

Abstract
The human apolipoprotein B mRNA editing enzyme catalytic peptide-like 3 (APOBEC3;A3) family of proteins (A3A-H) are known to restrict various retroviruses and retroelements, but the full complement of rhesus macaque A3 proteins remains unclear. We report the isolation and characterization of the hA3A homologue from rhesus macaques (rhA3A) and show that the rhesus macaque and human A3 genes are orthologous. RhA3A is expressed at high levels in activated CD4+ T cells, is widely expressed in macaque tissues, and is degraded in the presence of the human immunodeficiency virus (HIV-1) and simian-human immunodeficiency virus (SHIV) genomes. Our results indicate that rhA3A is a potent inhibitor of SHIVΔvif and to a lesser extent HIV-1Δvif. Unlike hA3A, rhA3A did not inhibit adeno-associated virus 2 (AAV-2) replication and L1 retrotransposition. These data suggest an evolutionary switch in primate A3A virus specificity and provide the first evidence that a primate A3A can inhibit lentivirus replication.
INTRODUCTION
In the last decade several host restriction factors have been discovered that can restrict the replication of HIV-1. One of these restriction factors, Apolipoprotein B mRNA-editing catalytic polypeptide-like 3 (APOBEC3; A3), has been shown to inhibit a wide range of retroviruses and other viruses such as the parvoviruses, papillomaviruses, and hepadnaviruses (Abe et al., 2009; Baumert et al., 2007; Bonvin et al., 2006, 2007; Chen et al., 2006; Henry et al., 2009; Jarmuz et al., 2002; Köck et al., 2008; Mahieux et al., 2005; Narvaiza et al., 2009; Noguchi et al., 2007; Paprotka et al., 2010; Sheehy et al., 2002; Strebel et al., 2005; Turelli et al., 2004; Vartanian et al., 2008; Zhang et al., 2008). A3 proteins comprise a family of seven cytidine deaminases (A3A, A3B, A3C, A3D, A3F, A3G, and A3H) that catalyze the deamination of cytidine to uracil on single-stranded DNA or RNA through a highly conserved Zn+2-binding motif (C/H-X-E-X23-28-P-C-X2-4-C) (Chiu et al., 2009; Goila-Gaur and Strebel., 2008; Huthoff et al., 2005; Yu et al., 2004). These proteins also contain a key glutamate required for proton shuttling during catalysis and two aromatic amino acids residues required for RNA substrate binding (Jarmuz et al., 2002).
The A3 proteins can be divided into those with a single (A3A, A3C, A3H) or double (A3B, A3D, A3F, and A3G) cytidine deaminase domains (CDA). The double CDA proteins A3D, A3F, and A3G have been shown to restrict the replication of HIV-1 isolates that do not express a functional Vif protein while A3B could inhibit HIV-1 independent of Vif (Dang et al., 2006; Doehle et al., 2005; Kao et al., 2003; Liddament et al., 2004; Sheehy et al., 2002; Smith et al., 2010; Wiegand et al., 2004; Yu et al., 2004; Zheng et al., 2004). The Vif protein is known to bind to select A3 proteins and shunt the A3 proteins to the proteasome for degradation (Liu et al., 2004; Marin et al., 2003; Mehle et al., 2004; Sheehy et al., 2003; Yu et al., 2003). Mutagenesis studies have shown that the N-terminal CDA lacks catalytic activity and is responsible for binding the RNA while the C-terminal CDA mediates sequence-specific deamination of single-stranded DNA (Yu et al., 2004). In addition to deaminase dependent functions, hA3G and hA3F inhibit virus replication by deaminase-independent mechanisms (Holmes et al., 2007a,b). Human A3G (hA3G) and hA3F can also inhibit DNA synthesis, which may involve interference of t-RNA Lys3 primer annealing, initiation and elongation of DNA synthesis and minus or plus strand DNA transfers (Bishop et al., 2008; Guo et al., 2006, 2007; Holmes et al., 2007a; Iwatani et al., 2007; Li et al., 2007; Luo et al., 2007; Newman et al., 2005; Ooms et al., 2010). Of the single CDA A3 proteins, hA3A has no activity against HIV-1 while hA3C can be incorporated into virions but only has weak activity against HIV-1 and foamy virus (Langlois et al., 2005; Perkovic et al., 2009). Human A3H is now known to have seven haplotypes, several of which can be stably expressed (Harai et al., 2009; Ooms et al., 2010; Wang et al., 2011). However, attempts to detect hA3H in isolated peripheral blood mononuclear cells (PBMC) has been unsuccessful (Li et al., 2010).
Previous research has shown that human APOBEC3A (hA3A) lacks antiviral activity against HIV-1, but inhibits the replication of adeno-associated virus 2 (AAV-2), intracisternal A particles (IAP), and long interspersed nuclear element 1 (L1) (Bogerd et al., 2006a,b; Chen et al., 2006; Muckenfuss et al., 2006). Although hA3A contains one CDA, the deaminase activity of hA3A is not required for its inhibitory role against AAV or retrotransposons, suggesting that a deaminase-independent mechanism of inhibition is occurring (Narvaiza et al., 2009). Human A3A also contains significant homology to the N-terminal half of hA3G, does not associate with the viral nucleocapsid complex (NPC) of HIV-1 but is readily incorporated into virions (Goila-Gaur et al., 2007).
Simian immunodeficiency (SIV) and simian-human immunodeficiency (SHIV) viruses have been invaluable models for studying various aspects of HIV-1 pathogenesis. Sequencing of the rhesus macaque genome has led to the identification of rhesus homologues of A3B, A3C, A3D, A3F, A3G and A3H. Here we describe the identification of an A3A homologue in rhesus macaques (rhA3A) and present evidence that the human and macaque Apobec3 genes are orthologous. We show that the rhA3A protein is expressed well in activated macaque CD4+ T cells and other rhesus macaque tissues, is incorporated into virions, is degraded by both HIV-1 and SHIV in a Vif dependent manner, and restricts the replication of SHIV (expressing the SIVmac239 Vif) and HIV-1.
RESULTS
Molecular cloning of rhesus macaque A3A
Humans encode seven APOBEC3 (hA3) genes that are tandemly arrayed on chromosome 22, but there is uncertainty on whether a similar array of rhA3 genes is expressed in the syntenic region on rhesus macaque chromosome 10. In particular, no rhA3A gene has been cloned from rhesus macaques suggesting that this gene may have been deleted in the genome of this primate species (OhAinle et al., 2008; Virgen et al., 2007). However, a recent study showed that A3A is encoded in the genome of several monkey species, including African green and owl monkeys (Bulliard et al., 2011). The evolutionary conservation of A3A therefore suggested that the rhA3A gene was missed in previous cloning attempts. To determine whether rhA3A is expressed, we took advantage of the sequencing of the rhesus macaque genome (Rhesus Macaque Genome Sequencing and Analysis Consortium, 2007). BLAST analysis of the hA3A sequence revealed that the first exon has a counterpart on the rhesus macaque chromosome 10, thus providing a potential forward primer for PCR amplification. However, counterparts of other hA3A exons were not detected in the rhesus macaque genome immediately downstream of this putative exon 1 sequence. Thus, the last exon of hA3A was used as a reverse primer.
