

Abstract
Purpose
The epidermal growth factor receptor (EGFR) and cyclooxygenase-2 (COX-2) pathways are upregulated in head and neck squamous cell carcinoma (HNSCC). Preclinical models indicate synergistic anti-tumor activity from dual blockade. We conducted a randomized, double-blind, placebo-controlled window trial of erlotinib, an EGFR inhibitor; erlotinib plus sulindac, a non-selective COX inhibitor, vs. placebo.
Experimental Design
Patients with untreated, operable Stage II-IVb HNSCC were randomized 5:5:3 to erlotinib, erlotinib-sulindac, or placebo. Tumor specimens were collected before and after 7-14 days of treatment. The primary endpoint was change in Ki-67 proliferation index. We hypothesized an ordering effect in Ki-67 reduction: erlotinib-sulindac > erlotinib > placebo. We evaluated tissue microarrays by immunohistochemistry for pharmacodynamic modulation of EGFR and COX-2 signaling intermediates.
Results
From 2005-2009, 47 patients were randomized for the target 39 evaluable patients. Thirty-four tumor pairs were of sufficient quality to assess biomarker modulation. Ki-67 was significantly decreased by erlotinib or erlotinib-sulindac (omnibus comparison, two-sided Kruskal-Wallis, p=0.04). Wilcoxon pairwise contrasts confirmed greater Ki-67 effect in both erlotinib groups (erlotinib-sulindac vs. placebo p=0.043; erlobinib vs. placebo, p=0.027). There was a significant trend in ordering of Ki-67 reduction: erlotinib-sulindac > erlotinib > placebo (two-sided exact Jonckheere-Terpstra, p =0.0185). Low baseline pSrc correlated with greater Ki-67 reduction (R2 = .312, p = 0.024).
Conclusions
Brief treatment with erlotinib significantly decreased proliferation in HNSCC, with additive effect from sulindac. Efficacy studies of dual EGFR-COX inhibition are justified. pSrc is a potential resistance biomarker for anti-EGFR therapy, and warrants investigation as a molecular target.
Keywords: erlotinib, sulindac, phase 0, head and neck cancer, pSrc
Introduction
The epidermal growth factor receptor (EGFR), a transmembrane receptor tyrosine kinase (RTK) propagating proliferative and anti-apoptotic signals, is amplified and/or overexpressed in approximately 90% of head and neck squamous cell carcinoma (HNSCC).(1, 2) EGFR expression is higher in cancers driven by environmental carcinogens than by human papillomavirus (HPV)(3), and correlates with increased stage, reduced survival, and radioresistance.(1, 2, 4) EGFR signaling is also deregulated by HPV oncoproteins independent of EGFR expression level.(5) EGFR has been validated as a therapeutic target in phase III trials enrolling both HPV-positive and negative patients.(6, 7) Yet absolute benefit is low, and intrinsic resistance to the monoclonal antibody, cetuximab, or small molecule RTK inhibitors (gefitinib, erlotinib) is implied by low monotherapeutic response rates.(8, 9) Predictive biomarkers for sensitivity to EGFR inhibition represent a major unmet need in HNSCC. Activating mutations in the EGFR kinase domain, associated with clinical response in lung cancer, are rarely identified in HNSCC.(10) Furthermore, no alternative biomarker has been found.(11, 12)
In HNSCC, an important mechanism of EGFR activation is autocrine secretion of EGFR ligands. G protein-coupled receptors (GPCRs) transactivate EGFR by triggering proteolytic shedding of membrane-bound proligands.(13, 14) GPCR-EGFR crosstalk may represent a targetable upstream resistance mechanism to EGFR blockade.(15-17) Cyclooxygenase-2 (COX-2), an inflammatory enzyme which synthesizes the GPCR ligand PGE2, is upregulated in HNSCC and amenable to targeting by non-steroidal anti-inflammatory drugs (NSAIDs). Available NSAIDs, including the non-selective COX inhibitor sulindac, inhibit HNSCC cell growth in vitro, and suppress formation of preneoplastic lesions in a transgenic HNSCC model.(18) Concurrent EGFR-COX targeting demonstrates therapeutic synergism in HNSCC cell line and xenograft models.(17, 19, 20)
We conducted a randomized, double-blind, placebo-controlled window trial of erlotinib-sulindac, erlotinib or placebo-control in patients with HNSCC planned for definitive surgery. We analyzed paired, pre- and post-treatment tumor specimens for modulation of the Ki-67 proliferation index, to test the hypothesis that dual EGFR-COX targeting would potentiate anti-tumor effects relative to EGFR inhibition alone. We studied associated pharmacodynamic changes in EGFR-GPCR signaling intermediates, to identify predictive biomarkers for anti-EGFR therapy which may translate to the clinic.
Materials and Methods
Ethics Statement
This multicenter trial was approved by the Institutional Review Boards of the University of Pittsburgh (UP), Oregon Health Sciences University, and the Portland Veterans Administration Medical Center. The trial was nationally registered at clinicaltrials.gov (NCT01515137). All subjects provided written, informed consent.
Eligibility Criteria
Eligible subjects met the following key inclusion criteria: histologically confirmed, previously untreated HNSCC (Stage II-IVA) of the oral cavity, oropharynx, hypopharynx, or larynx, as defined by the American Joint Committee on Cancer Staging Handbook, 6th edition; planned complete resection of the primary tumor; age ≥18 years; Eastern Cooperative Oncology Group performance status 0-1; adequate hematologic, hepatic and renal function. Key exclusion criteria included: prior history of HNSCC; hypersensitivity to NSAIDs; interstitial lung disease; chronic use of NSAIDs or steroids.
Treatment
Subjects were randomized by the UP Biostatistics Facility to one of three neoadjuvant treatment groups: erlotinib (150 mg daily), erlotinib (150 mg daily) plus sulindac (150 mg twice daily), or placebo (for erlotinib) in a 5:5:3 ratio. There was no placebo for sulindac. Pre-operative therapy was administered in double-blind fashion for 7-14 days and discontinued 24-36 hours prior to surgery. Concomitant use of non-study NSAIDs or steroids was disallowed. Toxicities were described according to NCI Common Terminology Criteria for Adverse Events, version 3.0. Pre- and post-treatment tumor specimens were obtained at the time of diagnostic evaluation and definitive resection. Specimens were formalin-fixed and paraffin-embedded (FFPE), then shipped to UP for centralized analyses.
