

Abstract
Waves of cyclin synthesis and degradation regulate the activity of Cdc2 protein kinase during the cell cycle. Cdc2 inactivation by Wee1B-mediated phosphorylation is necessary for arrest of the oocyte at G2-prophase, but it is unclear whether this regulation functions later during the metaphase to anaphase transition. We show that reactivation of a Wee1B pathway triggers the decrease in Cdc2 activity during egg activation. When Wee1B is downregulated, oocytes fail to form a pronucleus in response to Ca2+ signals. Calcium-calmodulin-dependent kinase II (CaMKII) activates Wee1B, and CaMKII-driven exit from metaphase II (MII) is inhibited by Wee1B downregulation, demonstrating that exit from metaphase requires not only a proteolytic degradation of cyclin B, but also the inhibitory phosphorylation of Cdc2 by Wee1B.
Mature oocytes are arrested in metaphase of the second meiosis (MII). Fertilization triggers a series of rapid, transient increases in the concentration of intracellular Ca2+. These Ca2+ signals drive resumption of the cell cycle by inactivating maturation-promoting factor (MPF), a complex of Cdc2 and cyclin B (1). During the entry into mitosis, the activity of MPF is regulated by the inhibitory phosphorylation of Cdc2, which is catalyzed by the Wee1 kinase family (2). However, the generally accepted view is that proteolytic degradation of cyclin B controls MPF inactivation and M-phase exit (1, 3). In oocytes, cyclin B degradation occurs through the activation of the anaphase-promoting complex (APC) in association with its coactivator Cdc20 (APCcdc20). However, the activation of APC through the overexpression of Cdc20 is not sufficient to overcome the MII arrest (4, 5), suggesting that the inactivation of MPF might be initiated by mechanisms other than cyclin B degradation. We show that the inhibitory phosphorylation of Cdc2 through the reactivation of Wee1B is necessary to initiate MPF inactivation during exit from meiosis II in mouse oocytes.
To investigate Wee1B function during meiosis, we examined whether Wee1B is expressed during the various stages of oocyte to embryo transition. Unlike many maternally-expressed mRNAs that are degraded during the onset of oocyte maturation (6), Wee1B mRNA was continuously expressed during the meiotic cell cycle and was replaced by somatic Wee1 (hereafter termed Wee1A) during the 2- or 4-cell embryo mitotic divisions (Fig.1A). In contrast, expression of the Myt1 protein kinase remained stable throughout oocyte and preimplantation development. Consistent with the mRNA data, Wee1B protein abundance increased during the meiotic cell cycle (Fig.1B). These results corroborate that Wee1B is expressed in oocytes at the germinal vesicle (GV) and MII stages and remains stable during the meiotic cell cycle (7).
Fig.1. Expression of Wee1B during the oocyte-to-embryo transition.
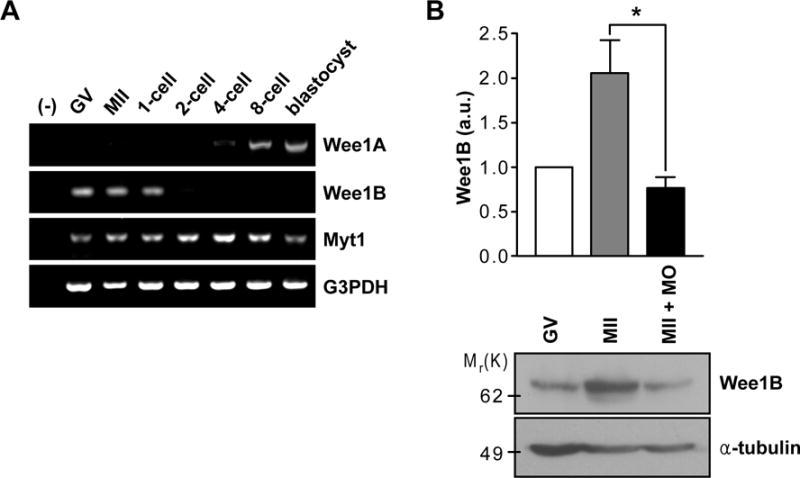
(A) RT-PCR of Wee1 kinases in oocyte and preimplantation embryos at different stages of development. G3PDH was used as an internal control. (B) GV and MII oocytes along with Wee1B MO-injected MII oocytes were immunoblotted for Wee1B and α-tubulin. Each lane contains 100 oocytes. Band intensities for Wee1B were quantified and normalized to α-tubulin level. a.u., arbitrary units. *p=0.0283.
