

Abstract
The simian-human immunodeficiency virus (SHIV)/macaque model for human immunodeficiency virus type 1 has become a useful tool to assess the role of accessory genes in lentiviral pathogenesis. In this study, we introduced two amino acid changes in the highly conserved SLQYLA domain (to AAQYLA) of the SIV Vif protein. The resulting virus, SHIVVifAAQYLA, was used to infect three macaques, which were followed for over six months. Plasma viral loads and circulating CD4+ T cell levels were assessed during the course of infection. The three macaques inoculated with SHIVVifAAQYLA did not develop significant CD4+ T cell loss over the course of their infection, had plasma viral RNA loads that were over 100-fold lower than macaques inoculated with parental SHIVKU-1bMC33, and developed no histological lesions in lymphoid tissues. DNA and RT-PCR analysis revealed that only a select number of tissues were infected with this virus. Sequence analysis indicates that the site-directed changes were stable during the first three weeks after inoculation but thereafter the S147A amino acid substitution changed to a threonine in two of three macaques. The L148A substitution remained stable in the vif amplified from the PBMC of all three macaques. Sequence analysis of vif, vpu, env and nef genes revealed G-to-A mutations in the genes amplified from macaques inoculated with SHIVVifAAQYLA, which were higher than in a macaque inoculated with parental SHIVKU-1bMC33. We found that the majority (>85%) of the G-to-A mutations were in the context of 5′-TC (minus strand) and not 5′-CC, suggestive that one or more of the rhesus APOBEC3 proteins may be responsible for the observed mutational patterns with rhesus APOBEC3G for a minority of the mutations since its GG-to-AG mutational pattern was infrequently detected. Finally, macaques inoculated with SHIVVifAAQYLA developed immunoprecipitating antibody responses against the virus. The results from this study provide the first in vivo evidence of the importance of the SLQYLA domain in viral pathogenesis and show that targeted mutations in vif can lead to a persistent infection with G-to-A changes accumulating in the viral genome.
INTRODUCTION
Human immunodeficiency virus type 1 (HIV-1) as well as other lentiviruses encode for a Vif protein, which has been shown to be essential for HIV-1 replication in certain cell types. The Vif protein of HIV-1 was first shown to interact with apolipoprotein B mRNA editing enzyme catalytic polypeptide-like 3G (APOBEC3G; hA3G) (Sheehy et al., 2002). This protein was found to provide cells with an innate intracellular anti-retroviral activity that is associated with hypermutation of the viral genome through cytidine deamination (Harris et al., 2003; Lecossier et al., 2003; Mangeat et al., 2003; Zhang et al., 2003). This hA3G-induced cytidine deamination results in cytidine to uridine changes during minus strand DNA synthesis, which ultimately leads to G-to-A mutations in the plus strand (Yu et al., 2004a). Several groups subsequently showed that the Vif protein can prevent hypermutation by binding to hA3G and targeting this protein for degradation via the proteasome (Conticello et al., 2003; Kao et al., 2004; Mariani et al., 2003; Marin et al., 2003; Mehle et al., 2004; Sheehy et al., 2003; Stopak et al., 2003; Yu et al., 2003; 2004). In addition to A3G, humans have six other A3 genes: hA3A, hA3B, hA3C, hA3DE, hA3F, and hA3H (Jarmuz et al., 2002). The members of this family either have one (hA3A, hA3C, and hA3H) or two (hA3B, hA3DE, hA3F and hA3G) Zn+2 coordinating deaminase domains organized as H-x1-E-x25-31-C-x 2-4-C (with x being a non- conserved position) (Chiu and Greene, 2008). In addition to the well studied hA3G, other family members such as hA3B, hA3DE, hA3F, and hA3H have been shown to also inhibit the replication of ΔvifHIV-1 (Dang et al., 2007; Doehle et al., 2005; Wiegand et al., 2004; Yang et al., 2007; Yu et al., 2004; Zheng et al., 2004). Moreover, ΔvifSIVmac239 is potently restricted by hA3G, hA3F, and hA3H and to a lesser extent by A3B and A3C and A3DE (Dang et al., 2007; 2008; Mariani et al., 2003; Virgen and Hatziianou; Yu et al., 2004). While fewer studies have been performed on the APOBEC3 family members in macaques, one study has also shown that rhesus macaque rA3G and rA3F inhibit the replication of ΔvifSIVmac (Zennou and Bieniasz, 2006; Virgen and Hatziianou, 2007).
Sequence analysis of Vif proteins from different lentiviruses reveals that there is a highly conserved SLQ(Y/F)LA domain near the carboxyl terminus. The introduction of mutations in this domain results in decreased binding of Vif to Elongin C of the Cul5/Elongin B/C E3 ligase complex and increases A3G incorporation into virions resulting in G-to-A hypermutation (Kobayashi et al., 2005). However, no studies have assessed the role of this domain using a non-human primate model of HIV-1 pathogenesis. Our laboratory has been using the chimeric simian-human immunodeficiency (SHIV)/macaque model to study the role of Vpu and its various domains in CD4+ T cell loss, virus release and pathogenesis (Stephens et al., 2002; Singh et al., 2003; Hout et al., 2005; 2006; Hill et al., 2008). In this study, we constructed a simian-human immunodeficiency virus (SHIV) with amino acid changes in the highly conserved SLQYLA (SHIVVifAAQYLA) domain and assessed it for pathogenesis in macaques. Our results show for the first time that the SLQYLA domain has determinants that contribute to the pathogenicity of SHIV in macaques and that mutations in this domain result in the accumulation of G-to-A mutations in the viral genome.
RESULTS
Replication of SHIVVifAAQYLA in APOBEC3G positive and negative cell lines
We performed assays to examine the replication of parental SHIVKU-1bMC33 and SHIVVifAAQYLA in A3G/F positive (CEM) and negative (CEM-SS) cell lines. Cells were infected with each of the two viruses and the levels of p27 Gag released into the culture medium were quantified using a commercial antigen capture assay. SHIVVifAAQYLA replicated in CEM-SS cells with similar kinetics as parental SHIVKU-1bMC33 (Figure 1A). However, SHIVVifAAQYLA released less than 0.5% p27 compared to the parental SHIVKU-1bMC33 in CEM cells (Figure 1B). We also examined virions for the incorporation of A3G following passage in CEM cells. Our results indicate that SHIVVifAAQYLA but not SHIVKU-1bMC33 incorporated A3G into virions (data not shown).
Figure 1.


Replication of SHIVKU-1bMC33 and SHIVVifAAQYLA in APOBEC3G positive (C8166) and negative (CEM-SS) cell lines. Cells were inoculated with equal amounts of each virus and levels of p27 in the culture supernatants determined at various times post-inoculation. Panel A. Replication of SHIVKU-1bMC33 and SHIVVifAAQYLA in CEM-SS cells. Panel B. Replication of SHIVKU-1bMC33 and SHIVVifAAQYLA in C8166 cells. (●) SHIVKU-1bMC33; (■) SHIVVifAAQYLA.