Statistical Methods
The reported window trial was originally nested in a single-arm, phase II study of adjuvant, open-label erlotinib following primary surgery and (chemo)radiation for locally advanced HNSCC. All subjects entering the parent trial were first randomized to one of three pre-operative treatment groups. After enrollment of 30 patients, the parent trial was discontinued due to non-feasibility.(21) At the time of discontinuation, and prior to analysis of biospecimens, the nested window trial was redesigned as an independent study. Patient allocation and randomization were unaffected. The primary endpoint was defined as change in the Ki-67 proliferation index (ΔKi-67) in pre and post-treatment tumor specimens, as validated in neoadjuvant breast cancer trials.(22, 23) We hypothesized differential ΔKi-67 according to treatment group. With 39 patients randomized 5:5:3, we had 92% power at α=0.05 for a two-sided Kruskal-Wallis test to detect an omnibus between-group difference of 1 log in ΔKi-67. Over-sampling was permitted to reach the target of 39 evaluable patients, defined as being randomized, undergoing neoadjuvant treatment, and providing at least one tissue specimen. We further hypothesized an ordering in Ki-67 down-modulation, with erlotinib-sulindac > erlotinib > placebo; an exact, two-sided Jonckheere-Terpstra test formally evaluated for this trend. Pairwise contrasts were evaluated by two-sided Wilcoxon tests. Exact two-sided Kruskal-Wallis tests evaluated for randomization imbalances. For the observed randomization imbalance in baseline Ki-67 (Kruskal-Wallis, p = 0.022), we used analysis of covariance to model the within-group association between baseline and change. Using adjusted values from this model we verified that the baseline imbalance did not confound analysis of treatment group differences in ΔKi-67 (Supplemental Figure 1). Thus, the presented analysis is based upon observed data.
Secondary endpoints included modulation of 25 candidate protein biomarkers, including 21 empirically selected from pre-clinical signaling models and 4 from mass spectrometry (MS) discovery. Prior to analysis in February 2013, we proposed two biomarker tiers, with specialized alpha spending for priority, hypothesis-driven analytes. For tests of trend, we designated three priority analytes: ΔpSrc, ΔpAkt, and ΔpSTAT.(17, 24-28) We hypothesized that phosphorylated forms of these key EGFR signaling nodes mediated short-term changes in proliferative signaling. We used the Jonckheere-Terpstra statistic to test the hypothesis that a valid biomarker would recapitulate the ordering of Ki-67 down-modulation, that is, erlotinib-sulindac > erlotinib > placebo. For analyses of covariance, we defined four priority baseline proteins which could mechanistically explain resistance to Ki-67 modulation: pSrc, pAkt, pSTAT and COX-2. We first estimated flexible parametric regression models to evaluate the association of each baseline protein with ΔKi-67 across treatment groups. Significant baseline proteins were then tested for treatment group differences with analysis of covariance. For priority analytes, significance was set at α=0.05. The remaining analytes were exploratory; multiple testing was corrected for false discovery by the method of Benjamini and Hochberg.(29)
Specimen Analysis
The primary method for analyzing biomarker response was immunohistochemical (IHC) analysis of tissue microarrays (TMA). Using guiding H&E staining from FFPE blocks, 0.6 mm cores were transferred in triplicate from each pre- and post-treatment block to a blank recipient block. TMAs were assembled and stained after all tumor specimens had been submitted to minimize technical variation.
Pharmacodynamic modulation of Ki-67 and candidate biomarkers was evaluated by IHC of the TMA. Antibodies, clones, company, dilution and retrieval technique are summarized in Supplemental Table 1. Biomarkers, including Ki-67, were scored quantitatively with Aperio computer-assisted digital analysis by the research pathologist (LW), who was blinded to patient identity, treatment assignment, and specimen time-point. In keeping with international consensus guidelines for Ki-67, at least 5000 tumor cells/specimen were counted. The percentage of positive tumor cells was represented as the proliferation index.(30) Tumors were stained for p16, a recognized surrogate for HPV in the oropharynx, and classified as p16(+) if ≥ 70% of neoplastic cells demonstrated strong and diffuse nuclear and cytoplasmic staining.(31)
Mass Spectrometry
As an unbiased source of biomarker discovery, 10 post-treatment specimens (5 erlotinib-treated, 5 placebo-treated) were evaluated by MS for differential protein expression. Tissues were prepared and digested with trypsin as described.(32) A false peptide discovery rate of less than 2% was determined by searching the primary tandem MS data using the same criteria against a decoy database.(33) Differences in protein abundance were derived by spectral counting.(34) 7390 proteins were filtered for low variability; the remaining 610 proteins were analyzed by Wilcoxon test for differential expression. Among 73 proteins with an unadjusted p-value < 0.10, four had a commercially available antibody for IHC, and were analyzed in trial specimens: gelsolin, calreticulin, desmoglein and GAPDH.
PIK3CA Mutation Testing
DNA was isolated from tumor cores and tested for mutations in exons 9 and 20 of the PIK3CA gene as described.(35)
Results
Enrollment and Baseline Characteristics
Between December 2005 and December 2008, 47 subjects enrolled across three study centers in order to meet the evaluable target of 39. Patient allocation is presented in the Consort Diagram (Figure 1). Baseline characteristics for the 46 subjects who received at least one dose of neoadjuvant study drug are summarized in Table 1. Subjects were well-balanced among groups with respect to age, gender, disease site, stage, and p16 status. This was a largely HPV-negative cohort; only 2 of 14 oropharynx tumors were p16(+).
Figure 1.

CONSORT diagram. This flow chart depicts the number of patients who signed consent, were randomized and treated, and ultimately provided paired tumor specimens of sufficient quality for biomarker analysis.