Human A3A is not expressed in resting peripheral blood mononuclear cells (PBMC), but is substantially induced by cellular activation and cytokines (Koning et al., 2009; Refsland et al., 2010). We therefore utilized cDNA from Concanavilin A-activated rhesus macaque PBMCs in an attempt to detect rhA3A. A dominant band of the correct size was amplified in the first attempt by PCR. BLAST analysis of the cloned amplicon revealed 89% nucleotide identity with hA3A. Alignment of the amino acid sequence with recently identified A3A sequences showed the highest identity (90%) with A3A from African green monkeys, which, together with Asian macaques, are considered as “Old World” monkeys (Fig. 1A). This alignment also revealed primate lineage-specific insertions/deletions (indels). In particular, we observed deletions at positions 27 to 29 in the hominid A3A genes. To further explore the clustering of A3A sequences with specific primate lineages, we constructed a phylogenetic tree that excluded the contribution of indels. As shown in Fig. 1B, A3A sequences clustered accordingly with their corresponding primate taxons. These analyses demonstrate that the cloned rhA3A gene is a bona fide member of the A3A gene family.
Fig. 1.
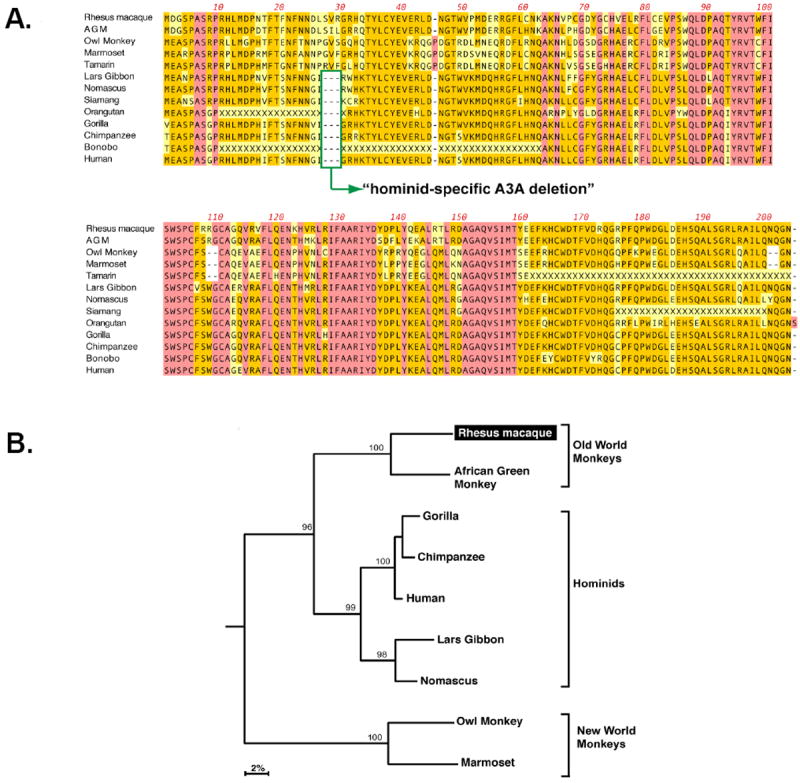
Sequence analysis of the rhA3A proteins. Panel A. Protein sequence alignment of primate A3A proteins. Dashes correspond to indels, while residues highlighted in pink or orange are conserved or matched the consensus, respectively. Some indels were associated with specific primate taxons. This includes a hominid-specific deletion at amino acid positions 27 to 29, and a deletion in residues 108-109 in New World monkeys. Panel B. A3A phylogeny. The amino acid sequence alignment was subjected to phylogenetic analyses using the neighbor joining method, excluding sequence gaps, and rooted using the C-terminal half of rhesus macaque A3G as an outgroup. Numbers correspond to the percentage of clustering following 1000 subreplicates. RhA3A clustered most significantly with A3A proteins from another Old World monkey species, African Green monkey (AGM).
Evidence that seven A3 genes are tandemly arrayed on chromosome 10 of rhesus macaques
While the sequence of the rhesus macaque genome has been “completed,” it still remains unclear why this did not reveal the rhA3A gene. We attempted to determine the position of the cloned rhA3A sequence in relation with other rhA3 genes. At the time that the analyses were performed, only the nucleotide sequences for rhesus macaque rhA3C, rhA3F, rhA3G and rhA3H had been deposited into GenBank. BLAST analysis of these cDNA sequences against the rhesus macaque genome revealed their distinct positions as shown in Fig. 2A. The position of rhesus macaque rhA3B and rhA3DE remains unknown, although their amino acid sequences were published (Virgen et al., 2007). We first attempted to determine the relative positions of rhA3B and rhA3D in chromosome 10 based solely on the genome sequence. Using the geneid program (http://genome.crg.es/geneid.html), genome sequences between rhA3C and rhA3F predicted rhA3D (Fig. S1), while genomic and previously unlocalized sequences upstream of rhA3C predicted rhA3B (Fig. S2).
Fig. 2.
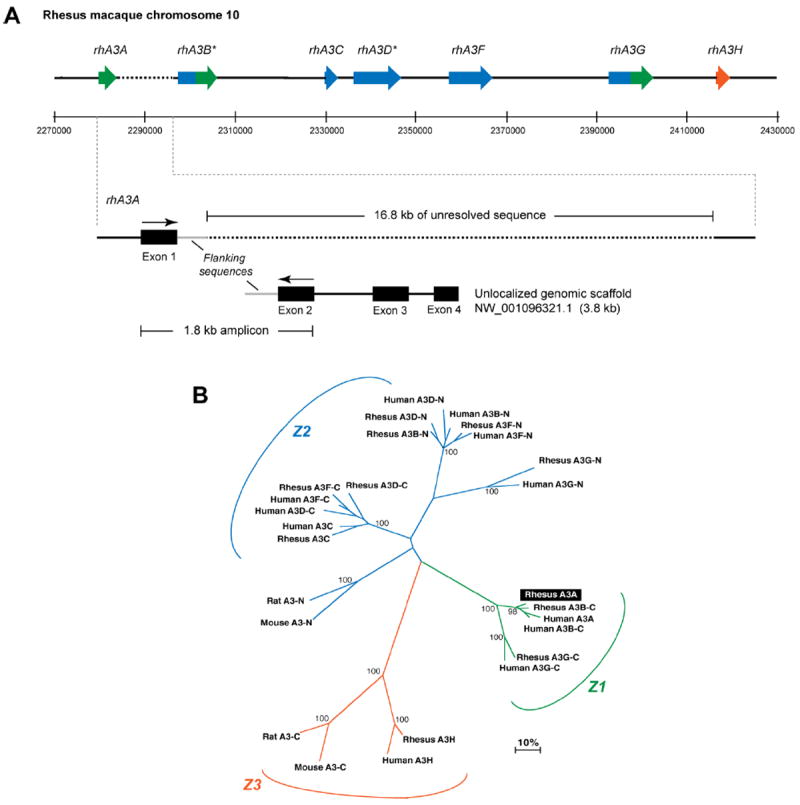
Sequence characterization of the rhesus macaque A3 locus. Panel A. Genome organization of rhesus macaque A3 genes. (Top) Published cDNA sequences for rhA3C, rhA3F, rhA3G and rhA3H were subjected to BLAST analysis against the rhesus macaque genome. Matching exons were plotted against assigned genome coordinates, shown here in 20 kb intervals. *RhA3B and rhA3DE were predicted by subjecting the genome sequences upstream of rhA3C or flanked by rhA3C and rhA3F to the geneid prediction program (http://genome.crg.es/geneid.html), respectively. RhA3A amplified from activated PBMCs were subjected to BLAST analysis. RhA3A exon 1 mapped in the genome directly upstream of A3B, but this was followed by 16.8 kb of unresolved sequence (dotted lines). (Bottom) A3A exons 2 to 4 matched sequences from an unlocalized chromosome 10 genomic scaffold sequence. Primers based on A3A exons 1 and 2 amplified a 1.8 kb product that matched flanking sequences after exon 1 in the genome and before exon 2 in the unlocalized genomic scaffold (gray lines). This provides strong evidence for the current placement of rhA3A in the rhesus macaque genome. Panel B. Phylogeny of rhesus macaque and human A3 proteins. Amino acid sequences from rhesus macaque and human A3 genes were aligned, with the double-CDA members split into an N- and C-terminal half. For example, A3B-C corresponds to the C-terminal half of A3B. RhA3A clustered with the C-terminal half of A3B and A3G, forming the Z1 clade. Each rhesus macaque A3 gene clustered significantly with the corresponding human A3 gene, suggesting that rhesus macaque and human A3 proteins are orthologous.