Disease in macaques inoculated with the SHIVVifAAQYLA
We inoculated three macaques with the SHIVVifAAQYLA. Prior to inoculation, macaques RAK10, RPL10, and RCS10 had circulating CD4+ cell counts of 2638, 2480, and 3206, respectively. As shown in Figure 2A, macaques RAK10, RCS10, and RPL10 developed a slight decrease in the levels of circulating CD4+ T cell levels at one week post-inoculation but these rebounded to near pre-inoculation levels by four weeks post-inoculation. These macaques maintained high levels of circulating CD4+ T cells throughout the course of their infection. All three macaques were euthanized at 28 weeks post-inoculation in excellent condition. At necropsy, RAK10, RPL10, and RCS10 had circulating CD4+ T cell counts of 2427, 1656, and 3021, respectively. This contrasts with macaques inoculated with parental SHIVKU-1bMC33 (Figure 2B), which developed a severe loss of circulating CD4+ T cells. Analysis of the plasma viral loads of the macaques inoculated with SHIVVifAAQYLA revealed that during the early peak (1 to 3 weeks) of viremia, the mean load was 8.52 × 104 copies per ml (Figure 3A), approximately 100-fold less than the macaques inoculated with parental SHIVKU-1bMC33 (Figure 3B). Following the first month of infection, the plasma viral loads in macaque RPL10 declined faster than macaques RAK10 and RCS10.
Figure 2.


Circulating CD4+ T cell levels in macaques inoculated with SHIVKU-1bMC33 and SHIVVifAAQYLA. Panel A. The levels of circulating CD4+ T cells in three macaques (RAK10,●; RCS10, □; and RPL10, ▲) following inoculation with SHIVVifAAQYLA. Panel B. The levels of circulating CD4+T cells in four macaques (2000, ●; 2001, ■; CM4G, ▲; and CM4K, □; RRH10;▼) following inoculation with SHIVKU-1bMC33.
Figure 3.


Plasma viral loads in macaques inoculated with SHIVKU-1bMC33 and SHIVVifAAQYLA. Panel A. Plasma viral loads in three macaques (RAK10, ●; RCS10, □; RPL10, ▲) following inoculation with SHIVVifAAQYLA. Panel B. Plasma viral loads in four macaques (2000, ●; CM4G, ■; CM4K, ▲; and RRH10, ▼) following inoculation with SHIVKU-1bMC33.
Stability of the vif gene mutations during the course of infection and at necropsy
We assessed the stability of the engineered mutations in the vif gene during the course of infection. DNA was extracted from PBMC samples at various times post-inoculation and from lymphoid tissues at necropsy. The vif sequence was amplified, directly sequenced and compared to the input vif sequence of SHIVVifAAQYLA. As shown in Figure 4, the vif mutations were stable from one to three weeks post-inoculation for all three macaques inoculated with SHIVVifAAQYLA, but by 4 weeks post-inoculation the S147A amino acid substitution changed to a threonine in two of the macaques RPL10 and RAK10. Interestingly, this amino acid substitution was mediated by a G-to-A mutation. DNA from lymphoid organs obtained at necropsy from RAK10 and RPL10 also showed the A147T mutation whereas macaque RCS10 maintained S147A amino acid substitution. The L148A substitution was found to be stable during the course of infection for all three macaques and in lymphoid organs analyzed at necropsy. There were no mutations in this region of vif from RRH10, which was inoculated with parental SHIVKU-1bMC33.
Figure 4.

Analysis of the vif gene for the stability of the engineered mutations. DNA was isolated from PBMC at 1, 3, 4, 8, 12 and 28 weeks post-inoculation and from lymphoid organs (thymus, mesenteric lymph node, and inguinal lymph node) at necropsy. The vif gene was amplified and directly sequenced. Shown at the top are the sequences for both parental SHIVKU-1bMC33 and SHIVVifAAQYLA.
Macaques inoculated with SHIVVifAAQYLA developed antibody responses against the virus
At necropsy, we analyzed the plasma for the presence of immunoprecipitating antibody responses. As shown in Figure 5, all three macaques developed immunoprecipitating antibodies against SHIVKU-1bMC33, although macaque RCS10 developed significantly lower antibody response compared to the other two macaques. In contrast, a macaque inoculated with parental SHIVKU-1bMC33 (RRH10) did not develop antibodies to the virus, which is common for macaques that develop severe CD4+ T cell loss during the acute phase (< 4 weeks) following inoculation with a pathogenic X4 SHIV.
Figure 5.

Macaques inoculated with SHIVVifAAQYLA developed antibody responses against the virus. C8166 cells were inoculated with SHIVKU-1bMC33 for 5 days, starved for methionine/cysteine and then radiolabeled overnight with 35S-methionine/cysteine. The culture medium was harvested and used in immunoprecipitation reactions with plasma from RAK10, RPL10, RCS10 and RRH10 as described in the materials and Methods section. The immunopreciptates were washed with 1X RIPA buffer, boiled in 1X sample reducing buffer and proteins separated on 10% SDS-PAGE gels.
Lane 1. SHIV proteins immunoprecipitated using from an uninfected culture. Lane 2. SHIV proteins immunoprecipitated from a positive control plasma sample. Lane 3. SHIV proteins immunoprecipitated using RAK10 plasma. Lane 4. SHIV proteins immunoprecipitated using RCS10 plasma. Lane 5. SHIV proteins immunoprecipitated using RPL10 plasma. Lane 6. SHIV proteins immunoprecipitated using RRH10 plasma. The molecular weight standards are shown on the left.
Histological examination of tissues
Tissues from macaques were examined for the presence of lesions consistent with pathogenic X4 SHIV (Stephens et al., 2002). Histological examination of tissues from macaque RRH10, which was inoculated with SHIVKU-1bMC33, revealed severe loss of CD4+ T cells, severe lymphoid depletion in both the thymus and lymph nodes and mild lymphoid depletion in the spleen (Figure 6). The lymphoid depletion observed in macaque RRH10 is typical of that observed following inoculation with the parental SHIVKU-1bMC33 (Stephens et al., 2002). The macaques inoculated with SHIVVifAAQYLA did not exhibit lesions in any of the 13 visceral organs and CNS. Micrographs of sections from the thymus, mesenteric lymph node, and spleen from macaque RAK10 are shown along with micrographs of histological sections from an uninfected macaque (Figure 6).
Figure 6.

Histopathology associated with SHIVVifAAQYLA infection of macaques. Hematoxylin and eosin stains of sections from the thymus (Panels A, B, C) and mesenteric lymph node (Panels D, E, F) and spleen (Panels G, H, I) from a SHIVVifAAQYLA-inoculated macaque RAK10 (Panels A, D, G), from an uninfected macaque (panels B, E, H) and from a SHIVKU-1bMC33- infected macaque RRH10 (Panels C, F, I).