Because Wee1B accumulates after resumption of meiosis, it is likely that this kinase has an additional role during progression through meiosis I or meiosis II, in addition to its function in maintaining arrest of oocytes at the GV stage (7, 8). To investigate this possibility, we injected Wee1B morpholino oligonucleotide (MO) into GV-stage oocytes, which were then allowed to mature in vitro for 20 hours (hereafter called ‘Wee1B knockdown oocytes’). Although Wee1B MO prevented the accumulation of Wee1B protein after meiotic resumption (Fig.1B), Wee1B knockdown oocytes progressed to MII stage (fig.S1). Spindle staining, chromosome spreads and MPF activity assays further confirmed that Wee1B knockdown oocytes normally establish MII arrest (Fig.2B and C and fig.S1C), suggesting that Wee1B is dispensable for transition from meiosis I to meiosis II.
Fig.2. Effect of down-regulation of Wee1B during egg activation.
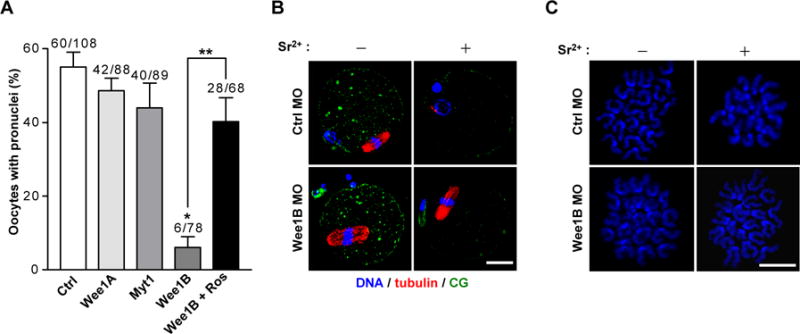
(A) GV oocytes injected with indicated MOs were in vitro matured for 20 hours. MII oocytes were activated with strontium with or without roscovitine (Ros) and oocytes with pronuclei were scored after 8 hours. Data are mean ± SEM of three independent experiments. The total number of oocytes used is reported above the bar. *p<0.0001 compared to control, **p<0.005. (B) In vitro matured MII oocytes injected with control (Ctrl) or Wee1B MO at GV stage were stained for DNA, tubulin and cortical granule (CG) before (−) or after (+) activation with strontium for 8 hours. (C) Chromosome spreads of Wee1B knockdown oocytes after strontium activation. Note that sister chromatids remained attached in Wee1B knockdown oocytes, whereas they had undergone disjunction in control oocytes. Bar, 20μm.
We examined whether Wee1B functions during the events associated with egg activation. Wee1B knockdown oocytes were parthenogenetically activated with strontium and scored for pronuclear formation, indicative of progression to interphase. Wee1B knockdown oocytes failed to form pronuclei, whereas oocytes injected with control, Myt1, or Wee1A MO were correctly activated (Fig.2A). This phenotype was rescued by inhibition of Cdc2 by roscovitine, suggesting that MPF inactivation is defective in Wee1B-downregulated oocytes. Similar results were obtained with RNAi-mediated depletion of Wee1B (fig.S2), excluding off-target effects of the MO. The Wee1B requirement at this stage was further confirmed by sperm-induced egg activation (fig.S3). Because Wee1B knockdown oocytes failed to activate, we examined whether these oocytes were arrested at MII stage. Even after activation, Wee1B knockdown oocytes still had intact spindles (Fig.2B), normal morphology of chromosomes (Fig.2C), and high MPF and mitogen-activated protein kinase (MAPK) activities (Fig.3D and fig.S4) similar to those of MII arrested oocytes. However, cortical granule (CG) release, one of the earliest Ca2+-induced events after egg activation (9), occurred normally, suggesting that Ca2+ signals are not impaired in oocytes deficient in Wee1B (Fig.2B).
Fig.3. Wee1B-dependent inactivation of MPF during egg activation.