The relative placement of rhA3B and rhA3D in the rhesus macaque genome suggested a similar A3 gene organization as found in humans, with the notable uncertainty of rhA3A. We therefore focused on genomic sequences immediately upstream of rhA3B. BLAST analysis of the cloned rhA3A sequence revealed an identical sequence corresponding to rhA3A exon 1 in the genome. This putative rhA3A exon 1 was immediately followed by 560 bp of CT-rich repeat sequences and 16.8 kb of unresolved sequence. It is possible that these repeat sequences interfered with accurate sequencing of this region. On the other hand, exons 2 to 4 of rhA3A mapped to an unlocalized chromosome 10 genomic scaffold sequence with GenBank Accession NW_001096321.1. The 3,764 bp NW_001096321.1 sequence could easily be accommodated in the 16.8 kb genomic gap. Notably, the NW_001096321.1 sequence contained 838 bp of intron sequence upstream of exon 2. We therefore investigated whether the NW_001096321.1 links directly with the genome by amplifying rhesus macaque genomic DNA with primers designed in exon 1 and 2 of rhA3A (Fig. 2A). Direct sequencing of the 1.8-kb amplicon revealed identity with the flanking sequences following the putative exon 1 in the genome and prior to exon 2 in NW_001096321.1 (Fig. 2A). These results provide strong evidence that rhA3A is likely encoded in the rhesus macaque genome immediately upstream of rhA3B. Overall, these analyses suggest that the seven rhA3 members are tandemly arrayed in the rhesus macaque genome, similar to humans.
The rhesus macaque and human A3 genes are orthologous
Rodents encode a single A3 gene, while humans encode seven. Other mammals, such as dogs, pigs, horses and sheep encode a variety of A3 genes that reveal substantial duplication, unequal cross-over and divergent evolution of individual members and are therefore considered paralogues (Conticello et al., 2005; LaRue et al., 2006). This pattern of “evolutionary shuffling” could be roughly inferred in phylogenetic trees, where A3 segments containing the canonical deoxycytidine deaminase motif can be classified into Z1, Z2 and Z3 clades (LaRue et al., 2006). To extend this classification to rhA3 genes, a phylogenetic tree comparing the human, rodent and rhesus macaque A3 amino acid sequences was constructed (Fig. 2B). The rhA3 genes consistently clustered with the hA3 counterparts (Fig. 2B). Thus, each of the seven rhesus macaque and human A3 genes appear to originate from a common ancestor, indicating that the genes are orthologous. In other words, we found no evidence for significant “shuffling events” in the A3 locus after the evolutionary split between monkeys and humans.
Expression of rhA3 genes in rhesus macaque tissues and activated CD4+ T cells
We next determined rhA3A mRNA expression in various perfused rhesus macaque organs and tissues. Specific rhA3A primers were used to determine expression in 14 visceral organs and 12 regions of the central nervous system (CNS) by RT-PCR. GAPDH was used to assess the integrity of RNA in the samples (data not shown). Our results indicate that rhA3A was not only widely expressed in the visceral organs but also in different regions of the CNS (Fig. 3A). We verified the specificity of the rhA3A amplification by directly sequencing the PCR amplicons (data not shown). Extensive detection of rhA3A in various lymphoid organs raised the possibility that rhA3A may also be expressed in CD4+ T cells, a primary cellular target of SHIV and SIV infection in vivo.
Fig.3.
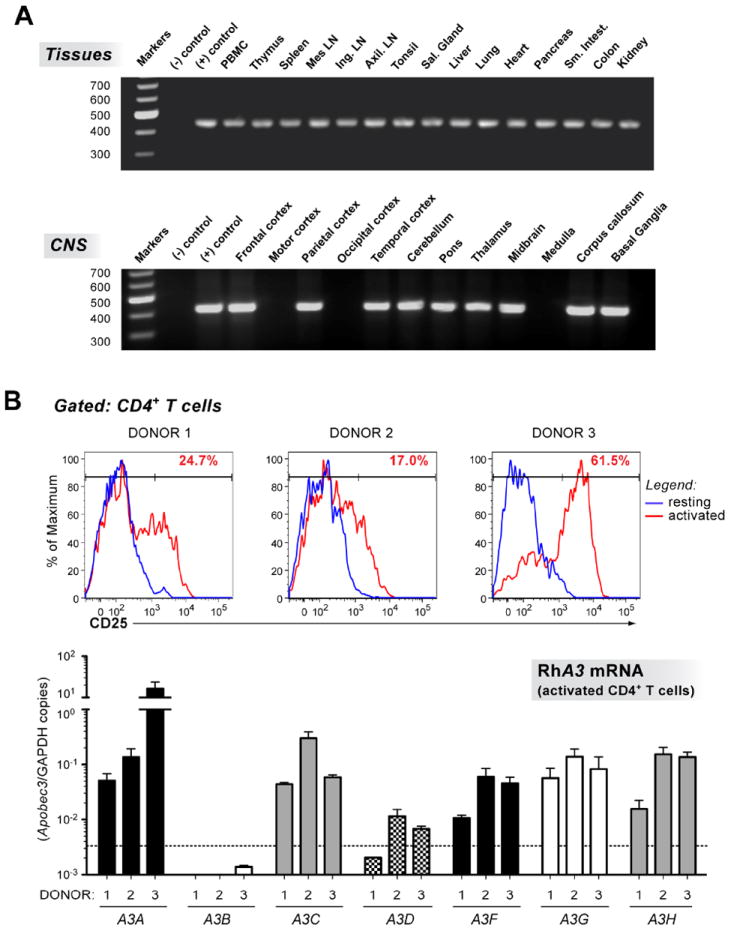
Expression of rhA3A in rhesus macaque tissues and CD4+ T cells. Panel A. RhA3A expression profile in visceral organs and the CNS. Multiple tissues were obtained from an SIV negative macaque. RNA from these tissues was analyzed for the presence of rhA3A mRNA by RT-PCR. Amplicons were run on 1.5% agarose gels and visualized by staining with ethidium bromide. Amplicons were directly sequenced and found to correspond to rhA3A. All samples were positive for GAPDH RNA to control for integrity of RNA in the samples (data not shown). Upper gel, rhA3A mRNA from 14 visceral organs. Lower gel, rhA3A mRNA from 12 regions of the central nervous system. The size markers (in base pairs) are shown to the left. Panel B. Expression of A3 genes in rhesus macaque CD4+ T cells. Upper, Activation of rhesus CD4+ T cells. Rhesus macaque PBMCs were activated with 2.5 μg/ml SEB overnight, and subjected to FACS analysis. FACS histograms of CD25 expression in CD3+ CD4+ cells before (resting; blue) and after SEB activation (activated; red) are shown. CD4+ T cells were magnetically purified by negative selection, and RNA was extracted for quantitative RT-PCR analyses. Lower, rhA3 mRNA levels in activated rhesus macaque CD4+ T cells. A3 cDNA was subjected to real-time quantitative RT-PCR using primers specific for individual rhA3 genes (Table 1). Dashed lines correspond to the assay limit of detection (5 input copies/reaction), while error bars correspond to standard deviations from triplicate determinations.