Table 1. Subject Characteristics by Treatment Group.
| Placebo (N = 12) |
Erlotinib (N = 18) |
Erlotinib + Sulindac (N = 16) |
Test of Equality1 | ||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Age | |||||||
| Median | 59 | 64 | 49 | .8976 | |||
| range | 33 - 86 | 44 -73 | 49 - 68 | ||||
|
| |||||||
| N | % | N | % | N | % | ||
|
| |||||||
| Gender | |||||||
| Female | 3 | 25 | 4 | 22 | 3 | 19 | .9924 |
| Male | 9 | 75 | 14 | 78 | 13 | 81 | |
|
| |||||||
| Tumor Site | |||||||
| Oral Cavity | 8 | 67 | 9 | 50 | 9 | 56 | .6606 |
| Oropharynx | 3 | 35 | 5 | 28 | 6 | 38 | |
| Hypopharynx | 0 | 0 | 2 | 11 | 0 | 0 | |
| Larynx | 1 | 8 | 2 | 11 | 1 | 6 | |
|
| |||||||
| Tumor (T) Stage2 | |||||||
| 1 | 1 | 9 | 0 | 0 | 0 | 0 | .2371 |
| 2 | 5 | 45 | 3 | 18 | 3 | 20 | |
| 3 | 3 | 28 | 4 | 24 | 5 | 33 | |
| 4 | 2 | 18 | 10 | 59 | 7 | 47 | |
|
| |||||||
| Nodal (N) Stage3 | |||||||
| 0 | 7 | 70 | 8 | 44 | 6 | 40 | .3389 |
| 1 | 1 | 10 | 4 | 22 | 6 | 40 | |
| 2 | 2 | 20 | 6 | 33 | 2 | 13 | |
| 3 | 0 | 0 | 0 | 0 | 1 | 7 | |
|
| |||||||
| p16 Status4 | |||||||
| Positive | 1 | 11 | 3 | 20 | 0 | 0 | .3579 |
| Negative | 10 | 89 | 12 | 80 | 11 | 100 | |
Wilcoxon Test for age, Fisher's exact two-tailed test for others
T classification was not available for 3 subjects.
N classification was not available for 3 subjects.
p16 status was not available for 9 patients. Among 14 oropharynx cases, 11 were p16(-), 2 were p16(+), and 1 was unknown.
Toxicity
Brief exposure to erlotinib, erlotinib-sulindac, or placebo was well tolerated in the pre-operative setting. Clinically significant toxicities attributed to study treatment are summarized in Table 2. Adverse events represented typical class toxicities for EGFR inhibitors, including rash and diarrhea.(36) One patient discontinued study treatment for grade 2 anxiety, and one required erlotinib dose-reduction for grade 2 mucositis. Median hospitalization for surgery was 9 days. No unusual rate or type of post-operative complication was observed; complications included fistula (2), wound infection (2), free flap necrosis (1), prolonged intubation (2), infection outside the surgical field (2) and bleeding (1).
Table 2. Toxicities.
| Nonhematologic Toxicity | Grade 1 | Grade 2 | Grade 3 | Grade 4 | Total Grade ≥ 3 |
|---|---|---|---|---|---|
| Allergy/Immunology | |||||
| Hypersensitivity | 1 (2%) | 0 | 0 | 0 | 0 |
| Constitutional | |||||
| Fatigue | 3 (7%) | 2 (4%) | 1 (2%) | 0 | 1 (2%) |
| Dermatologic | |||||
| Rash/desquamation | 11 (24%) | 5 (11%) | 2 (4%) | 0 | 2 (4%) |
| Gastrointestinal | |||||
| Anorexia | 0 | 1 (2%) | 0 | 0 | 0 |
| Diarrhea | 4 (9%) | 1 (2%) | 0 | 0 | 0 |
| Mucositis/Stomatitis | 1 (2%) | 1 (2%) | 0 | 0 | 0 |
| Nausea | 2 (4%) | 0 | 0 | 0 | 0 |
| Neurologic | |||||
| Anxiety | 0 | 1 (2%) | 0 | 0 | 0 |
Biomarker Modulation
Evaluable tissue for analysis of at least one paired-specimen biomarker was available from 34 of 39 patients (87%); and for at least one baseline protein was available from 35 of 39 patients (90%). Depending upon the biomarker assayed, 49 - 92% of the 34 paired specimens (median 78%) had measurable protein in both samples.
Ki-67
Twenty-seven patients (69%) had measurable Ki-67 in both samples. The primary, omnibus hypothesis test demonstrated a significant between-group difference in ΔKi-67 (Kruskal-Wallis, two-sided, p=0.04). Box-plots depicting treatment group medians and inter-quartile ranges are presented in Figure 2A. There was no change in Ki-67 attributable to placebo. As compared to placebo, Ki-67 was significantly modulated by erlotinib (p=0.04) or erlotinib-sulindac (p=0.03). There was a significant trend in ordering of Ki-67 down-modulation: erlotinib-sulindac > erlotinib > placebo (Exact Jonckheere-Terpstra, two-sided, p=0.02), indicating additive anti-proliferative effect from sulindac. A waterfall plot depicting per-patient percent change in Ki-67 is shown in Figure 2B.
Figure 2.
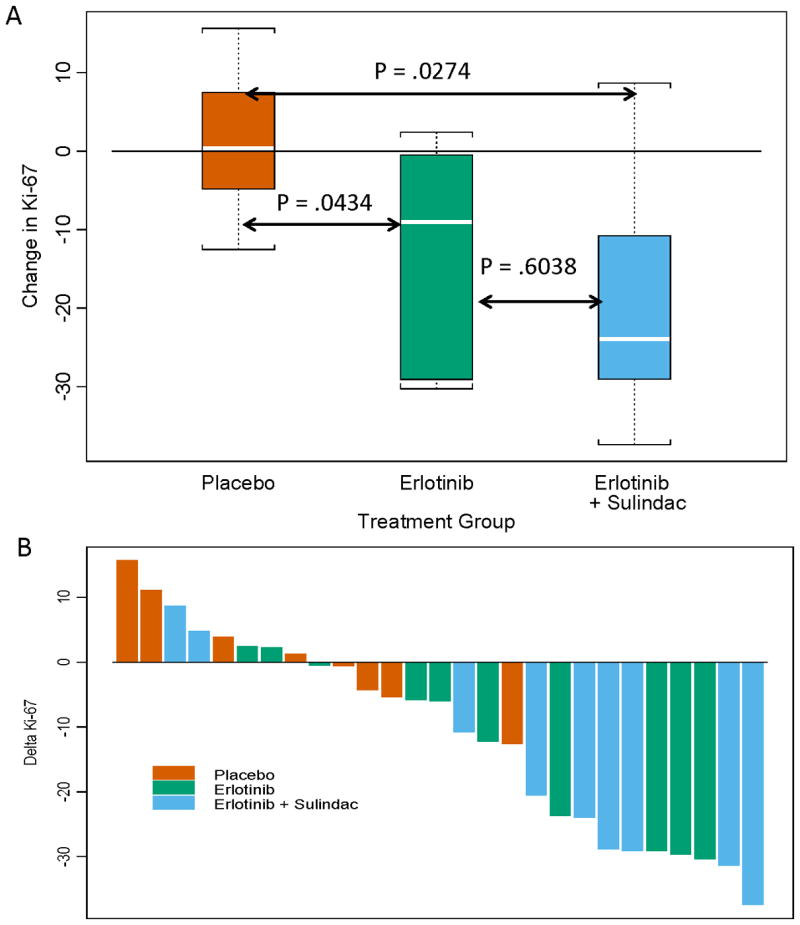
The Primary Endpoint, Ki-67 Proliferation Index. (A) The primary omnibus hypothesis demonstrated a significant difference in Ki-67 modulation among groups (two-sided Kruskal-Wallis, p=0.04). Depicted in this box and whisker plot are the Wilcoxon pairwise contrasts confirming a significant decrease in Ki-67 for patients treated with either erlotinib or erlotinib-sulindac vs. placebo. Box and whisker plots consist of the median (white line), the inter-quartile range (box) and the distance to observed values within 1.5 times the inter-quartile range (whiskers). (B) A waterfall plot depicts the intra- patient change in tumoral Ki-67 expression, pre and post-treatment. Bars are color-coded according to treatment arm.