Macaques inoculated with SHIVVifAAQYLA had a decreased tissue distribution of replicating virus
As the plasma viral loads were decreased in comparison to macaques inoculated with parental SHIVKU-1bMC33, we wanted to determine if this was due to infection of select cell populations in one or more organs, which may not express APOBEC3G. We first analyzed DNA isolated from 13 visceral organs from the SHIVVifAAQYA macaques for the presence of viral gag sequences by nested DNA PCR. We previously showed that these oligonucleotide primers were capable of detecting 1–5 copies of viral DNA (Joag et al., 1994). Generally, we find that nearly all visceral organs from a SHIVKU-1bMC33-inoculated macaques are positive for viral gag sequences in DNA and RNA samples (Figure 7A; macaque RRH10). For the macaques inoculated with SHIVVifAAQYLA, we found that 6 of 13, 8 of 13, and 12 of 13 organs from macaques RAK10, RCS10, and RPL10, respectively, were positive for viral gag sequences by nested DNA PCR (Figures 7B–D). The majority of the organs that were gag positive were lymphoid organs. We next analyzed RNA isolated from tissues for the presence of RNA using RT-PCR. The results of this analysis is shown in Figure 8. We found that had 5 of 13, 6 of 13, and 9 of 13 organs from macaques RAK10, RCS10, and RPL10 were positive for viral gag RNA, respectively. The control macaque (RRH10), had 12 of 13 and 11 of 13 organs positive for the presence of viral DNA and RNA gag sequences, respectively. Taken together, these results indicate that SHIVVifAAQYLA infection was less widespread in 2 of 3 macaques compared to macaques inoculated with parental SHIVKU-1bMC33.
Figure 7.

Macaques inoculated with SHIVVifAAQYLA had a decreased tissue distribution of virus. DNA was isolated from different organs as indicated at necropsy and amplified using nested DNA PCR using oligonucleotides specific for gag. Samples were run on a 1.5% agarose gel, stained with ethidium bromide and photographed. Shown are the results of the nested DNA PCR using DNA from macaques RRH10 (panel A), RAK10 (panel B), RCS10 (Panel C) and RPL10 (Panel D). The tissue DNAs analyzed are noted above each lane. The positive control was gag amplified from the lymph node tissue DNA from macaque 50O that died of neuroAIDS and the negative control was lymph node tissue DNA from an uninfected macaque. The markers to the right are in base pairs.
Figure 8.

Macaques inoculated with SHIVVifAAQYLA had a decreased tissue distribution of viral RNA. RNA was isolated from different organs as indicated at necropsy, DNase I treated to remove residual DNA and used in nested RT-PCR using oligonucleotides described in the Materials and Methods. Samples were run on a 1.5% agarose gel, stained with ethidium bromide and photographed. The results are shown as follows: Panel A: macaques RRH10; Panel B: RAK10; Panel C: RCS10; and Panel D: RPL10. The tissue RNAs analyzed are noted above each lane. The positive control was amplification of gag from the lymph node tissue RNA from macaque 50O that died of neuroAIDS. The negative control was lymph node tissue RNA from an uninfected macaque. The markers to the right are in base pairs.
Sequence changes vif, vpu, env and nef in primary and secondary lymphoid organs at necropsy
A well documented feature of ΔvifHIV-1 infection is the incorporation of select APOBEC3 proteins into viral particles leading to cytosine deamination during minus strand DNA synthesis. This results in hypermutation (guanosine to adenosine transitions) of the viral genome and inhibition of virus replication. We examined the extent of G-to-A mutations in viral DNA amplified from one primary lymphoid organ (thymus) and several secondary lymphoid organs (mesenteric, axillary, and inguinal lymph nodes) from the three macaques inoculated with SHIVVifAAQYLA and one macaque inoculated with SHIVKU-1bMC33. We analyzed sequences from the vif, vpu, env, and nef genes. The results of the sequence analyses are shown in Table 1. The salient features of these analyses were that: a) with the exception of env, the thymus consistently had the lowest percentage of G-to-A mutations; b) not all lymphoid organs had significant G-to-A mutations; and c) greater than 85% of the G-to-A mutations were in the context of TCT and not CC. The sequence of vpu amplified from the lymphoid tissues of macaque RPL10 is shown as an example (Figure 9). Macaque RRH10, (inoculated with SHIVKU-1bMC33) had the fewest G-to-A mutations in all lymphoid organs, which is similar to other macaques inoculated with this virus (unpublished observations). Generally, the number of mutations was approximately <0.2–0.41% of the nucleotides analyzed. The highest percentage of G-to-A mutations found in the macaques inoculated with SHIVVifAAQYLA was in the nef gene amplified from macaque RCS10 (5.0%).
Table 1.
Results of viral DNA sequence analysis from tissues at necropsy. The ratio of the number of G-to-A nucleotide substitutions/number of bases sequenced is shown.
|
|
||||
|---|---|---|---|---|
| Tissue | ||||
| Macaque/gene | Thymus | MLN | ALN | ILN |
| RRH10 | ||||
| vif | 1/466 (0.21%)a | 1/466 (0.21%) | 1/466 (0.21%) | 1/466 (0.21%) |
| vpu | 2/246 (0.0%) | 1/246 ( 0.4%) | 1/246 ( 0.4%) | 1/246 (0.4%) |
| env | 2/490 (0.41%) | 0/490 (0.0%) | 0/480 (0.0%) | 0/480 (0/0%) |
| nef | 1/400 (0.25%) | 1/400 (0.25%) | 2/400 (0.50%) | 0/400 (0.0%) |
| RAK10 | ||||
| vif | 0/466 (0.00%) | 16/466 (3.4%) | 7/400 (1.50%) | 1/466 (0.21%) |
| vpu | 1/246 (0.4%) | 4/246 (0.4%) | 4/246 (1.63%) | 4/246 (1.63%) |
| env | 12/490 (2.44%) | 10/490 (2.04%) | 12/490 (2.44%) | 10/490 (2.04%) |
| nef | 2/400 (0.50%) | 1/400 (0.25%) | 17/400 (4.25%) | 1/400 (0.25%) |
| RCS10 | ||||
| vif | 0/466 (0.0%) | 1/466 (0.21%) | 8/466 (3.25%) | 1/466 (0.21%) |
| vpu | 1/246 (0.4%) | 1/246 (0.4%) | 4/246 (1.62%) | 2/246 (0.8%) |
| env | 10/490 (2.04%) | 8/490 (1.63%) | 10/490 (2.04%) | 12/490 (2.44%) |
| nef | 1/400 (0.25%) | 20/400 (5.00%) | 2/400 (0.5%) | 1/400 (0.25%) |
| RPL10 | ||||
| vif | 3/466 (0.64%) | 6/466 (1.28 %) | 9/466 (1.93%) | 8/466 (1.72%) |
| vpu | 3/246 (1.2%) | 6/246 (2.4%) | 9/246 (3.70%) | 8/246 (3.3%) |
| env | 9/490 (1.83%) | 8/490 (1.63%) | 8/490 (1.63%) | 8/490 (1.63%) |
| nef | 1/400 (0.25%) | 2/400 (0.5%) | 6/400 (1.25%) | 4/400 (1.0%) |
The percentage of G to A substitutions are in parentheses.
Figure 9.