To determine the relative expression of rhA3A against other rhA3 members, we developed Taqman-based real-time PCR assays to specifically quantify each of the seven rhA3 genes (Table 1). To evaluate specificity, we first performed an end-point PCR and determined by agarose gel electrophoresis whether the primers could amplify other rhA3 cDNAs. If some cross-reactivity with the primers was observed, we subjected 108 rhA3 cDNA copies in a real-time PCR reaction that also includes the probe. The results of these analyses are presented in Table 2. Most of the assays developed did not cross-react with other A3 family members. In those instances where some cross-reactivity was observed, detection of similar A3 members were least 104 fold lower in magnitude than what would be expected for the gene of interest. Thus, the real-time PCR methods could be used to distinguish the relative levels of the individual A3 members.
Table 1.
Rhesus macaque A3 real-time PCR primers and probes.
| Apobec3 | Primers* | Primer Size (nt) | PCR product (nt) | Ta (°C) |
|---|---|---|---|---|
| rhA3A | Forward: CAAGGCTAAGAATGTTCCCT | 20 | 201 | 59 |
| Reverse: ATGCGCAGTCTCACGTGTTT | 20 | |||
| Probe: [6-FAM]TGAGGTTCCTTCTTGGCAGT[TAMRA-6~FAM] | 20 | |||
| rhA3B | Forward: TGGGGTAGAGATTGGCAGAG | 20 | 224 | 59 |
| Reverse: TTGGAAGTGAACGTGTCTGG | 20 | |||
| Probe: [6-FAM]CCGCATGCTAAAGGAGATTC[TAMRA-6~FAM] | 20 | |||
| rhA3C | Forward: AGGTGCTTCCTCTCCTGGTT | 20 | 197 | 59 |
| Reverse: CCCTGCTGGTAATTCGGATA | 20 | |||
| Probe: [6-FAM]GGCACAACAATGTGATGCTC[TAMRA-6~FAM] | 20 | |||
| rhA3DE | Forward: GTCTTCCGAAACCAGGTTGA | 20 | 245 | 59 |
| Reverse: AGCCCCTCCTGGTAATCTGT | 20 | |||
| Probe: [6-FAM]CTCTTGGTTCTGCGACAACA[TAMRA-6~FAM] | 20 | |||
| rhA3F | Forward: GAAGGTGGCCGAATTCCTA | 19 | 264 | 60 |
| Reverse: TTTCTGAGAATCTCCTTTAGCTTGT | 25 | |||
| Probe:[6-FAM]ACTGGGAAACAGATTATCGCA[TAMRA-6~FAM] | 21 | |||
| rhA3G | Forward: GCCATTTAAGCCTTGGAACA | 20 | 230 | 60.5 |
| Reverse: GAGCCTGGTTGCGTAGAAAG | 20 | |||
| Probe: [6-FAM]AACCTTGGGTCAGTGGACAG[TAMRA-6~FAM] | 20 | |||
| rhA3H | Forward: AAGGACCATGCAGAAATTCG | 20 | 239 | 59 |
| Reverse: ACCTGGGATCCACACAGAAG | 20 | |||
| Probe: [6-FAM]AACTATCAGGAGGGGCTGCT[TAMRA-6~FAM] | 20 |
PCR conditions were as follows: 95°C for 10 min followed by 40 cycles of 95°C for 15 s and the corresponding annealing temperature (Ta) for 45 s.
Table 2.
Specificity evaluation of rhApobec3 primers and probes.
| Primer-probe set | Template cross-reactivity (input: 5 × 108 copies) | ||||||
|---|---|---|---|---|---|---|---|
| rhA3A | rhA3B* | rhA3C | rhA3DE* | rhA3F | rhA3G | rhA3H | |
| rhA3A | NT | — | NT | 9.9×101 | 2.7×103 | — | |
| rhA3B | — | — | 2.0×103 | — | — | — | |
| rhA3C | — | NT | NT | — | — | 1.5×104 | |
| rhA3D | — | 1.5×102 | — | — | — | — | |
| rhA3F | 6.5×103 | NT | — | NT | 3.4×101 | — | |
| rhA3G | — | NT | — | NT | 5.1×104 | — | |
| rhA3H | — | NT | 1.7×103 | NT | — | — | |
rhA3B (340nt) and rhA3DE (370nt) templates were synthetic oligos based on predicted sequences. —, no cross-reactivity observed based on endpoint-PCR and gel electrophoresis; NT: not tested.
Rhesus macaque PBMCs from three uninfected donors were stimulated with Staphylococcal Enterotoxin B (SEB), previously determined to be a potent activator of rhesus macaque T cells (Kakimoto et al., 1999). However, SEB-stimulation still did not result in consistent activation, since the levels of CD25 expression in CD4+ T cells varied between the 3 donors (Fig. 3B; upper panel A). Total RNA was extracted from magnetically purified CD4+ T cells from these activated PBMCs, and subjected to real-time RT-PCR. Consistent with our failure to clone rhA3B from activated PBMCs, rhA3B mRNA was not detected in any donor (Fig. 3B; lower panel). With one exception (rhA3A donor 3), the expression of individual rhA3 genes between the donors did not vary more than 10-fold, and did not appear to correlate with CD25+ expression levels. Since rhA3G is well accepted as a biologically relevant restriction factor against SIV (Schmitt et al., 2009; 2010; Schrofelbauer 2006; Sui et al., 2010), we compared the expression of other rhA3 genes relative to rhA3G. Setting the average rhA3G level from the 3 donors as 1, only rhA3A (excluding donor 3), rhA3C and rhA3H exhibited mRNA expression levels that are similar in magnitude (1.0-1.5 fold) to that of rhA3G. In contrast, rhA3F and rhA3D were on average expressed at 2.4- and 13.6-fold lower than rhA3G, respectively. Thus, rhA3A, rhA3C and rhA3H mRNAs are expressed to similar levels as rhA3G in SEB-activated rhesus macaque CD4+ T cells.