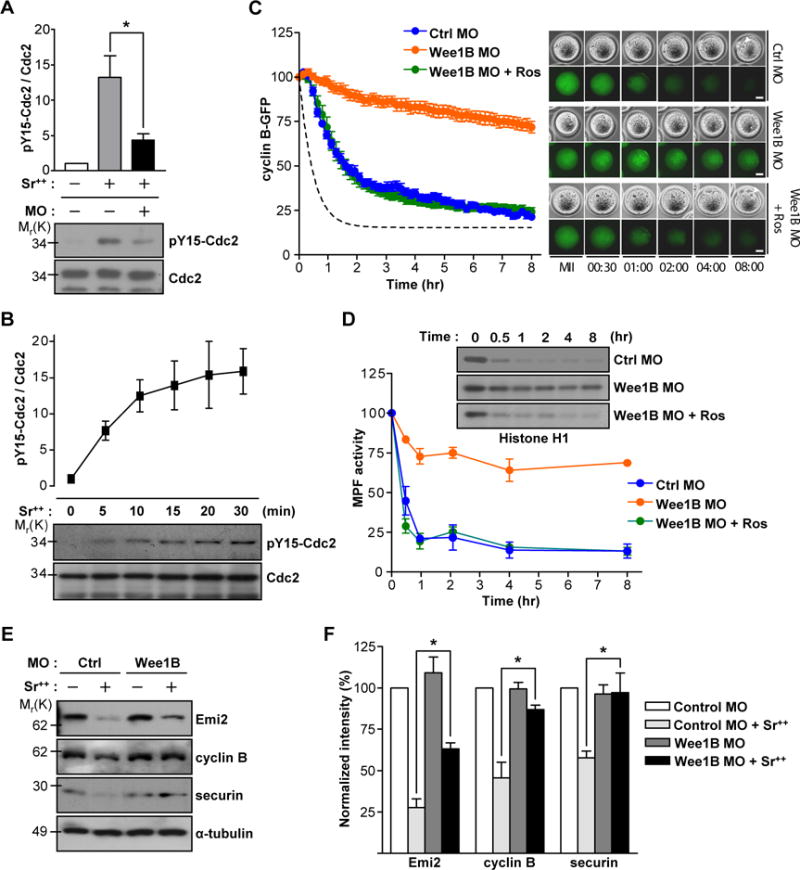
(A) Non-injected or Wee1B MO injected MII oocytes were activated with strontium for 20 min and subjected to SDS-PAGE followed by immunoblotting for Cdc2 and tyrosine 15 phosphorylation (pY15) of Cdc2. (B) MII oocytes activated with strontium were prepared at different time after activation and subjected to SDS-PAGE followed by immunoblotting for Cdc2 and pY15 Cdc2. Data are the mean ± SEM from three independent experiments. A representative image is shown. (C and D) GV oocytes injected with Wee1B MO and cyclin B-GFP mRNA were in vitro matured and activated with strontium at time t=0. (C) GFP levels were measured every 10 min for 8 hours. Average of MPF activity in control oocytes from (D) is shown with dotted line. Data are mean ± SEM of ten oocytes from two independent experiments. Representative images are shown in right panel. Pronucleus is indicated by arrowheads. Bar, 20μm. (D) Oocyte lysates were incubated with [γ-32P]ATP and either Histone H1 or myelin basic protein (MBP) (see fig.S4). Phosphorylation was detected by SDS-PAGE and autoradiography. Representative images from two independent experiments are shown. (E) In vitro matured MII oocytes injected with control or Wee1B MO at GV stage were incubated with (+) or without (−) strontium for 2 hours. Emi2, cyclin B and securin levels were determined by immunoblots. Each lane contains 100 oocytes. Representative images from three independent experiments are shown. (F) Band intensities of Emi2, cyclin B and securin were quantified and normalized to controls. Data are the mean ± SEM from three independent experiments. *p<0.05.