Sequence of the vpu gene amplified from lymphoid tissues of macaque RPL10. Shown above is the input sequence of the vpu from SHIVKU-1bMC33. The (.) represents identity with nucleotide changes from the input sequence shown. The underlined sequences represent the context in which the G-to-A mutations occurred.
Pooled plasma from the two macaques inoculated with SHIVVifAAQYLA contained low levels of infectious virus when transfused into a naive macaque
To determine if infectious virus was present in plasma that was not detected in our infectivity assays, plasma from macaques RAK10, and RPL10 (2ml each) were pooled and intravenously inoculated into a naive macaque, I95. As shown in Figure 10A, we could detect viral gag sequences in DNA isolated from PBMC at 0, 1, 2, 3, 4, 5, 6, 9, 20 weeks post-inoculation. We also assessed plasma for viral loads using real time PCR. As shown in figure 11B, viral gag sequences could not be detected in plasma by real time RT-PCR (detection limit 100 copies) from weeks 0 to 20. The circulating CD4+ T cell levels did not decrease from pre-inoculation levels (Figure 10B) and we could not detect antibodies in the plasma from this macaque at week 9 (data not shown). At 20 weeks post-inoculation, macaque I95 was sacrificed. DNA was isolated from 10 visceral organs and used in nested DNA PCR to detect viral gag sequences. As shown in Figure 11A, viral gag sequences were amplified from all 10 organs examined. We also isolated RNA from the same organs for use in RT-PCR to detect viral gag RNA sequences. As shown in Figure 11B, the viral RNA was detected in six organs (liver, kidney, axillary lymph node, small intestine (ileum), spleen, and tonsil). This indicates that some level of viral replication was ongoing although we were unable to isolate infectious virus on CEM-SS or SupT1 cells. Taken together, these results indicate that pooled plasma contained only low levels of infectious virus that was controlled by the macaque I95.
Figure 10.
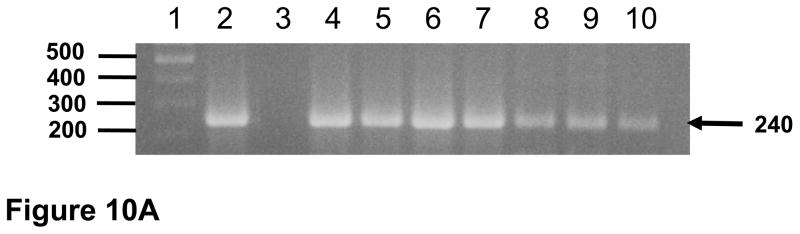

Viral loads and circulating CD4+ T cell levels in macaque I95 that was intravenously inoculated with pooled plasma from macaques RAK10 and RPL10 obtained at necropsy (2 ml each). Blood was obtained at 0 (just prior to inoculation), 1, 2, 3, 4, 5, 6, 9, and 20 weeks after inoculation. The plasma was retained and the PBMC purified over Ficoll-Hypaque gradients. The DNA was isolated from the PBMC. Panel A. Results of the nested DNA PCR for viral gag using DNA isolated from PBMC. Lane 1. 100 base pair markers. Lane 2. Positive control. Lanes 3–10 represent gag sequences amplified from PBMC isolated from I95 at 0, 1, 2, 3, 4, 5, 6, 9, and 20 weeks post-inoculation, respectively. Panel B. Results of the real time PCR for viral gag sequences from plasma samples (▲; scale to left) and circulating CD4+ T cell levels (●; scale to right).
Figure 11.
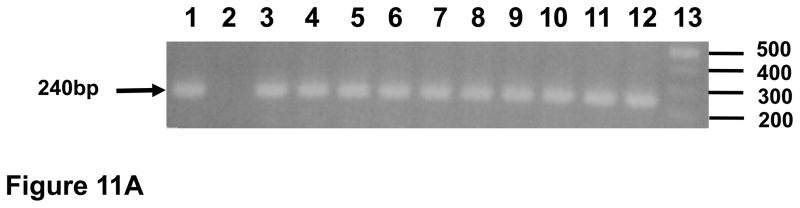

Distribution of viral RNA and DNA sequences in tissues from macaque I95. At necropsy, tissues were harvested and DNA or RNA isolated and used in nested DNA (panel A) or nested RT-PCR (panel B). Panel A. Results of the nested DNA PCR for viral gag using isolated DNA. Lane 1. Positive control. Lane 2. Negative control. Lane 3. Liver. Lane 4. Lung. Lane 5. Kidney. Lane 6. Mesenteric lymph node. Lane 7. Axillary lymph node. Lane 8. Inguinal lymph node. Lane 9. Small intestine (ileum). Lane 10. Spleen. Lane 11. Thymus. Lane 12. Tonsil. Lane 13. 100 base pair markers. Panel B. Results of the nested RT- PCR for viral gag using isolated RNA. Lane 1. 100 base pair markers. Lane 2. Positive control. Lane 3. Negative control. Lane 4. Liver. Lane 5. Lung. Lane 6. Kidney. Lane 7. Mesenteric lymph node. Lane 8. Axillary lymph node. Lane 9. Inguinal lymph node. Lane 10. Small intestine (ileum). Lane 11. Spleen. Lane 12. Thymus. Lane 13. Tonsil.
DISCUSSION
The results presented here are the first to evaluate the role of domain specific mutations in the BC box domain of the vif gene in a non-human primate model of HIV-1/AIDS. Previous studies have shown that Vif interacts with elongin C, which has a consensus sequence of (A,P,S,T)1L2xxx(C,A,S)6xxx(A,I,L,V)10 with the leucine at position 2 invariant (Luo et al., 2007). The invariant leucine at position 2 of this box is critical because mutation of this amino acid to an alanine residue prevents the interaction of Vif with elongin C (Kobayashi et al., 2005; Yu et al., 2003 Yu et al., 2004). Since the S147 and L148 amino acids of the SLQYLA domain are conserved among the primate lentivirus Vif proteins, we chose to change these amino acids to alanine residues. Our results differ from those previously reported by Desrosiers and colleagues (1998), who reported transient replication of a SIVΔVif in macaques before it was undetectable in the plasma. At first glance the results presented here and those reported by Desrosiers and colleagues (1998) might appear to be conflicting. However, there are important differences in the viruses used in each study. In the study of Desrosiers and colleagues (1998), the vif was truncated (only 28 amino acids were expressed) and the virus was SIVmac239, which expresses a R5 envelope glycoprotein. The virus used in this study had site-directed mutations in vif and the viral background was SHIVKU-1bMC33, which is based on SIVmac239 but expresses an X4 envelope glycoprotein. It is known that SIVmac239 targets infection of the gut mucosal CD4+ cells, (Veazey et al., 1998) while previous studies have shown that pathogenic X4 SHIVKU-1bMC33 targets the thymus during the acute phase of infection. Thus, it is possible that each virus is targeting different populations of cells in the early stage of infection, which may express differing amounts of the APOBEC3 family members.