Nucleocytoplasmic localization of HA-tagged rhA3A
Previous studies have shown that the cytoplasmic localization of hA3G and the nucelocytoplasmic localization of hA3A could partially explain the differences in the range of retroviruses or retrotransposons that were inhibited by these proteins (Bogerd et al., 2006a,b; Chen et al., 2006; Giola-Gaur et al., 2007; Muckenfuss et al., 2006). To determine whether rhA3A exhibited a similar subcellular localization, we generated a molecular clone of rhA3A with an HA-tag at the amino terminal end for detection purposes. A plasmid expressing either rhA3G or rhA3A (Fig. 4A) was transfected into 293 cells and the expression examined by Western blot analysis using an antibody directed against the HA tag. As shown in Fig. 4A, 293 cells transfected with the vector expressing rhA3G expressed a protein with an Mr of 45kD while cells transfected with the vector expressing rhA3A expressed a protein with an Mr of 24kD, which reflects the size of the two different proteins (363 amino acids for rhA3G versus 202 amino acids for rhA3A). Using confocal microscopy, we found that rhA3G was observed exclusively in the cytoplasm (Fig.4B-D) and that rhA3A was detected in both the cytoplasm and nucleus as it co-localized with a nuclear marker (Fig. 4E-G). Similar to other studies, hA3A was observed rhA3A in both the nucleus and cytoplasm (Fig 4H-J). Thus any differences in the retroviral restriction properties of hA3A and rhA3A could not be explained by the subcellular localization.
Fig. 4.
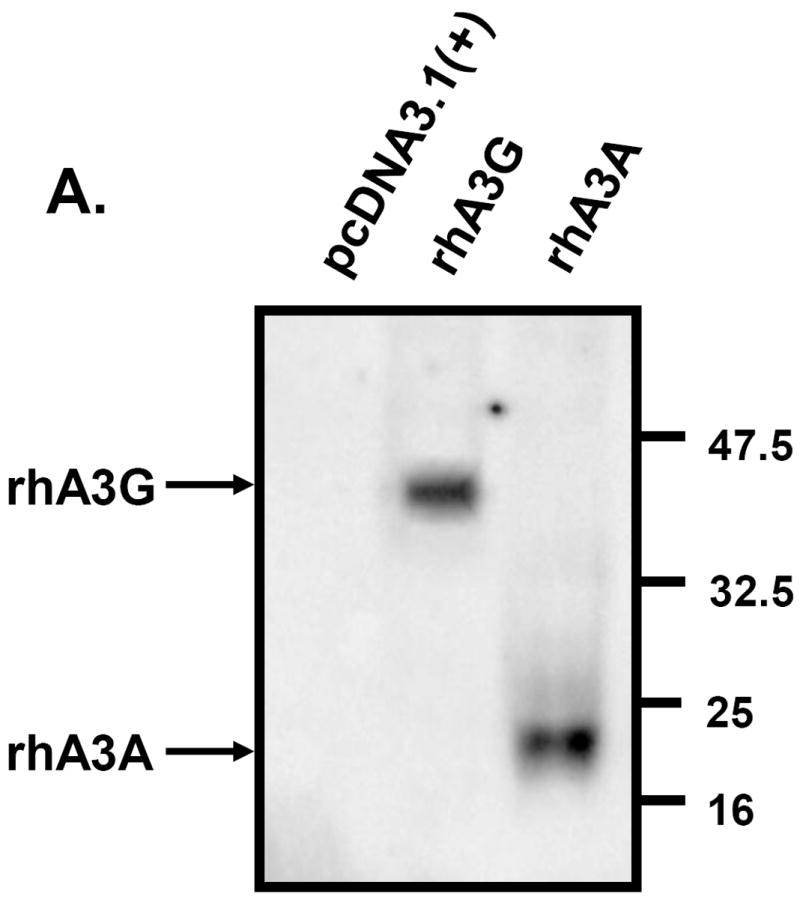
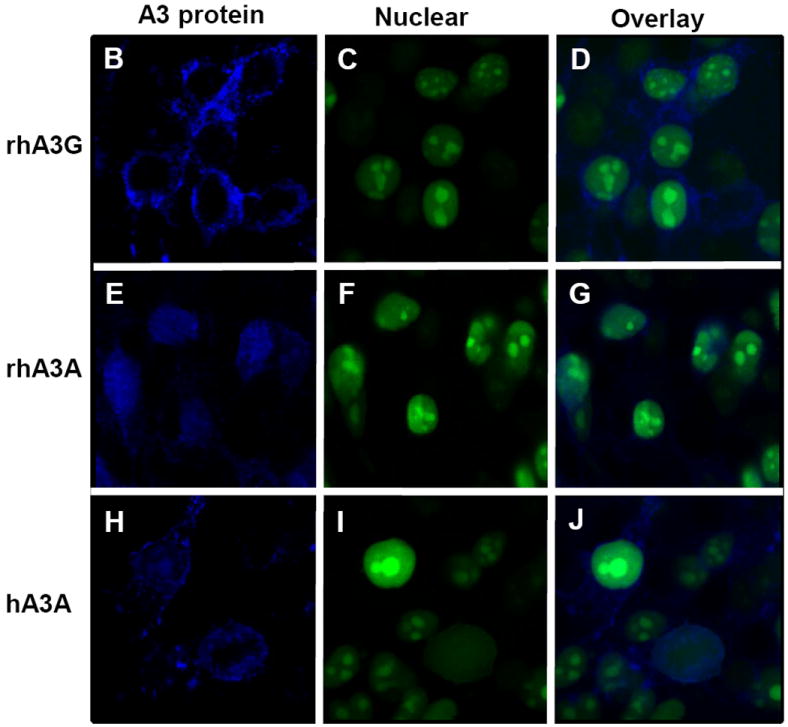
Expression of an HA-tagged rhA3A. Panel A. 293 cells were transfected with 1.5 μg HA-tagged rhA3A, rhA3G, or the empty expression vectors. At 48 h, cells were lysed and proteins separated by SDS-PAGE and analyzed by Western blot using an antibody directed against the HA-tag. Sizes of the molecular standards are to the right. RhA3A migrates at about half the molecular weight than rhA3G, consistent with their amino acid sequences. Panel B-J. Subcellular localization of rhA3G, hA3A, and rhA3A. 293 cells were transfected with HA-tagged vectors expressing rhA3A, hA3A, or rhA3G and a vector expressing an eGFP tagged nuclear marker (___, Clontech). At 48 h, cells were fixed and stained with anti-HA for 1 hr, washed and detected using a horse anti-rabbit-Ig Cy5 antibody. Cells were examined under a Nikon A1 fluorescent microscope and micrographs taken using a 100× objective with a 2× digital zoom using the E2-C1 software package as described in the Materials and Methods.
Minimal inhibition of foreign DNA reporter gene expression by rhA3A and hA3A
In a recent study, hA3A was shown to inhibit the expression of transfected plasmid DNA by deamination of the incoming plasmid DNA (Stenglein et al., 2010). Co-transfection of an eGFP reporter plasmid with hA3A in 293 cells resulted in approximately 20% reduction in the number of cells expressing eGFP signal 2 days. In these investigators’ model, the incoming DNA induces an interferon-induced up-regulation of hA3A expression, the cytidine residues are deaminated resulting in uracils, which are excised by UNG2. This will eventually result in cleavage and degradation of the DNA backbone (Stenglein et al., 2010). This phenomenon could significantly confound experiments that involve co-transfection of A3A with viral plasmid constructs.