Given that inhibitory phosphorylation of Cdc2 on tyrosine 15 is mediated by Wee kinases (2), these findings raised the possibility that Wee1B-mediated phosphorylation of Cdc2 is required for pronuclear formation. Indeed, tyrosine 15 phosphorylation of Cdc2, which is undetectable during MII arrest, appeared immediately after egg activation (Fig.3B), and was reduced after Wee1B downregulation (Fig.3A). Oocytes expressing a constitutively active Cdc2 with mutations that preclude phosphorylation by Wee1B were resistant to stimulation by increased Ca2+ concentrations (fig.S5). Entry and exit from mitosis are disturbed either when the inhibitory phosphorylation of Cdc2 is removed in frog egg (10) or when Wee1 and Cdc25 are absent in fission yeast cells (11). Moreover, in frog eggs, tyrosine 15 phosphorylation of Cdc2 rapidly appears after stimulation of eggs to promote accumulation of Ca2+ (12). Thus, our results indicate that Wee1B-mediated inhibition of Cdc2 is required for pronuclear formation in mouse oocytes.
Wee1B knockdown oocytes have high MPF activity consistent with the absence of inhibitory phosphorylation of Cdc2. However, because MPF is a heterodimer of Cdc2 and cyclin B, we tested whether this high MPF activity was also caused by the decreased degradation of cyclin B during egg activation. We monitored the stability of cyclin B fused to green fluorescent protein (GFP) during egg activation. When the amounts of Wee1B were decreased, cyclin B degradation was reduced and occurred with a slower time course (Fig.3C). Treatment of Wee1B knockdown oocytes with roscovitine reactivated cyclin B degradation, further documenting that Cdc2 inhibition is a prerequisite for cyclin B destabilization (Fig.3C). Time courses comparing MPF activity and cyclin B degradation showed that MPF activity declined ahead of cycin B degradation (Fig.3D). Considering that cyclin B is degraded by the APCcdc20 during egg activation, these findings indicate that the APC pathway is not properly activated in response to Ca2+ signals when Wee1B is downregulated and Cdc2 remains activated. Indeed, endogenous cyclin B and securin were not degraded in Wee1B knockdown oocytes (Fig.3E and F). Moreover, Emi2, which negatively regulates APC activity and is degraded during egg activation (13), was partially stabilized in Wee1B knockdown oocytes (Fig.3E and F). Consistent with this, the activation of APC is impaired when constitutively active Cdc2 is overexpressed in mitotic cells (12, 14). Taken together, these data indicate that Wee1B-mediated inhibition of Cdc2 is required for the timely activation of the APC, which further promotes MPF inactivation through the degradation of cyclin B.
In the reverse experiments, Wee1B overexpression in oocytes drove pronuclear formation. However, cortical granule exocytosis did not occur, indicating that pronuclear formation proceeds in the absence of Ca2+ signals (fig.S6).
A sperm-induced increase in the intracellular concentrations of Ca2+ switches on CaMKII which in turn activates the APC pathway, leading to resumption of the meiotic cell cycle (1). However, in frog oocytes, CaMKII directly inactivates the phosphatase Cdc25C, which acts in opposition to Wee1 kinases (15). Thus, we tested whether CaMKII might directly regulate Wee1B activity in mouse oocytes. We incubated ectopically expressed Wee1B with CaMKII in vitro. Wee1B was directly phosphorylated by CaMKII and this phosphorylation, although significantly reduced, was still present when catalytically inactive Wee1B (Wee1B-dead) was used (Fig.4A). This is consistent with Wee1B having autophosphorylation activity (7), but also indicates that CaMKII phosphorylates Wee1B. To identify the CaMKII phosphorylation site in Wee1B, we incubated several truncated forms of Wee1B fused to maltose-binding protein (MBP) with CaMKII. Phosphorylation was detected only in the N-terminus of Wee1B (amino acids 1–124) (Fig.4B). Sequence analysis of Wee1B predicted that the S15 residue of Wee1B is a potential CaMKII phosphorylation site. Indeed, disruption of this residue (S15A) almost completely abolished Wee1B phosphorylation by CaMKII (Fig.4C). Moreover, the tyrosine autophosphorylation of Wee1B was significantly increased by Ca2+ stimulation with A23187, and this phosphorylation was blocked by S15A mutation (Fig.4D). Taken together, these results demonstrate that Wee1B is a direct substrate of CaMKII in mouse oocytes and phosphorylation causes an increase in Wee1B activity. Next, we tested whether phosphorylation of the S15 of Wee1B by CaMKII mediated Wee1B activation during egg exit from MII. Injection of the S15D mutant that mimics phosphorylation status induced pronuclear formation more efficiently than wild-type Wee1B (Fig.4E). On the other hand, pronuclear formation was significantly decreased when the S15A mutant was injected. Thus, we conclude that phosphorylation of Wee1B at the S15 residue by CaMKII activates the dormant Wee1B during egg activation.