The results from this study indicate that macaques inoculated with SHIVVifAAQYLA developed no significant loss of circulating CD4+ T cells and had a 100-fold lower plasma viral loads during the acute phase of infection, when compared to macaques inoculated with SHIVKU-1bMC33. Additionally, our analyses revealed fewer tissues positive for viral gag DNA and RNA sequences in 2 of 3 macaques in this group, suggesting that SHIVVifAAQYLA was for the most part controlled by the host. This could be due to the presence of susceptible cells expressing one of the APOBEC3 family members that could restrict a virus with these mutations in Vif. The presence of plasma viral loads in these macaques indicates that virus may have persisted in populations of cells that probably either did not express A3G/F/DE/C/B or expressed these proteins at very low levels. These results correlate with our previous studies that showed restricted expression of A3G in brain and kidney tissues (Hill et al., 2006; 2007), both of which are targets for HIV-1, SIV and SHIV (Gattone et al., 1998; Khan et al., 2006; Letendre et al., 2008; Raghavan et al., 1997; Stephens et al., 2000; Zink and Clements, 2002). Thus, a major question that emerged was, “Are the viruses produced from these cellular reservoirs infectious, replication competent viruses?” The gold standard for detecting small amounts of virus is by inoculation into a naive, susceptible animal. Passage of plasma from two macaques into a naive macaque (I95) revealed the presence of virus in PBMCs of this macaque at 1 to 20 weeks and in the 10 tissues examined at necropsy, although the plasma viral loads were below the limits of detection (100 copies per ml). These results suggest that plasma from macaques had infectious virus but it was readily controlled by the macaques.
While none of the three macaques exhibited significant histological lesions, there were two incongruent observations. The first was that macaque RPL10 had the lowest viral loads but had the most widespread tissue distribution of viral sequences. While the reason is unclear, it could be due to greater viral distribution during the early stage of infection. The second observation was that macaque RCS10 developed less immunoprecipitating antibodies compared with macaques RPL10 and RAK10. Whether this relates to the inability to select for the A147T amino acid substitution is currently unknown. Perhaps the A147T substitution permitted more replication in macaques RAK10 and RPL10 resulting in a better antigenic stimulus. It should be noted that virus with the A147T substitution could not be isolated from the two macaques in which it was observed. Thus, we do not know if a virus with a TAQYLA motif is capable of causing CD4+ T cell loss, histological lesions and high virus burdens.
Our sequence analysis indicated that not all viral genes amplified from the lymphoid tissues had G-to-A mutations that were above background levels. For the three macaques, a total of 12 tissues (axillary, mesenteric, and inguinal lymph nodes, thymus from each animal) and four genes (vif, vpu, env and nef) analyzed. Approximately half (25) of the 48 sequences had G-to-A mutations that were above 1.2% with the majority of the mutations in the context of 5′-TC. Potential reasons for this may include that the aliquot of tissue that was examined may have had archived virus from the initial round of replication, the viruses amplified were from a cell type that did not express APOBEC3G/F, or that the domain-specific mutations may have crippled but not completely abolished Vif antiviral activity. Alternatively, it is possible that both deaminase-dependent and deaminase-independent mechanisms may be present to decrease virus replication in macaques. The context in which the G-to-A mutations were found can give clues as to what APOBEC3 member involved in the cytidine deamination. Human A3G is known to cause cytidine deamination with a base preference of 5′-CC (minus strand), A3F a preference for 5′-TC, and A3DE a preference for T/A-T/A-C-G/T (Dang et al., 2007; Liddament et al., 2004; Yu et al., 2004). Previous studies showed that A3G and A3F were widely expressed while a more recent study showed that A3F may not be as widespread as previously reported (Wiegand et al., 2004; Dang et al., 2007). A3DE was also shown to be expressed in the same tissues as A3G (Dang et al., 2007). Of those tissues that showed significant G-to-A mutations, over 85% of the G-to-A mutations were in the context of 5′-TC, suggesting that one or more APOBEC3 proteins were probably responsible for the majority of the observed mutational patterns with APOBEC3G and its GG to AG mutational pattern accounting for a minority (<15%) of the observed G to A mutations.
Attenuated viruses have been used to prevent many viral diseases in humans (smallpox, poliovirus, measles, mumps, rubella, chickenpox etc.). It is known that live, attenuated virus vaccines generally result in better immune responses (both humoral and cell mediated) that are longer lived when compared to killed virus vaccines. However, the major drawback of attenuated virus vaccines is that live viruses are genetic elements and replication can introduce mutations that result in reversion of the attenuated phenotype to a pathogenic phenotype. The prime example of this is the live attenuated poliovirus vaccine (Kew et al., 2005). The major concern for the use of attenuated lentiviruses has been that it involved the removal of “non-essential” viral genes such as vpr, vpx, nef or vpu, which could allow for low level virus replication and the accumulation of compensatory mutations in other gene and potentially a reversion to a pathogenic phenotype. The work by Daniel and colleagues (1992) showed that deletion of the nef from the pathogenic SIVmac239 resulted in an attenuated virus that resisted challenge against pathogenic parental virus. However, the hope that an attenuated vaccine approach was soon diminished by the studies showing that inoculation of a SIVmac239 derivative with deletions in vpr, nef and the negative regulatory element (NRE) could cause AIDS in neonatal macaques (Baba et al., 1995). This was followed with a study showing that the same virus could cause disease in both neonatal and adult macaques (Baba et al., 1999). Despite these setbacks, the vaccines against SIV (or HIV-1) infection that have shown the most promise have been those that have deleted or inactivated one or more genes of the virus (Koff et al., 2006). In contrast to Vpr, Vpx, Vpu and Nef, the Vif protein is required for replication in CD4+ T cells and macrophages, the two major cell populations that HIV-1 infects productively (Chiu and Greene, 2008). This suggests that mutations that target one or more conserved (and functional) domains within Vif may permit limited replication and generation of effective immune responses against the virus prior to “self inactivation” through the activities of various APOBEC3 family members and may represent a novel means to attenuate lentiviruses for candidate vaccines.
MATERIALS AND METHODS
Cells, plasmids, and viruses
The CEM, and CEM-SS lymphocyte cell lines were used for transfections and as indicator cells to measure infectivity and cytopathicity of the viruses used in this study. CEM and CEM-SS cell lines (obtained from the NIH AIDS Research and Reagents Program) and C8166 cells were maintained in RPMI-1640, supplemented with 10mM Hepes buffer pH 7.3, 2 mM glutamine, 5 μg per ml gentamicin and 10% fetal bovine serum (R10FBS). The derivation of SHIVKU-1bMC33 has been previously described (McCormick-Davis et al., 2000b; Stephens et al., 2002).