Since our virus experiments involving rhA3A or hA3A lasted no longer than 48 h, we determined whether rhA3A, hA3A or rhA3G interfered with the expression of enhanced green fluorescent protein (eGFP) from a foreign plasmid. Plasmids expressing either rhA3A, hA3A or rhA3G were co-transfected into 293 cells 24 h prior to transfection with the plasmid expressing eGFP under the control of the CMV promoter. At 2 days post-transfection of eGFP, the cells were analyzed for fluorescence by flow cytometry. We detected 14.1, 14.5, 11.2, and 14.6% eGFP positive cells in the presence of empty vector, rhA3A, hA3A, or rhA3G, respectively (Fig. 5A). These results indicate that hA3A had minimal impact (<20%) while rhA3A had no impact on reporter gene expression from foreign plasmids at 48 h.
Fig. 5.
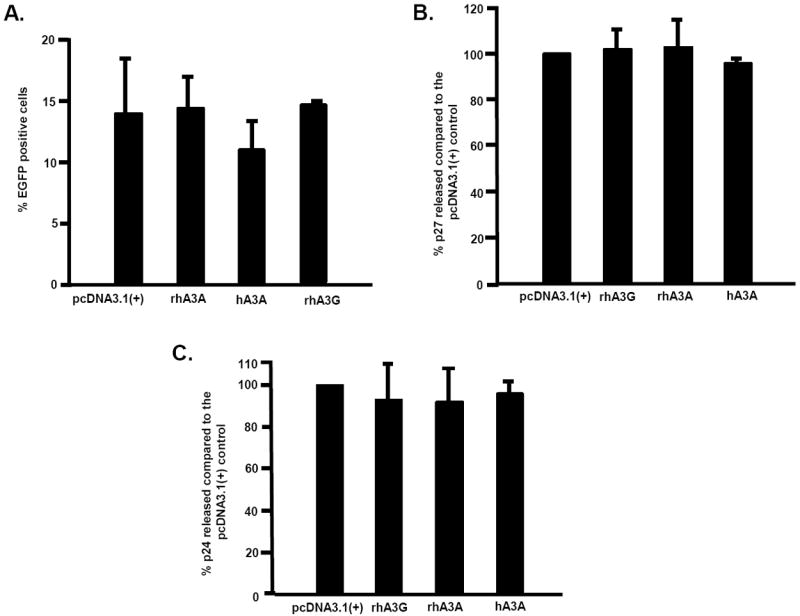
Minimal restriction of foreign DNA by rhA3A and hA3A. Panel A. Expression constructs (3 μg) of HA-tagged rhA3A, hA3A and rhA3G were first transfected into 293 cells for 24 h. At 24 h post-transfection, the 293 cells were re-transfected with a plasmid encoding a CMV-driven eGFP reporter and assayed for reporter expression. The cells were removed from the plate and fixed. All samples were analyzed by a flow cytometer and the mean fluorescence intensity (MFI) for eGFP positive cells was calculated. The MFI ratio and percentage of eGFP cells were calculated for each samples and the experiment was run in triplicate. All groups were compared to the pcDNA3.1(+) plus EGFP control using a Student’s t-test with p<0.05 considered significant. Panel B. SHIVΔVif production in the presence of rhA3A, hA3A and rhA3G. Cells were first transfected with 0.5 μg of plasmids expressing HA-tagged rhA3A, hA3A, rhA3G or empty vector. At 24 h post-transfection, the 293 cells were re-transfected with a plasmid containing the SHIVΔvif. Virus supernatants were harvested at 2 days post-transfection with the viral genome. Mean Gag p27 levels as measured using a commercially available ELISA kit (Zeptometrix) are shown. In all panels, error bars correspond to standard deviations from triplicate determinations. Panel C. HIV-1Δvif production in the presence of hA3A and rhA3A. Cells were first transfected with 0.5 μg of plasmids expressing HA-tagged rhA3A, hA3A, rhA3G or empty vector. At 24 h post-transfection, the 293 cells were re-transfected with a 1 μg plasmid containing the HIV-1Δvif. Virus supernatants were harvested at 2 days post-transfection with the viral genome. Mean Gag p24 levels as measured using a commercially available ELISA kit (Zeptometrix) are shown. In all panels, error bars correspond to standard deviations from triplicate determinations.
Human and rhesus macaque A3A does not inhibit SHIV or HIV-1 production in 293 cells
It can be argued that the slight inhibition of plasmid reporter gene expression by hA3A may be amplified with typically longer plasmids containing a full-length viral genome. Similarly, the absence of an effect by rhA3A in plasmid reporter expression may be enhanced in the context of a full-length viral molecular clone. We conjectured that if A3A can degrade viral infectious molecular clones, then virus production would be significantly inhibited. We therefore investigated whether virus production was inhibited in cells transfected with rhA3A or hA3A. 293 cells were transfected with the plasmids expressing hA3A, rhA3A, rhA3G or the empty plasmid 24 hours prior to transfection with plasmids with the SHIVΔvif or HIV-1Δvif constructs. At 2 days post-transfection with the viral genomes, the culture medium was assayed for either p27 (SHIV) or p24 (HIV-1). We did not observe a significant decrease in the level of SHIVΔvif p27 released into the culture medium in cells co-transfected with empty vector, rhA3A, hA3A, or rhA3G at 5 days post-transfection (Fig. 5B). Similar findings were observed for HIV-1Δvif (Figure 5C). These data suggests that rhA3A and hA3A likely have no post-entry restriction activity against SHIV or HIV-1 and reinforce the conclusion that rhA3A and hA3A have minimal impact on plasmid DNA stability at 2 days post-transfection. Thus, our subsequent experiments, which involve analyzing cells within 48 h, should not be influenced by potential plasmid-specific restriction.
Rhesus macaque A3A inhibits the infectivity of SHIVΔvif and HIV-1Δvif
We next determined if rhA3A could inhibit the infectivity of SHIV and SHIVΔvif. For comparison, we also co-transfected these viral constructs with plasmids expressing rhA3G, rhA3C, or hA3A. Virion infectivity was measured by taking the ratio of infectious titer in TZM-bl cells and comparing to the wild-type SHIV construct co-transfected with empty vector control. Our results indicate that rhA3A and rhA3G reduced the level of infectious SHIVΔvif nearly 20-fold compared to the parental SHIV, while hA3A reduced the infectivity of SHIVΔvif approximately 10-fold (Fig. 6A). In contrast, rhA3C did not inhibit the level of infectious virus released from cells. SHIV virions produced in the presence of rhA3A, hA3A or rhA3G were purified and concentrated by ultracentrifugation and analyzed by Western blot. Consistent with the virion infectivity data, rhA3A was incorporated into SHIVΔvif and to a lesser extent in SHIV (Fig. 6B). However, we were unable to detect hA3A in either SHIV or SHIVΔvif virions.
Fig. 6.