Biomarker Intermediates
We hypothesized that pSrc, pAkt, or pSTAT mediated the significant reduction in Ki-67 by erlotinib or erlotinib-sulindac. If valid signaling intermediates of reduced proliferation, changes in these proteins would directly correlate with the ordering of ΔKi-67. As shown in Figures 3A and 3B, none of the three priority analytes, nor 22 exploratory biomarkers, demonstrated a significant ordering effect.
Figure 3.
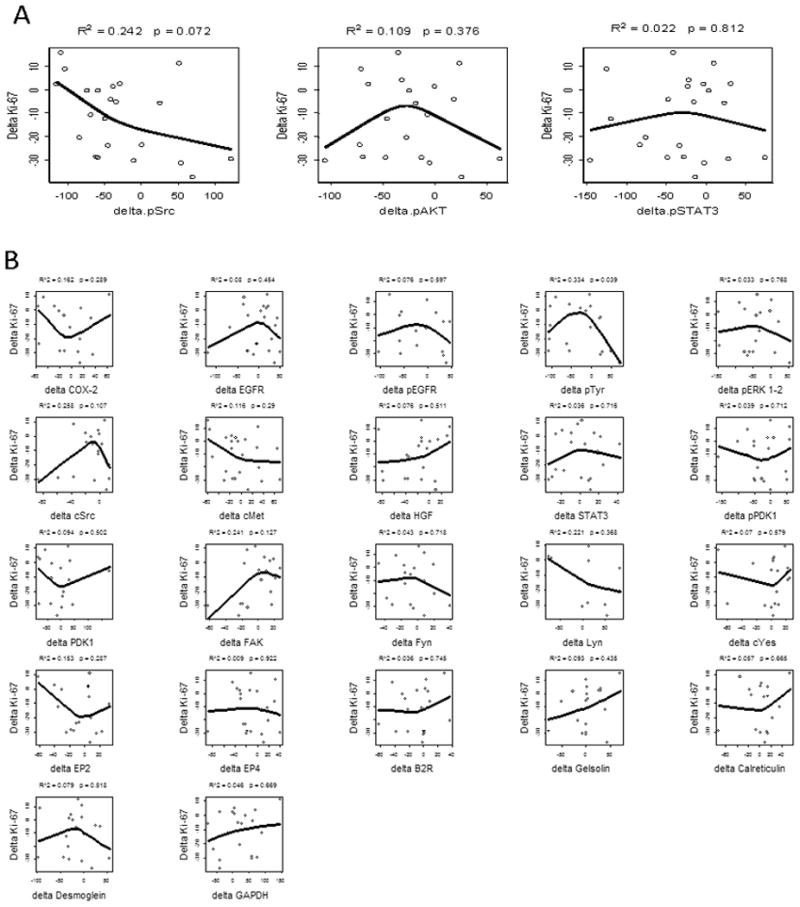
Biomarker Intermediates and ΔKi-67. We hypothesized that constituents of the EGFR or COX-2 signaling pathways may mediate the observed change in proliferation. The line represents restricted cubic-spline fit for the relationship between Δanalyte and ΔKi-67. No significant correlation was identified between ΔKi-67 and the priority analytes, ΔpSTAT3, ΔpSrc, or ΔpAkt (A) or 21 other candidate biomarkers (B).
Resistance Biomarkers
We hypothesized that increased baseline pSrc, pAkt, pSTAT or COX-2 might mediate resistance to Ki-67 modulation. We first evaluated the association between baseline expression of these candidate resistance biomarkers and ΔKi-67, independent of randomized treatment assignment (Figure 4A). Higher pre-treatment pSrc was associated with a smaller decrease in Ki-67 (R2 =0.3, p = 0.04). To understand whether these findings might represent erlotinib resistance, we plotted pSrc against ΔKi-67 within each treatment group. As displayed in Figure 4B, resistance to Ki-67 modulation was demonstrated only in the active treatment groups (tests that slopes differ from 0: placebo, p=0.8775; erlotinib, p=0.0024; erlotinib-sulindac, p=0.0150). Collectively, these findings implicate high baseline pSrc expression as a resistance biomarker to erlotinib, irrespective of co-treatment with sulindac. No significant association between baseline expression of 21 exploratory biomarkers and Ki-67 modulation was discovered (Supplemental Figure 2).
Figure 4.