Construction of SHIVVifAAQYLA
For the construction of SHIVVifAAQYLA, a Pac I/SphI fragment (nucleotides 4609 to 5901) from the p5′-SHIV4 was subcloned into pGEMT-EZ vector (a Pac I site was introduced into the multi-cloning site). The serine to alanine substitution at position 147 of Vif was introduced using oligonucleotides (only sense strand shown) 5′-AAGTACCAGGTACCAGCCCTACAGTACTTA-3′ and the Quick-Change Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. The resulting plasmid, pGEMPAC-SphALQYLA, was used to change the leucine at position 148 to an alanine using oligonucleotides (sense strand shown) 5′-ACCAGGTACCAGCCGCACAGTACTTAGCAC-3′ and the Quick- Change Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. The resulting plasmid, pGEMPAC-SphAAQYLA, was isolated and digested with PacI/SphI. The isolated fragment was subcloned into p5′SHIV-4 to generate 5′-SHIVVifAAQYLA and was sequenced to determine if the desired mutations were introduced as expected. To generate virus, p5′SHIVVifAAQYLA and p3′SHIVKU-1bMC33 were digested with Sph I and the two plasmids ligated together with T4 DNA ligase. The resulting ligation was transfected into CEM-SS cells as previously described (McCormick-Davis et al., 2000b; Stephens et al., 2002; Hout et al., 2004; 2005). Stocks of SHIVKU-1bMC33 and SHIVVifAAQYLA were prepared and titered in CEM-SS cells and stored at −86C.
Replication of SHIVVifAAQYLA in APOBEC3G positive and negative cell lines
We assessed the replication of SHIVVifAAQYLA in C8166 (A3G/F positive) and CEM-SS (A3G/F negative) cell lines. Cells were infected with equivalent levels (50 ng) of parental SHIVKU-1bMC33 or SHIVVifAAQYLA for 4 hours at 37C. The cells were centrifuged and washed 3 times to remove the inoculum and incubated in fresh medium at 37C for 9 days. Aliquots of culture supernatants were obtained at 0, 1, 3, 5, 7, and 9 days post-inoculation and the levels of p27 assessed using commercial p27 antigen capture kits.
Macaques analyzed in this study
Five 1–1.5 year old pig-tailed macaques (Macaca nemestrina: 2000Macaca nemestrina: 2001; CM4G; CM4K; RRH10) were intravenously inoculated with 1 ml of undiluted supernatant from C8166-grown stocks of SHIVKU-1bMC33 containing 104 TCID50 per milliliter. Three additional macaques (RAK10; RPL10; RCS10) were inoculated with 104 TCID50 SHIVVifAAQYLA (titered in CEM-SS). At necropsy, 2 ml of plasma from macaques RPL10 and RAK10 were used to inoculate a naive macaque, I95. The animals were housed in the AAALAC-approved animal facility at the University of Kansas Medical Center. Heparinized blood was collected weekly for the first 4 weeks, then at 2 week intervals for the next month, and thereafter at monthly intervals.
Assays for circulating CD4+ T cells
Alterations in the levels of CD4+ T cells after experimental inoculations were monitored sequentially by FACS analysis (Becton Dickinson). T cell subsets were labeled with OKT4 (CD4; Ortho Diagnostics Systems, Inc), SP34 (CD3; Pharmingen) or FN18 (CD3; Biosource International) monoclonal antibodies. T cell subsets from a normal uninfected macaque were always performed at the same time to serve as a control for the FACS analysis.
Processing of tissue samples at necropsy
At the time of euthanasia (28 weeks for those macaques inoculated with SHIVVifAAQYLA and 20 weeks for macaque I95), all animals in this study were anesthetized by administration of 10 mg/kg ketamine (IM) followed by intravenous administration of sodium phenobarbital at 20–30 mg/kg. A laparotomy was performed and the animal exsanguinated by aortic canulation and perfused with one liter of cold Ringer’s saline. All aspects of the animal studies were performed according to the institutional guidelines for animal care and use at the University of Kansas Medical Center. Lymphoid and non lymphoid tissues (heart, kidneys, liver, lungs, mesenteric, inguinal and axillary lymph nodes, pancreas, salivary gland, small intestine, spleen, thymus, tonsils) were obtained and aliquots of tissue snap frozen for DNA and RNA assays. Portions of lymphoid tissues were immersed in HBSS to quantify levels of infectious virus in tissues.
Analysis of tissues for viral RNA and DNA sequences
DNA was extracted from the visceral organs listed above as previously described (Hill et al., 2008) and used in nested PCR to amplify SIV gag sequences. The sensitivity of detection of gag was 1–5 copies and have been previously described (Joag et al., 1994). The amplified product was 240 base pairs. The amplified product was visualized by electrophoresis through 1.5% agarose gels and staining with ethidium bromide.
We also analyzed tissues for the presence of viral RNA, indicative of actively replicating virus. RNA was extracted from approximately 30 mg of each visceral or CNS tissue using the RNAEasy kit (Qiagen) and the manufacturer’s instructions. RNA samples were digested with DNase I for 30 minutes. Samples were run on agarose formaldehyde gels before and after DNase I treatment to check for the presence of contaminating DNA. RT-PCR amplification of the extracted RNA was performed using the Titan One RT-PCR kit (Roche) and the manufacturer’s instructions. Each reaction used one microgram of total RNA and was amplified using oligonucleotide primers specific for the SIV gag gene. The oligonucleotides used for the first round of amplification were 5′-CGTCATCTGGTGCATTCACG-3′ (sense) and 5′-CTGATTAATGTCATAGGGGGTGC-3′ (antisense), which are complementary to bases 1343–1362 and 1636–1658 of SIVmac239, respectively. The nested SIVmac239 primers used were 5′-CACGCAGAAGAGAAAGTGAAACAC-3′ (sense) and 5′-GGTGCAACCTTCTGACAGTGC-3′ (antisense), which are correspond to bases 1359–1382 and 1620–1640, respectively. As a control, β-actin was amplified using the oligonucleotides 5′-TCATGTTTGAGACCTTCAACACCCCAG-3′ (sense) and 5′-CCAAGAAGGAAGGCTGGAAGAGTGCC-3′ (antisense). The reactions were performed with an Applied Systems 2720 Thermal Cycler using the following thermal profile: 42°C for 30 min, 1 cycle; 94°C for 2 min, 1 cycle; 94°C for 30 seconds, 55°C for 30 seconds, and 68°C for 45 seconds, 10 cycles; 94°C for 30 seconds, 55°C for 30 seconds, and 68°C for 2 minutes, 25 cycles. One microliter of the initial reaction mixture was then added to a nested PCR mixture containing gag primers and performed with the following thermal profile: 95°C for 1 minute, 48°C for 2 minute, and 72°C for 3 minutes, 35 cycles. The amplified gag fragment is 281 base pairs.
Sequence analysis of the vif, vpu, env, and nef genes
To determine the stability of the mutations that were introduced into vif, the vif gene was amplified from PBMCs at different time points during infection and from several tissue DNA samples taken at necropsy. One microgram of extracted genomic DNA was used in a DNA polymerase chain reaction (Takara, Madison, WI) following the manufacturer’s instructions. Oligonucleotides were employed that amplified the entire vif, vpu, and nef genes and a region of env from nucleotides 704–1530 in gp120. For sequence analysis, the PCR products from three separate PCR reactions were pooled and separated by electrophoresis in a 1.5% agarose gel, isolated, and each PCR reaction directly sequenced. Cycle sequencing reactions using the BigDye Terminator Cycle Sequencing Ready Reaction Kit with AmpliTaq DNA polymerase, FS (PE Applied Biosystems, Foster City, CA) sequence detection was conducted with an Applied Biosystems 377 Prism XL automated DNA sequencer and visualized using the ABI Editview program. Sequences were compared to the sequence of SHIVKU-1bMC33. A total of 466, 246, 490, and 400 nucleotides were analyzed from the vif, vpu, env, and nef genes, respectively using the SE Central Software package.