Fig.4. Phosphorylation of Wee1B by CaMKII.
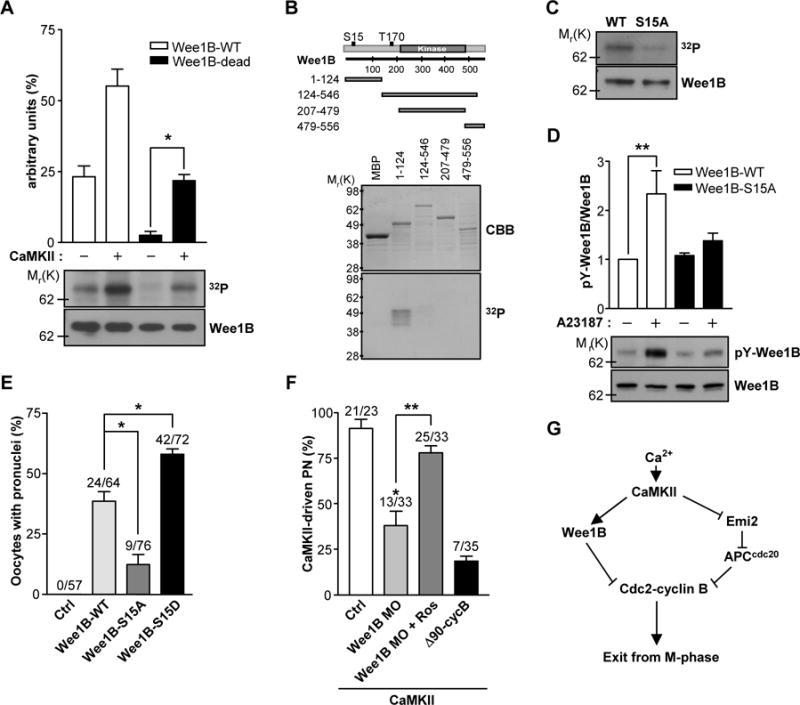
(A) Wee1B was overexpressed in HEK293 cells, immunoprecipitated, and incubated with or without CaMKII in the presence of [γ-32P]ATP for 10 min. The phosphorylation state was detected by SDS-PAGE and autoradiography. The amounts of loaded protein were determined by immunoblot. A representative experiment of the three performed is shown. (B) MBP-tagged truncated forms of Wee1B were generated and expressed in Escherichia coli. After purification by affinity chromatography, fusion proteins were incubated with CaMKII in the presence of [γ-32P]ATP for 10 min. The radiolabeled fusion proteins were detected by autoradiogram. The expression and purification of MBP fusion proteins were monitored by Coomassie brilliant blue (CBB) staining. (C) Immunoprecipitated Wee1B-WT or -S15A mutant were incubated with CaMKII in the presence of [γ-32P]ATP for 10 min, and incorporation of 32P was detected by autoradiography. The amounts of loaded protein were determined by immunoblot. (D) HEK293 cells overexpressing Wee1B-WT or -S15A mutant were treated with 10μM of A23187 for 20 min. The activity of Wee1B was measured by the autophosphorylation of tyrosine residues. A representative image of three independent experiments performed is reported. (E) MII oocytes were injected with mRNAs encoding the indicated Wee1Bs, and oocytes with a pronucleus were counted after 6 hours. (F) MII oocytes injected with Wee1B MO or Δ90cyclin B mRNA were microinjected with constitutively active CaMKII (CA-CaMKII) and pronuclear formation scored after 8 hours. *p<0.005 and **p<0.05 (G) Proposed model of mouse egg activation.