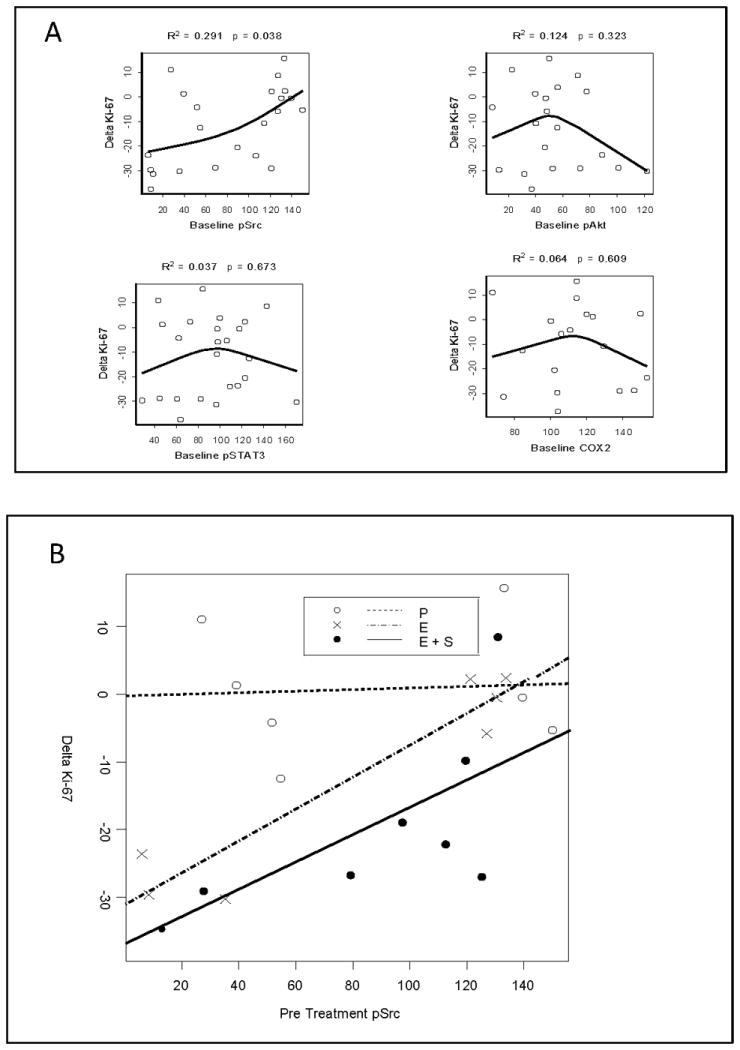
Baseline pSrc and ΔKi-67. (A) Four priority baseline analytes (pSrc, pAkt, pSTAT3 and COX-2) were evaluated for association with Ki-67 modulation, with the line representing restricted cubic-spline fit. Higher pre-treatment pSrc was associated with a smaller decrease in Ki-67 following neoadjuvant treatment. (B) Baseline pSrc was plotted against ΔKi-67 for each color-coded treatment group. A significant association was noted only in the active treatment groups (tests that slopes differ from 0: placebo, p=0.8775; erlotinib, p=0 .0024; erlotinib-sulindac, p=0.0150), forwarding baseline pSrc as a candidate biomarker of erlotinib resistance.
PIK3CA Mutations
Because PIK3CA mutations are associated with chemopreventive benefit from aspirin in patients with colorectal cancer(37), we conducted a post hoc correlation between PIK3CA mutation status and ΔKi-67 in the erlotinib-sulindac arm. Two of 9 patients harbored non-common exon 9 mutations (p.E522K, c.1564G>A; p.A533V, c.1598C>T AND p.I543I, c.1629C>T) while one bore a canonical exon 20 mutation (H1047L, c.3140 A>T). There was no relationship between mutation status and ΔKi-67 (data not shown).
Discussion
While EGFR has been validated as the first molecular target in HNSCC, absolute improvement in a clinically reliable endpoint following exposure to an EGFR inhibitor is limited to 10-20% of patients, implicating intrinsic resistance despite EGFR over-expression in the vast majority.(6, 7, 9) Predictive biomarkers for anti-EGFR therapy in HNSCC represent a major unmet need. The current trial took advantage of a window design to investigate mechanistic signaling hypotheses regarding response and resistance to short-term erlotinib, with or without sulindac, in HNSCC. The study met its primary endpoint. First, we observed differential down-modulation of the Ki-67 proliferation index across treatment groups, attributable to erlotinib or erlotinib-sulindac as compared to placebo. Second, we confirmed that sulindac potentiates the anti-proliferative effect of erlotinib in a formal test of trend, indicating that the forward feedback loop between COX-2 and EGFR is a relevant clinical target. Finally, we identified baseline pSrc expression as a potential resistance biomarker to erlotinib. Study results were strengthened by incorporation of a placebo-control. Lack of significant change in Ki-67 in placebo-treated patients raised confidence in its validity as a primary biomarker endpoint. Conversely, recognition of background changes in GPCR-EGFR signaling intermediates in placebo-treated patients avoided over-stated mechanistic conclusions.
Ki-67 is a nuclear non-histone protein expressed in proliferating human tissue.(38) While recognized as a poor prognostic marker in breast cancer, the Ki-67 proliferation index is inconsistently prognostic in HNSCC.(39, 40) ΔKi-67 is a validated surrogate biomarker in neoadjuvant studies of targeted therapy in breast cancer(22, 23), however relevance to HNSCC window trials is unknown as Ki-67 modulation has not been evaluated directly against clinical outcome.(41-43) Here, the non-feasibility of adjuvant erlotinib in the parent trial precluded our planned correlation of ΔKi-67 and 3-year PFS. An alternate surrogate biomarker, the TUNEL apoptotic index, was evaluated in a randomized window trial of lapatinib vs. placebo in HNSCC.(41) Although apoptosis was increased by lapatinib, the increase was not significant against placebo, underscoring the value of placebo-control in short-term biomarker modulation studies. Notably, the Ki-67 proliferation index was significantly decreased by lapatinib vs. placebo, in line with our results. Collectively, these data suggest that proliferation is a more robust short-term endpoint than apoptosis for assessing response to targeted therapy in HNSCC.
The major secondary objective of this window study was identification of GPCR-EGFR signaling intermediates responsible for pharmacodynamic change in Ki-67. However, among 25 protein candidates, none demonstrated an ordering effect consistent with ΔKi-67. Preclinical HNSCC models demonstrate at-least additive downregulation of multiple phospho-proteins (eg. pSrc, pAkt, and pSTAT3) by GPCR-EGFR co-inhibition relative to EGFR inhibition alone.(17, 20) Our hypothesis test requiring linear ordering of valid intermediates may not reflect in vivo complexity of the GPCR-EGFR signaling network. The candidate biomarker approach was further limited by availability of validated commercial antibodies. Of note, multiple signaling proteins changed significantly in placebo-treated patients, raising questions regarding tissue heterogeneity, stability of signaling patterns during placebo treatment, and assay reliability. While we attempted to mitigate these variables by standardized tissue collection/processing, single-batch antibody staining and blinded, centralized review, methodologic limitations for quantifying protein expression changes must be emphasized. This parallels our recent report demonstrating no significant differences between treatment and placebo groups in serum protein expression from this trial.(44) As multiplex proteomics technologies are piloted for pharmacodynamic assessment, placebo-controlled data will be critical to avoiding Type I error.