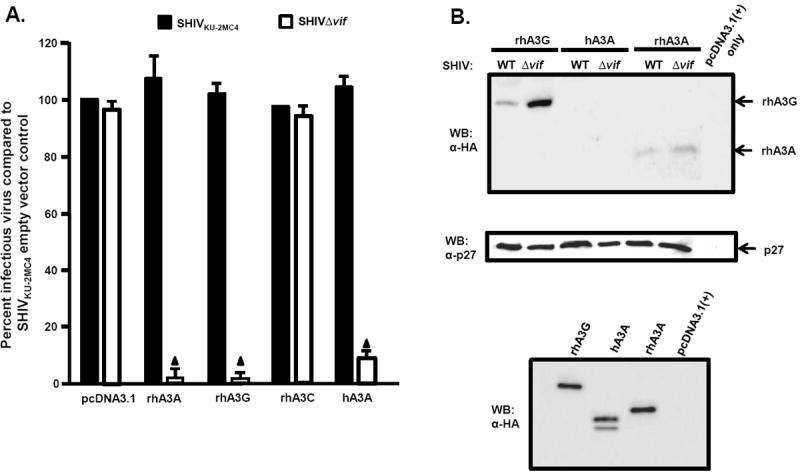
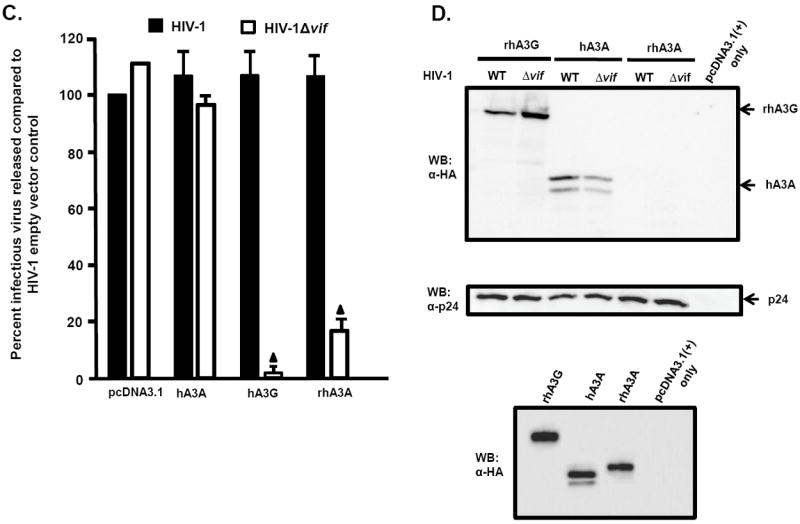
Inhibition of SHIVΔVif and HIVΔVif virion infectivity by rhA3A. Panel A. RhA3A restricts SHIVΔvif. 293 cells were co-transfected with 1 μg wild-type SHIV or SHIVΔvif molecular clones and 0.5 μg plasmids expressing either empty vector (pcDNA3.1), rhA3A, hA3A, or rhA3G. At 48 h, the culture medium was harvested and virion infectivity measured by taking the ratio of infectious viral titers on TZM-bl cells. Shown is the percentage virion infectivity with the empty plasmid control normalized to 100%. Panel B. 293 cells were transfected with wild-type SHIV or SHIVΔvif molecular clones and HA-tagged rhA3A, hA3A, or rhA3G. At 48 h, the culture medium was collected, clarified by low speed centrifugation, and concentrated by ultracentrifugation through a 20%/60% (w/v) step-gradient. Each sample was resuspended in 2X sample reducing buffer, boiled, and the proteins were separated by SDS-PAGE. The presence of rhA3G, hA3A, or rhA3A was detected by Western blot using an antibody directed against the HA-tag (upper) and the presence of p27 using an antibody directed against p27 (middle). The lower panel are 293 cells transfected with vectors expressing rhA3G, hA3A, rhA3A or pcDNA3.1(+) vector in the absence of the viral genome. Panel C. RhA3A restricts HIV-1Δvif virions. HIV-1 or HIV-1Δvif genomes were co-transfected with HA-tagged hA3A, hA3G or rhA3A as described for the SHIV experiments in Panel A. Shown is the percentage virion infectivity with the empty plasmid control normalized to 100%. Panel D. Undetectable rhA3A in HIV-1Δvif virions. Virions were purified as described for the SHIV experiments in Panel B, and hA3G, hA3A and rhA3A incorporation were detected by Western blot using an anti-HA antibody, normalizing for Gag p24 levels. The lower panel are 293 cells transfected with vectors expressing rhA3G, hA3A, rhA3A or pcDNA3.1(+) vector in the absence of the viral genome. Mean values in triplicate experiments are shown, and statistical differences with the wild-type control were evaluated using a two-tailed Student’s t-test, with p<0.05 (▲) considered significant.
We also tested whether rhA3A had activity against HIV-1. HIV-1(NL4-3) and HIV-1Δvif constructs were co-transfected with plasmids expressing rhA3A, hA3A, or hA3G. Consistent with previous reports, hA3A did not inhibit infectivity of HIV-1Δvif virions, while hA3G reduced HIV-1Δvif virion infectivity by 10-fold. Surprisingly, rhA3A reduced HIV-1Δvif virion infectivity approximately 3-fold (Fig. 6C). HIV-1 virions released from 293 cells expressing either hA3G, hA3A or rhA3A were partially purified by ultracentrifugation The resulting virions were analyzed by Western blot using an anti-HA antibody. Consistent with previously published data, hA3A was detected in both HIV-1 and HIVΔvif virions. However, rhA3A was not detected in HIV-1 and HIVΔvif virions (Fig. 6D). Together, these findings indicate that unlike hA3A, rhA3A reduces the infectivity of both SHIVΔvif and HIV-1Δvif.
Deletion of amino acids 27-29 of rhA3A abolishes its anti-viral activity
Hominid and monkey A3As can be distinguished by a 3 amino-acid indel between residues 27 to 29. We hypothesize that this region may contribute to the differential activity of rhA3A and hA3A against SHIVΔvif and HIV-1Δvif. To test this hypothesis, we constructed a rhA3A mutant in which amino acids 27-29 (SVR) were deleted. The resulting mutant, rhA3AΔSVR, was found to be stable in cells (data not shown). Interestingly, rhA3AΔSVR was inactive against SHIVΔvif (Fig. 7A) and HIV-1Δvif (Fig. 7B). These findings suggested that these monkey-specific A3A residues were critical for lentivirus inhibition, but was functionally lost in the hominid lineage.
Fig. 7.