Because Wee1B is a CaMKII substrate, CaMKII-induced MII exit should be blocked by Wee1B downregulation. To test this possibility, we injected constitutively active CaMKII, which is known to induce the completion of meiosis, into Wee1B knockdown oocytes (16). CaMKII-driven MII exit was significantly reduced in Wee1B knockdown oocytes but was restored in oocytes treated with roscovitine (Fig.4F). Comparable inhibition was obtained when nondegradable cyclin B (Δ90cyclin B) was overexpressed, suggesting that inhibitory phosphorylation and cyclin B degradation are equally important. These data, along with the observations that MPF activity remains high and the degradation of cyclin B does not properly occur when Wee1B is downregulated, indicate that Wee1B is activated by CaMKII. This activation is responsible for the initiation of MPF inactivation, which is further promoted by the degradation of cyclin B through the APC activation. Thus, we propose that M-phase exit is tightly regulated not only by the proteolytic degradation of cyclin B, but also by the kinase-dependent inhibition of Cdc2 to ensure rapid and irreversible exit from meiosis in mouse oocytes (Fig.4G).
Supplementary Material
Acknowledgments
We thank Seung Jin Han for Wee1B plasmids, Keith T. Jones for cyclin B plasmids and Thierry Lorca for CaMKII plasmid. This work was supported by NIH Grant GM080527 and HD052909.
Footnotes
Author contributions: JO designed, performed and analyzed most of the experiments in this study. AS performed immunostaining. MC conceived the project and supervised the study. JO and MC wrote the manuscript.
Supporting Online Material: www.sciencemag.org, Materials and Methods, Figs. S1 to S6, References
References
- 1.Jones KT. Reproduction. 2005 Dec;130:813. doi: 10.1530/rep.1.00710. [DOI] [PubMed] [Google Scholar]
- 2.Lew DJ, Kornbluth S. Curr Opin Cell Biol. 1996 Dec;8:795. doi: 10.1016/s0955-0674(96)80080-9. [DOI] [PubMed] [Google Scholar]
- 3.Wasch R, Cross FR. Nature. 2002 Aug 1;418:556. doi: 10.1038/nature00856. [DOI] [PubMed] [Google Scholar]
- 4.Tsurumi C, Hoffmann S, Geley S, Graeser R, Polanski Z. J Cell Biol. 2004 Dec 20;167:1037. doi: 10.1083/jcb.200405165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin S, et al. Cell Cycle. 2007 Dec 1;6:2990. doi: 10.4161/cc.6.23.4993. [DOI] [PubMed] [Google Scholar]
- 6.Bachvarova R, De Leon V, Johnson A, Kaplan G, Paynton BV. Dev Biol. 1985 Apr;108:325. doi: 10.1016/0012-1606(85)90036-3. [DOI] [PubMed] [Google Scholar]
- 7.Han SJ, Chen R, Paronetto MP, Conti M. Curr Biol. 2005 Sep 20;15:1670. doi: 10.1016/j.cub.2005.07.056. [DOI] [PubMed] [Google Scholar]
- 8.Oh JS, Han SJ, Conti M. J Cell Biol. 2010 Jan 25;188:199. doi: 10.1083/jcb.200907161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kline D, Kline JT. Dev Biol. 1992 Jan;149:80. doi: 10.1016/0012-1606(92)90265-i. [DOI] [PubMed] [Google Scholar]
- 10.Pomerening JR, Kim SY, Ferrell JE., Jr Cell. 2005 Aug 26;122:565. doi: 10.1016/j.cell.2005.06.016. [DOI] [PubMed] [Google Scholar]
- 11.Sveiczer A, Novak B, Mitchison JM. J Cell Sci. 1999 Apr;112(Pt 7):1085. doi: 10.1242/jcs.112.7.1085. [DOI] [PubMed] [Google Scholar]
- 12.D’Angiolella V, Palazzo L, Santarpia C, Costanzo V, Grieco D. PLoS One. 2007;2:e247. doi: 10.1371/journal.pone.0000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shoji S, et al. Embo J. 2006 Feb 22;25:834. doi: 10.1038/sj.emboj.7600953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pomerening JR, Ubersax JA, Ferrell JE., Jr Mol Biol Cell. 2008 Aug;19:3426. doi: 10.1091/mbc.E08-02-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hutchins JR, Dikovskaya D, Clarke PR. Mol Biol Cell. 2003 Oct;14:4003. doi: 10.1091/mbc.E03-02-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madgwick S, Levasseur M, Jones KT. J Cell Sci. 2005 Sep 1;118:3849. doi: 10.1242/jcs.02506. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.