The paucity of predictive biomarkers for EGFR targeting in HNSCC impairs selection of patients who will benefit, and redirection of those who will not. This study identified high baseline pSrc expression as a potential resistance biomarker for erlotinib, an observation consistent with mechanistic preclinical data. Src family kinases are activated in response to EGFR signaling.(13, 45) GPCRs also activate pSrc upstream of EGFR, recruiting pSrc to the complex mediating EGFR transactivation.(17) Finally, pSrc drives ligand-independent activation of cMet, a major resistance mechanism to erlotinib in HNSCC models.(46, 47) Because sulindac reduces PGE2-GPCR activation, and as a consequence pSrc(17), we hypothesized that the association between pSrc and resistance to Ki-67 modulation would be muted in the sulindac group. This was not the case, suggesting that baseline pSrc is not driven dominantly by PGE2, but the convergent influence of multiple GPCR ligands and accessory RTKs. Contrary to our hypothesis, no other GPCR-EGFR signaling protein was associated with ΔKi-67, including the PI3K/Akt and STAT3 resistance nodes described in preclinical models.(17, 20) Although we recently reported that PIK3CA mutations are associated with erlotinib resistance in HNSCC cell lines(48), this has not been observed in HNSCC clinical cohorts. Here, PIK3CA mutation status in the erlotinib-sulindac arm did not correlate with ΔKi-67; this post-hoc finding warrants cautious interpretation due to small sample size and the potential interaction between genomic activation of PIK3CA and NSAIDs.(37)
This study has several important limitations. First, the CONSORT diagram (Figure 1) highlights the challenges in executing window studies. Although 35 of 39 patients had sufficient quality tissue for analysis of one biomarker, only 27 (69%) pairs were analyzable for the primary endpoint. Second, a sulindac-alone arm was not incorporated. This decision was based upon two factors: 1) EGFR is an established therapeutic target in HNSCC whereas NSAIDs alone have not proven effective; 2) our biomarker hypothesis emphasized the potentiation of EGFR inhibition by sulindac, in the setting of GPCR-EGFR transactivation. Third, a potential criticism is use of Ki-67 IHC as the primary endpoint, given the observed heterogeneity of tumor protein expression and unknown reliability of many IHC antibodies. Although Ki-67 is subject to variability from pre-analytic processing and inter-observer scoring, the MIB1 antibody has been well-validated, is tolerant of a range of fixation times, demonstrates durable antigenicity, and is the subject of international consensus standards followed here.(30) Finally, the absence of a clinical endpoint limits interpretation of our mechanistic findings. Correlation of ΔKi-67 to radiologic response or disease outcome will be critical to validating Ki-67 modulation as a short-term biomarker endpoint in HNSCC window trials.
Despite acknowledged limitations, we affirmed that erlotinib significantly reduced proliferation in operable HNSCC after short-term exposure, an effect potentiated by sulindac. Efficacy studies evaluating dual EGFR-COX-2 targeting are justified, particularly in light of a phase I clinical trial of gefitinib and celecoxib, demonstrating a 22% response rate in recurrent/metastatic HNSCC.(49) Such a strategy may be more fruitful in patients with low tumoral pSrc expression, based upon our identification of baseline pSrc as a candidate resistance biomarker for erlotinib. Given pSrc's central role in activating compensatory pathways, pSrc also warrants investigation as a bona fide co-target in HNSCC. Although the Src family kinase inhibitor, dasatinib, demonstrated limited single agent activity in HNSCC(50), dual EGFR-Src inhibition remains of interest. Two translational studies are ongoing in HNSCC, including the combination of erlotinib-dasatinib in the window setting, and cetuximab-dasatinib in the recurrent/metastatic setting (NCT00779389; NCT01488318).
Supplementary Material
Translational Relevance.
The epidermal growth factor receptor (EGFR) and cyclooxygenase-2 (COX-2) pathways are upregulated in head and neck squamous cell carcinoma (HNSCC). Crosstalk potentiates growth, proliferation and invasion. Preclinical models indicate synergistic anti-tumor activity from dual blockade. We conducted a phase 0 trial of erlotinib, an EGFR inhibitor; erlotinib plus sulindac, a non-selective COX inhibitor; vs. placebo in operable HNSCC in order to mechanistically characterize dual targeting. We demonstrated significant down-modulation of the tumor Ki-67 proliferation index in both groups treated with erlotinib, and a formal test of trend evidenced potentiation by sulindac. Further, we identified baseline pSrc expression as a candidate biomarker of erlotinib resistance in the clinic. This window trial provides the first in-human mechanistic data to justify efficacy trials of dual EGFR-COX inhibition in HNSCC. pSrc warrants investigation as a resistance biomarker for EGFR inhibition, and is now under investigation as a bona fide molecular target in HNSCC.
Acknowledgments
Financial support: This work was supported by grants from the National Institutes of Health [R01CA098372; the UPCI Head and Neck Cancer SPORE (P50CA097190), the UPCI Cancer Center Support Grant (P30CA47904)]; the American Cancer Society Distinguished Professorship (to JRG); and OSI Pharmaceuticals.
Footnotes
Authors declare no conflict of interest for the submitted work.