Plasma virus loads
Plasma viral RNA loads were determined on RNA extracted from 500μl of EDTA-treated plasma. Virus was pelleted in an ultracentrifuge (Beckman SW55Ti, 100,00xg, 2 hours) and RNA extracted using the Qiagen viral RNA kit (Qiagen, Valencia, CA). RNA samples were analyzed by real-time RT-PCR using gag primers and a 5′FAM and 3′TAMRA labeled Taqman probe that was homologous to the SIV gag gene as previously described (Hofmann-Lehmann et al., 2000). Standard curves were prepared using a series of six ten-fold dilutions of viral RNA of known concentration. The sensitivity of the assay was 100 RNA equivalents per ml. Samples were analyzed in triplicate and the number of RNA equivalents were calculated per ml of plasma.
Immunoprecipitation assays
To determine if the macaques developed antibodies to SHIV proteins following inoculation, the plasma obtained at necropsy was used in immunoprecipitation assays. C8166 cells were inoculated with approximately 104 TCID50 of SHIVKU-1bMC33 for 4 days. The cells were then incubated in methionine/cysteine-free media for 2 hours and radiolabeled labeled with 500
 Ci of 35Smethionine/cysteine for 15 hours. The cells were lysed in 1ml of 1X RIPA buffer and nuclei were removed by centrifugation. The cell lysates were incubated overnight at 4°C with 10 μl of plasma from each macaque and protein A Sepharose beads. The immunoprecipitates bound to the beads were washed three times in 1X RIPA, resuspended in 75
l of 2X sample reducing buffer, and boiled for 5 minutes. Proteins were separated on a 10% SDS-PAGE gel and proteins visualized by autoradiography. Controls included pooled prebleed plasma from macaques RAK10, RPL10, and RCS10 (negative control) and plasma from macaques that had been previously inoculated with non-pathogenic SHIV-4 (positive control).
Ci of 35Smethionine/cysteine for 15 hours. The cells were lysed in 1ml of 1X RIPA buffer and nuclei were removed by centrifugation. The cell lysates were incubated overnight at 4°C with 10 μl of plasma from each macaque and protein A Sepharose beads. The immunoprecipitates bound to the beads were washed three times in 1X RIPA, resuspended in 75
l of 2X sample reducing buffer, and boiled for 5 minutes. Proteins were separated on a 10% SDS-PAGE gel and proteins visualized by autoradiography. Controls included pooled prebleed plasma from macaques RAK10, RPL10, and RCS10 (negative control) and plasma from macaques that had been previously inoculated with non-pathogenic SHIV-4 (positive control).
Acknowledgments
The work reported here is supported by grants NIH grants AI 64019 and AI51981 to E.B.S. We thank Dr. Joyce Slusser for assistance with the flow cytometry. The anti-APOBEC3G was kindly provided by the NIH AIDS Research and Reference Reagent Program. We thank members of the KUMC Biotechnology Support Facility and the Northwestern Genomics Core Facility for their assistance with the sequence analysis and oligonucleotide synthesis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baba TW, Jeong YS, Pennick D, Bronson R, Greene MF, Ruprecht RM. Pathogenicity of live, attenuated SIV after mucosal infection of neonatal macaques. Science. 1995;267:1820–1825. doi: 10.1126/science.7892606. [DOI] [PubMed] [Google Scholar]
- Baba TW, Liska V, Khimani AH, Ray NB, Dailey PJ, Penninck D, Bronson R, Greene MF, McClure HM, Martin LN, Ruprecht RM. Live attenuated, multiply deleted simian immunodeficiency virus causes AIDS in infant and adult macaques. Nat Med. 1999;5:194–203. doi: 10.1038/5557. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Greene WC. The APOBEC3 cytidine deaminases: an innate defensive network opposing exogenous retroviruses and endogenous retroelements. Annu Rev Immunol. 2008;26:317–335. doi: 10.1146/annurev.immunol.26.021607.090350. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Soros VB, Kreisberg JF, Stopak K, Yonemoto W, Greene WC. Cellular APOBEC3G restricts HIV-1 infection in resting CD4+ T cells. Nature. 2005;435:108–114. doi: 10.1038/nature03493. [DOI] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Cur Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Dang Y, Wang X, Esselman WJ, Zheng Y-H. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2007;80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang Y, Siew LM, Wang X, Han Y, Lampen R, Zheng YH. Human cytidine deaminase APOBEC3H restricts HIV-1 replication. J Biol Chem. 2008;283:11606–11614. doi: 10.1074/jbc.M707586200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel MD, Kirchhoff F, Czajak SC, Sehgal PK, Desrosiers RC. Protective effects of a live attenuated SIV vaccine with a deletion in the nef gene. Science. 258:1938–1941. doi: 10.1126/science.1470917. [DOI] [PubMed] [Google Scholar]
- Desrosiers RC, Lifson JD, Gibbs JS, Czajak SC, Howe AY, Arthur LO, Johnson RP. Identification of highly attenuated mutants of simian immunodeficiency virus. J Virol. 1998;72:1431–1437. doi: 10.1128/jvi.72.2.1431-1437.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Dussart S, Courcoul M, Bessou G, Douaisi M, Duverger Y, Vigne R, Decroly E. The Vif protein of human immunodeficiency virus type 1 is posttranslationally modified by ubiquitin. Biochem Biophys Res Com. 2004;315:66–72. doi: 10.1016/j.bbrc.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Gattone VH, 2nd, Tian C, Zhuge W, Sahni M, Narayan O, Stephens EB. SIV-associated nephropathy in rhesus macaques infected with lymphocyte-tropic SIVmac239. AIDS Res Hum Retroviruses. 1998;14:1163–1180. doi: 10.1089/aid.1998.14.1163. [DOI] [PubMed] [Google Scholar]
- Harris RS, Bishop KN, Sheehy AM, Craig HM, Petersen-Mahrt SK, Watt IN, Neuberger MS, Malim MH. DNA deamination mediates innate immunity to retroviral infection. Cell. 2003;113:803–809. doi: 10.1016/s0092-8674(03)00423-9. [DOI] [PubMed] [Google Scholar]
- Hill MS, Mulcahy ER, Gomez ML, Pacyniak E, Berman NEJ, Stephens EB. APOBEC3G expression is restricted to neurons in the brains of pig-tailed macaques. AIDS Res Human Retro. 2006;22:541–550. doi: 10.1089/aid.2006.22.541. [DOI] [PubMed] [Google Scholar]
- Hill MS, Ruiz A, Gomez LM, Miller JM, Berman NEJ, Stephens EB. APOBEC3G expression is restricted to epithelial cells of the proximal convoluted tubules and is not expressed in the glomeruli of macaques. JHistochem Cytochem. 2007;55:63–70. doi: 10.1369/jhc.6A7054.2006. [DOI] [PubMed] [Google Scholar]
