio.2004.08.007. [DOI] [PubMed] [Google Scholar]
- D'Agostino DM, Bernardi P, Chieco-Bianchi L, Ciminale V. Mitochondria as functional targets of proteins coded by human tumor viruses. Adv Cancer Res. 2005;94:87–142. doi: 10.1016/S0065-230X(05)94003-7. [DOI] [PubMed] [Google Scholar]
- Estabrook RW. Mitochondrial respiratory control and the polarographic measurements of ADP/O rations. Methods Enzymol. 1967;10:41–47. [Google Scholar]
- Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. Viral control of mitochondrial apoptosis. PLoS Pathog. 2008;4:e1000018. doi: 10.1371/journal.ppat.1000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickish T, Robertson D, Clarke P, Hill M, di Stefano F, Clarke C, Cunningham D. Ultrastructural localization of BHRF1: an Epstein-Barr virus gene product which has homology with bcl-2. Cancer Res. 1994;54:2808–2811. [PubMed] [Google Scholar]
- Honda A, Arai Y, Hirota N, Sato T, Ikegaki J, Koizumi T, Hatano M, Kohara M, Moriyama T, Imawari M, Shimotohno K, Tokuhisa T. Hepatitis C virus structural proteins induce liver cell injury in transgenic mice. J Med Virol. 1999;59:281–9. doi: 10.1002/(sici)1096-9071(199911)59:3<281::aid-jmv4>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Yi M, Li K, Lemon SM. Selectable subgenomic and genome-length dicistronic RNAs derived from an infectious molecular clone of the HCV-N strain of hepatitis C virus replicate efficiently in cultured Huh7 cells. J Virol. 2002;76:2997–3006. doi: 10.1128/JVI.76.6.2997-3006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irshad M, Dhar I. Hepatitis C virus core protein: an update on its molecular biology, cellular functions and clinical implications. Med Princ Pract. 2006;15:405–416. doi: 10.1159/000095485. [DOI] [PubMed] [Google Scholar]
- Korenaga M, Wang T, Li Y, Showalter LA, Chan T, Sun J, Weinman SA. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J Biol Chem. 2005;280:37481–37488. doi: 10.1074/jbc.M506412200. [DOI] [PubMed] [Google Scholar]
- Kunkel M, Watowich SJ. Biophysical characterization of hepatitis C virus core protein: implications for interactions within the virus and host. FEBS Lett. 2004;557:174–180. doi: 10.1016/s0014-5793(03)01486-8. [DOI] [PubMed] [Google Scholar]
- Lerat H, Honda M, Beard MR, Loesch K, Sun J, Yang Y, Okuda M, Gosert R, Xiao SY, Weinman SA, Lemon SM. Steatosis and liver cancer in transgenic mice expressing the structural and nonstructural proteins of hepatitis C virus. Gastroenterology. 2002;122:352–365. doi: 10.1053/gast.2002.31001. [DOI] [PubMed] [Google Scholar]
- Li K, Prow T, Lemon SM, Beard MR. Cellular response to conditional expression of hepatitis C virus core protein in Huh7 cultured human hepatoma cells. Hepatology. 2002;35:1237–1246. doi: 10.1053/jhep.2002.32968. [DOI] [PubMed] [Google Scholar]
- Li Y, Boehning DF, Qian T, Popov VL, Weinman SA. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007;21:2474–2485. doi: 10.1096/fj.06-7345com. [DOI] [PubMed] [Google Scholar]
- Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, Carney DS, Wang T, Ishida H, Yoneyama M, Fujita T, Saito T, Lee WM, Hagedorn CH, Lau DT, Weinman SA, Lemon SM, Gale M., Jr. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103:6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradpour D, Penin F, Rice CM. Replication of hepatitis C virus. Nat Rev Microbiol. 2007;5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- Moriya K, Fujie H, Shintani Y, Yotsuyanagi H, Tsutsumi T, Ishibashi K, Matsuura Y, Kimura S, Miyamura T, Koike K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat Med. 1998;4:1065–7. doi: 10.1038/2053. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci U S A. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda M, Li K, Beard MR, Showalter LA, Scholle F, Lemon SM, Weinman SA. Mitochondrial injury, oxidative stress and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology. 2002;122:366–375. doi: 10.1053/gast.2002.30983. [DOI] [PubMed] [Google Scholar]
- Otani K, Korenaga M, Beard MR, Li K, Qian T, Showalter LA, Singh AK, Wang T, Weinman SA. Hepatitis C virus core protein, cytochrome P450 2E1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology. 2005;128:96–107. doi: 10.1053/j.gastro.2004.10.045. [DOI] [PubMed] [Google Scholar]
- Rahmani Z, Huh KW, Lasher R, Siddiqui A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J Virol. 2000;74:2840–2846. doi: 10.1128/jvi.74.6.2840-2846.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray RB, Meyer K, Ray R. Hepatitis C virus core protein promotes immortalization of primary human hepatocytes. Virology. 2000;271:197–204. doi: 10.1006/viro.2000.0295. [DOI] [PubMed] [Google Scholar]
- Thomson BJ, Finch RG. Hepatitis C virus infection. Clin Microbiol Infect. 2005;11:86–94. doi: 10.1111/j.1469-0691.2004.01061.x. [DOI] [PubMed] [Google Scholar]
- Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A. 2005;102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]