References
- 1.Rubin Grandis J, Melhem MF, Gooding WE, Day R, Holst VA, Wagener MM, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–32. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 2.Chung CH, Ely K, McGavran L, Varella-Garcia M, Parker J, Parker N, et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J Clin Oncol. 2006;24:4170–6. doi: 10.1200/JCO.2006.07.2587. [DOI] [PubMed] [Google Scholar]
- 3.Troy JD, Weissfeld JL, Youk AO, Thomas S, Wang L, Grandis JR. Expression of EGFR, VEGF, and NOTCH1 Suggest Differences in Tumor Angiogenesis in HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma. Head Neck Pathol. 2013;7:344–55. doi: 10.1007/s12105-013-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ang KK, Berkey BA, Tu X, Zhang HZ, Katz R, Hammond EH, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–6. [PubMed] [Google Scholar]
- 5.Spangle JM, Munger K. The HPV16 E6 oncoprotein causes prolonged receptor protein tyrosine kinase signaling and enhances internalization of phosphorylated receptor species. PLoS Pathog. 2013;9:e1003237. doi: 10.1371/journal.ppat.1003237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med. 2008;359:1116–27. doi: 10.1056/NEJMoa0802656. [DOI] [PubMed] [Google Scholar]
- 7.Bonner JA, Harari PM, Giralt J, Azarnia N, Shin DM, Cohen RB, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–78. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 8.Perez CA, Song H, Raez LE, Agulnik M, Grushko TA, Dekker A, et al. Phase II study of gefitinib adaptive dose escalation to skin toxicity in recurrent or metastatic squamous cell carcinoma of the head and neck. Oral Oncol. 2012;48:887–92. doi: 10.1016/j.oraloncology.2012.03.020. [DOI] [PubMed] [Google Scholar]
- 9.Vermorken JB, Herbst RS, Leon X, Amellal N, Baselga J. Overview of the efficacy of cetuximab in recurrent and/or metastatic squamous cell carcinoma of the head and neck in patients who previously failed platinum-based therapies. Cancer. 2008;112:2710–9. doi: 10.1002/cncr.23442. [DOI] [PubMed] [Google Scholar]
- 10.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Licitra L, Mesia R, Rivera F, Remenar E, Hitt R, Erfan J, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first-line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol. 2011;22:1078–87. doi: 10.1093/annonc/mdq588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Licitra L, Storkel S, Kerr KM, Van Cutsem E, Pirker R, Hirsch FR, et al. Predictive value of epidermal growth factor receptor expression for first-line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer. 2013;49:1161–8. doi: 10.1016/j.ejca.2012.11.018. [DOI] [PubMed] [Google Scholar]
- 13.Zhang Q, Thomas SM, Xi S, Smithgall TE, Siegfried JM, Kamens J, et al. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64:6166–73. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Thomas SM, Lui VW, Xi S, Siegfried JM, Fan H, et al. Phosphorylation of TNF-alpha converting enzyme by gastrin-releasing peptide induces amphiregulin release and EGF receptor activation. Proc Natl Acad Sci U S A. 2006;103:6901–6. doi: 10.1073/pnas.0509719103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gschwind A, Prenzel N, Ullrich A. Lysophosphatidic acid-induced squamous cell carcinoma cell proliferation and motility involves epidermal growth factor receptor signal transactivation. Cancer Res. 2002;62:6329–36. [PubMed] [Google Scholar]
- 16.Gschwind A, Hart S, Fischer OM, Ullrich A. TACE cleavage of proamphiregulin regulates GPCR-induced proliferation and motility of cancer cells. Embo J. 2003;22:2411–21. doi: 10.1093/emboj/cdg231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas SM, Bhola NE, Zhang Q, Contrucci SC, Wentzel AL, Freilino ML, et al. Cross-talk between G protein-coupled receptor and epidermal growth factor receptor signaling pathways contributes to growth and invasion of head and neck squamous cell carcinoma. Cancer Res. 2006;66:11831–9. doi: 10.1158/0008-5472.CAN-06-2876. [DOI] [PubMed] [Google Scholar]
- 18.Zhang W, Bhola N, Kalyankrishna S, Gooding W, Hunt J, Seethala R, et al. Kinin b2 receptor mediates induction of cyclooxygenase-2 and is overexpressed in head and neck squamous cell carcinomas. Mol Cancer Res. 2008;6:1946–56. doi: 10.1158/1541-7786.MCR-07-2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moraitis D, Du B, De Lorenzo MS, Boyle JO, Weksler BB, Cohen EG, et al. Levels of cyclooxygenase-2 are increased in the oral mucosa of smokers: evidence for the role of epidermal growth factor receptor and its ligands. Cancer Res. 2005;65:664–70. [PubMed] [Google Scholar]
- 20.Chen Z, Zhang X, Li M, Wang Z, Wieand HS, Grandis JR, et al. Simultaneously targeting epidermal growth factor receptor tyrosine kinase and cyclooxygenase-2, an efficient approach to inhibition of squamous cell carcinoma of the head and neck. Clin Cancer Res. 2004;10:5930–9. doi: 10.1158/1078-0432.CCR-03-0677. [DOI] [PubMed] [Google Scholar]
- 21.Grandis JR, Lai SY, Kim S, Ferris RL, Gibson MK, Johnson R, et al. Erlotinib as adjuvant treatment for patients with resected head and neck squamous cell carcinoma with evaluation of neoadjuvant biomarker modulation with erlotinib with or without sulindac: a feasibility report. J Clin Oncol. 2008;26(15S):6037. [Google Scholar]
- 22.Baselga J, Semiglazov V, van Dam P, Manikhas A, Bellet M, Mayordomo J, et al. Phase II randomized study of neoadjuvant everolimus plus letrozole compared with placebo plus letrozole in patients with estrogen receptor-positive breast cancer. J Clin Oncol. 2009;27:2630–7. doi: 10.1200/JCO.2008.18.8391. [DOI] [PubMed] [Google Scholar]
- 23.Smith IE, Walsh G, Skene A, Llombart A, Mayordomo JI, Detre S, et al. A phase II placebo-controlled trial of neoadjuvant anastrozole alone or with gefitinib in early breast cancer. J Clin Oncol. 2007;25:3816–22. doi: 10.1200/JCO.2006.09.6578. [DOI] [PubMed] [Google Scholar]
- 24.Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses Garcia A, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964–73. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- 25.Bonner JA, Yang ES, Trummell HQ, Nowsheen S, Willey CD, Raisch KP. Inhibition of STAT-3 results in greater cetuximab sensitivity in head and neck squamous cell carcinoma. Radiother Oncol. 2011;99:339–43. doi: 10.1016/j.radonc.2011.05.070. [DOI] [PubMed] [Google Scholar]