- Hofmann-Lehmann R, Swenerton RK, Liska V, Leutenegger CM, Lutz H, McClure HM, Ruprecht RM. Sensitive and robust one-tube real-time reverse transcriptase-polymerase chain reaction to quantify SIV RNA load: comparison of one- versus two-enzyme systems. AIDS Res Hum Retro. 2000;16:1247–1257. doi: 10.1089/08892220050117014. [DOI] [PubMed] [Google Scholar]
- Hout DR, Gomez ML, Pacyniak E, Gomez LM, Inbody SH, Mulcahy ER, Culley N, Pinson DM, Powers MF, Wong SW, Stephens EB. Scrambling of the amino acids within the transmembrane domain of Vpu results in a simian-human immunodeficiency virus (SHIVTM) that is less pathogenic for pig-tailed macaques. Virology. 2005;339:56–69. doi: 10.1016/j.virol.2005.04.038. [DOI] [PubMed] [Google Scholar]
- Hout DR, Gomez ML, Pacyniak E, Gomez LM, Fegley B, Mulcahy ER, Hill MS, Culley N, Pinson DM, Nothnick W, Powers MF, Wong SW, Stephens EB. Substitution of the transmembrane domain of Vpu in simian-human immunodeficiency virus (SHIVKU1bMC33) with that of M2 of influenza A results in a virus that is sensitive to inhibitors of the M2 ion channel and is pathogenic for pig-tailed macaques. Virology. 2006;344:541–559. doi: 10.1016/j.virol.2005.08.022. [DOI] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Joag SV, Stephens EB, Adams RJ, Foresman L, Narayan O. Pathogenesis of SIVmac infection in Chinese and Indian rhesus macaques: effects of splenectomy on virus burden. Virology. 1994;200:436–446. doi: 10.1006/viro.1994.1207. [DOI] [PubMed] [Google Scholar]
- Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew OM, Sutter RW, de Gourville EM, Dowdle WR, Pallansch MA. Vaccine-derived polioviruses and the endgame strategy for global polio eradication. Annu Rev Microbiol 2005. 2005;59:587–635. doi: 10.1146/annurev.micro.58.030603.123625. [DOI] [PubMed] [Google Scholar]
- Khan S, Haragsim L, Laszik ZG. HIV-associated nephropathy. Adv Chronic Kidney Dis. 2006;13:307–313. doi: 10.1053/j.ackd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C complex is essential for Vif function. J Biol Chem. 2005;280:18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- Koff WC, Johnson PR, Watkins DI, Burton DR, Lifson JD, Hasenkrug KJ, McDermott AB, Schultz A, Zamb TJ, Boyle R, Desrosiers RC. HIV vaccine design: insights from live attenuated SIV vaccines. Nat Immunol. 2006;7:19–23. doi: 10.1038/ni1296. [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Lee YN, Bieniasz PD. Reconstitution of an infectious human endogenous retrovirus. PLoS Pathog. 2007;3:e10. doi: 10.1371/journal.ppat.0030010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letendre S, McCutchan JA, Ellis RJ. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2008;16:15–22. [PubMed] [Google Scholar]
- Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Cur Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci U S A. 2005;102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Mariani R, Chen D, Schrofelbauer B, Navarro F, Konig R, Bollman B, Munk C, Nymark-McMahon H, Landau NR. Species-specific exclusion of APOBEC3G from HIV-1 virions by Vif. Cell. 2003;114:21–31. doi: 10.1016/s0092-8674(03)00515-4. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mbisa JL, Barr R, Thomas JA, Vandegraaff N, Dorweiler IJ, Svarovskaia ES, Brown WL, Mansky LM, Gorelick RJ, Harris RS, Engelman A, Pathak VK. Human immunodeficiency virus type 1 cDNAs produced in the presence of APOBEC3G exhibit defects in plus-strand DNA transfer and integration. J Virol. 2007;81:7099–7110. doi: 10.1128/JVI.00272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick-Davis C, Dalton SB, Hout DR, Singh DK, Berman NE, Yong C, Pinson DM, Foresman L, Stephens EB. A molecular clone of simian-human immunodeficiency virus ( vpuSHIVKU-1bMC33) with a truncated, non-membrane-bound vpu results in rapid CD4+ T cell loss and neuroAIDS in pig-tailed macaques. Virology. 2000;272:112–126. doi: 10.1006/viro.2000.0333. [DOI] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. NatMed. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Singh DK, Griffin DM, Pacyniak E, Jackson M, Werle MJ, Wisdom B, Sun F, Hout DR, Pinson DM, Gunderson RS, Powers MF, Wong SW, Stephens EB. The presence of the casein kinase II phosphorylation sites of Vpu enhances the CD4+ T cell loss caused by the simian-human immunodeficiency virus SHIVKU-lbMC33 in pig-tailed macaques. Virology. 2003;313:435–451. doi: 10.1016/s0042-6822(03)00339-8. [DOI] [PubMed] [Google Scholar]
- Stephens EB, Tian C, Dalton SB, Gattone VH., 2nd Simian-human immunodeficiency virus-associated nephropathy in macaques. AIDS Res Hum Retroviruses. 2000;16:1295–1306. doi: 10.1089/08892220050117050. [DOI] [PubMed] [Google Scholar]
- Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, Nothnick W, Wong SW, Gunderson R, Berman NE, Singh DK. Deletion of the vpu sequences prior to the env in a simian-human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology. 2002;293:252–261. doi: 10.1006/viro.2001.1244. [DOI] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif blocks the antiviral activity of APOBEC3G by impairing both its translation and intracellular stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig MM, Johnson RP, Desrosiers RC, Lackner AA. Gastrointestinal tract as a major site of CD4+ T cell depletion and viral replication in SIV infection. Science. 1998;280:427–431. doi: 10.1126/science.280.5362.427. [DOI] [PubMed] [Google Scholar]
- Virgen CA, Hatziioannou T. Antiretroviral activity and Vif sensitivity of rhesus macaque APOBEC3 proteins. J Virol. 2007;81:13932–13937. doi: 10.1128/JVI.01760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif proteins. EMBO J. 2004;23:2451–2488. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Guo F, Cen S, Kleiman L. Inhibition of initiation of reverse transcription in HIV-1 by human APOBEC3F. Virology. 2007;365:92–100. doi: 10.1016/j.virol.2007.03.022. [DOI] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Yu Q, Chen D, Konig R, Mariani R, Unutmaz D, Landau NR. APOBEC3B and APOBEC3C are potent inhibitors of simian immunodeficiency virus replication. J Biol Chem. 2004;279:53379–53386. doi: 10.1074/jbc.M408802200. [DOI] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zennou V, Bieniasz PD. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology. 2006;349:31–40. doi: 10.1016/j.virol.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Kurosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink MC, Clements JE. A novel simian immunodeficiency virus model that provides insight into mechanisms of human immunodeficiency virus central nervous system disease. J Neurovirol Suppl. 2002;2:42–48. doi: 10.1080/13550280290101076. [DOI] [PubMed] [Google Scholar]