- 26.Leeman-Neill RJ, Seethala RR, Singh SV, Freilino ML, Bednash JS, Thomas SM, et al. Inhibition of EGFR-STAT3 signaling with erlotinib prevents carcinogenesis in a chemically-induced mouse model of oral squamous cell carcinoma. Cancer Prev Res (Phila) 2011;4:230–7. doi: 10.1158/1940-6207.CAPR-10-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kijima T, Niwa H, Steinman RA, Drenning SD, Gooding WE, Wentzel AL, et al. STAT3 activation abrogates growth factor dependence and contributes to head and neck squamous cell carcinoma tumor growth in vivo. Cell Growth Differ. 2002;13:355–62. [PubMed] [Google Scholar]
- 28.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, et al. Loss of PTEN/MMAC1/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–22. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 29.Hochberg Y, Benjamini Y. More powerful procedures for multiple significance testing. Stat Med. 1990;9:811–8. doi: 10.1002/sim.4780090710. [DOI] [PubMed] [Google Scholar]
- 30.Dowsett M, Nielsen TO, A'Hern R, Bartlett J, Coombes RC, Cuzick J, et al. Assessment of Ki67 in breast cancer: recommendations from the International Ki67 in Breast Cancer working group. J Natl Cancer Inst. 2011;103:1656–64. doi: 10.1093/jnci/djr393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jordan RC, Lingen MW, Perez-Ordonez B, He X, Pickard R, Koluder M, et al. Validation of methods for oropharyngeal cancer HPV status determination in US cooperative group trials. Am J Surg Pathol. 2012;36:945–54. doi: 10.1097/PAS.0b013e318253a2d1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alkhas A, Hood BL, Oliver K, Teng PN, Oliver J, Mitchell D, et al. Standardization of a sample preparation and analytical workflow for proteomics of archival endometrial cancer tissue. J Proteome Res. 2011;10:5264–71. doi: 10.1021/pr2007736. [DOI] [PubMed] [Google Scholar]
- 33.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 34.Liu H, Sadygov RG, Yates JR., 3rd A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 35.Chiosea SI, Grandis JR, Lui VW, Diergaarde B, Maxwell JH, Ferris RL, et al. PIK3CA, HRAS and PTEN in human papillomavirus positive oropharyngeal squamous cell carcinoma. BMC Cancer. 2013;13:602. doi: 10.1186/1471-2407-13-602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shepherd FA, Rodrigues Pereira J, Ciuleanu T, Tan EH, Hirsh V, Thongprasert S, et al. Erlotinib in previously treated non-small-cell lung cancer. N Engl J Med. 2005;353:123–32. doi: 10.1056/NEJMoa050753. [DOI] [PubMed] [Google Scholar]
- 37.Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012;367:1596–606. doi: 10.1056/NEJMoa1207756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerdes J, Li L, Schlueter C, Duchrow M, Wohlenberg C, Gerlach C, et al. Immunobiochemical and molecular biologic characterization of the cell proliferation-associated nuclear antigen that is defined by monoclonal antibody Ki-67. Am J Pathol. 1991;138:867–73. [PMC free article] [PubMed] [Google Scholar]
- 39.Liu M, Lawson G, Delos M, Jamart J, Ide C, Coche E, et al. Predictive value of the fraction of cancer cells immunolabeled for proliferating cell nuclear antigen or Ki67 in biopsies of head and neck carcinomas to identify lymph node metastasis: comparison with clinical and radiologic examinations. Head Neck. 2003;25:280–8. doi: 10.1002/hed.10218. [DOI] [PubMed] [Google Scholar]
- 40.Xie X, De Angelis P, Clausen OP, Boysen M. Prognostic significance of proliferative and apoptotic markers in oral tongue squamous cell carcinomas. Oral Oncol. 1999;35:502–9. doi: 10.1016/s1368-8375(99)00024-x. [DOI] [PubMed] [Google Scholar]
- 41.Del Campo JM, Hitt R, Sebastian P, Carracedo C, Lokanatha D, Bourhis J, et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy-naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer. 2011;105:618–27. doi: 10.1038/bjc.2011.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sen M, Thomas SM, Kim S, Yeh JI, Ferris RL, Johnson JT, et al. First-in-human trial of a STAT3 decoy oligonucleotide in head and neck tumors: implications for cancer therapy. Cancer Discov. 2012;2:694–705. doi: 10.1158/2159-8290.CD-12-0191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thomas F, Rochaix P, Benlyazid A, Sarini J, Rives M, Lefebvre JL, et al. Pilot study of neoadjuvant treatment with erlotinib in nonmetastatic head and neck squamous cell carcinoma. Clin Cancer Res. 2007;13:7086–92. doi: 10.1158/1078-0432.CCR-07-1370. [DOI] [PubMed] [Google Scholar]
- 44.Moskowitz HS, Gooding WE, Thomas SM, Freilino ML, Gross N, Argiris A, et al. Serum biomarker modulation following molecular targeting of epidermal growth factor and cyclooxygenase pathways: A pilot randomized trial in head and neck cancer. Oral Oncol. 2012;48:1136–45. doi: 10.1016/j.oraloncology.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xi S, Zhang Q, Dyer KF, Lerner EC, Smithgall TE, Gooding WE, et al. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278:31574–83. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 46.Sen B, Peng S, Saigal B, Williams MD, Johnson FM. Distinct interactions between c-Src and c-Met in mediating resistance to c-Src inhibition in head and neck cancer. Clin Cancer Res. 2011;17:514–24. doi: 10.1158/1078-0432.CCR-10-1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stabile LP, He G, Lui VW, Henry C, Gubish CT, Joyce S, et al. c-Src activation mediates erlotinib resistance in head and neck cancer by stimulating c-Met. Clin Cancer Res. 2013;19:380–92. doi: 10.1158/1078-0432.CCR-12-1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li H, Wawrose JS, Gooding WE, Garraway LA, Lui VW, Peyser ND, et al. Molecular Profiling of HNSCC Cells and Tumors Reveals a Rational Approach to Preclinical Model Selection. Mol Cancer Res. 2014 Jan 14; doi: 10.1158/1541-7786.MCR-13-0396. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wirth LJ, Haddad RI, Lindeman NI, Zhao X, Lee JC, Joshi VA, et al. Phase I study of gefitinib plus celecoxib in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2005;23:6976–81. doi: 10.1200/JCO.2005.02.4182. [DOI] [PubMed] [Google Scholar]
- 50.Brooks HD, Glisson BS, Bekele BN, Johnson FM, Ginsberg LE, El-Naggar A, et al. Phase 2 study of dasatinib in the treatment of head and neck squamous cell carcinoma. Cancer. 2011;117:2112–9. doi: 10.1002/cncr.25769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.