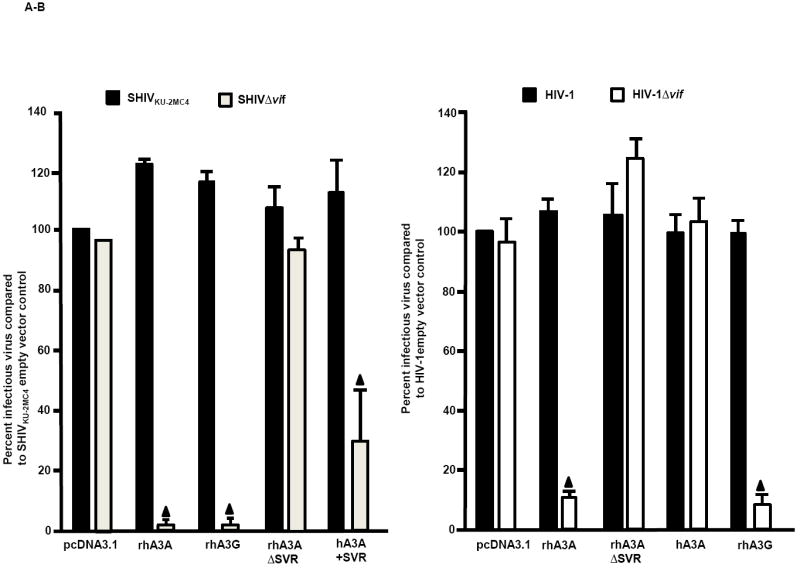
Deletion of three amino acids from the N-terminal region of rhA3A abolishes restriction. Panel A. RhA3AΔSVR is inactive against SHIVΔvif. 293 cells were co-transfected with (1.0 μg) SHIV and SHIVΔvif molecular clones with vectors expressing (0.5 μg) rhA3A, rhA3AΔSVR, rhA3C, rhA3G or empty vector (pcDNA3.1) in a 12-well plate. At 48 h, the culture medium was harvested, assessed for p27 and the amount of infectious virus titrated on the TZM.bl cells. Panel B. RhA3AΔSVR is inactive against HIV-1Δvif. 293 cells were co-transfected with (1.0 μg) HIV-1 and HIV-1Δvif molecular clones with vectors expressing (0.5 μg) rhA3A, rhA3AΔSVR, hA3A, rhA3G or empty vector (pcDNA3.1) in a 12-well plate. Virion infectivity was calculated normalizing to the wild-type HIV-1 empty vector control. In both panels A and B, the assays were run at least in triplicate. Significance in the restriction was determined with respect to the wild-type empty vector control using a two-tailed Student’s t-test (p<0.05; ▲).
RhA3A, but not hA3A, is sensitive to SIV and HIV-1 Vif-mediated degradation
We next determined the stability of rhA3A and hA3A in the presence of the SHIV, SHIVΔvif, HIV-1, or HIVΔvif genomes. Each full length molecular clone was co-transfected into 293 cells along with plasmids expressing HA-tagged rhA3A, rhA3G, hA3A, or rhA3AΔSVR. At 24 h, the cells were lysed, proteins precipitated, and analyzed by Western blot using an antibody specific to the HA-tag. Our results show that rhA3G was degraded in the presence of the parental SHIV but not in cells co-transfected with the SHIVΔvif genome. In contrast, hA3A was stable in the presence of parental SHIV or SHIVΔvif (the apparent difference of the hA3A in lanes 4 and 5 is due to less protein loaded in lane 4). Unlike hA3A, rhA3A was found to be degraded in the presence of the SHIV genome but stable in the presence of the SHIVΔvif genome (Fig. 8A). These results again suggest that rhA3A interacts with the SIV Vif protein. Finally, rhA3A appeared to be less stable in the presence of the HIV-1 genome and more stable in the presence of NL4-3Δvif (Fig. 8B). Similar results were observed at 48 h post-transfection (data not shown). These results suggest that the SIV and HIV-1 Vif proteins may interact with rhA3A promote its rapid turnover.
Fig. 8.
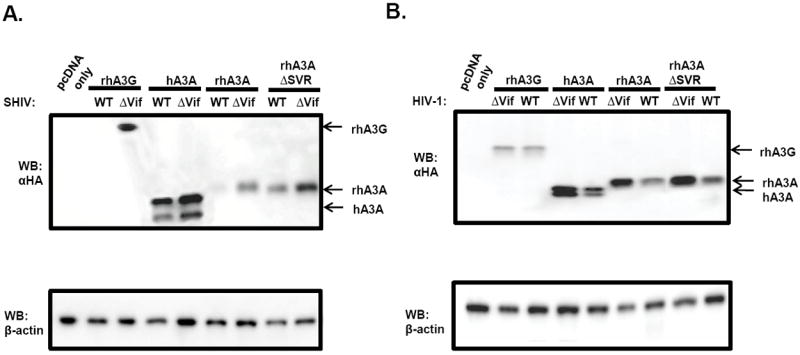
Sensitivity of rhA3A to lentivirus Vif-mediated degradation. Panel A. SHIV Vif mediates rhA3A degradation. SHIV and SHIVΔvif molecular clones were co-transfected with rhA3G, hA3A, rhA3A or rhA3AΔSVR. At 24 h post-transfection, cells were lysed and proteins were precipitated using methanol. Proteins were separated on a 12%-SDS-PAGE gel and probed using an antibody directed against the HA-tag. All samples were normalized to the amount of β-actin. Panel B. HIV-1 Vif mediates rhA3A degradation. HIV-1 and HIV-1Δvif molecular clones were co-transfected with rhA3G, hA3A, rhA3A and rhA3AΔSVR. In both panels, the blots were stripped and re-probed with an antibody against β-actin. All assays were performed twice.
RhA3A causes low levels of G-to-A mutations in nascent SHIVΔVif reverse transcripts
RhA3A incorporation into SHIVΔvif virions led to a significant decrease in infectivity (Fig. 6A and B). Our previous study revealed that rhA3G inhibition of SHIVΔvif virions in target cells was accompanied by substantial G-to-A mutations (Schmitt et al., 2010). To determine whether rhA3A has a similar effect, we amplified a 300 bp region in nef from target TZM-bl cells and sequenced 15 independent clones.
The results indicate that while rhA3G caused 33 G-to-A mutations in 15 clones (300 bases sequenced in each clone or total of 4,500 bp) in the nef gene of SHIVΔvif, most of which (29) were in the context of 5’-TCT-3’ (Schmitt et al., 2010), while only 7 such mutations were detected with rhA3A (Fig. 9A-B). As expected, the corresponding wild-type SHIV had significantly lower G-to-A mutation frequencies (Fig. 9C-D). These results suggest that while rhA3A and rhA3G appear to have similar antiviral potency against SHIVΔvif (Fig.6A), rhA3A produced an approximately 5-fold decrease in the number of G-to-A mutations in SHIVΔvif compared to rhA3G.
Fig. 9.
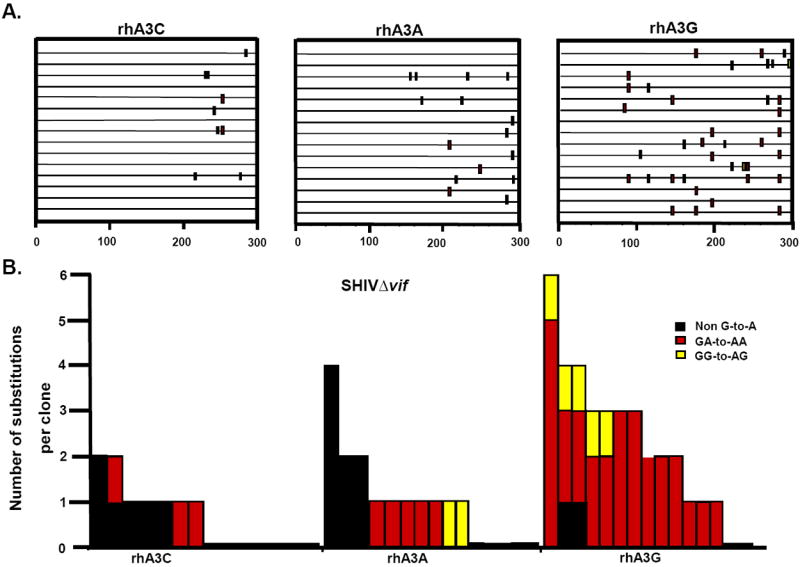
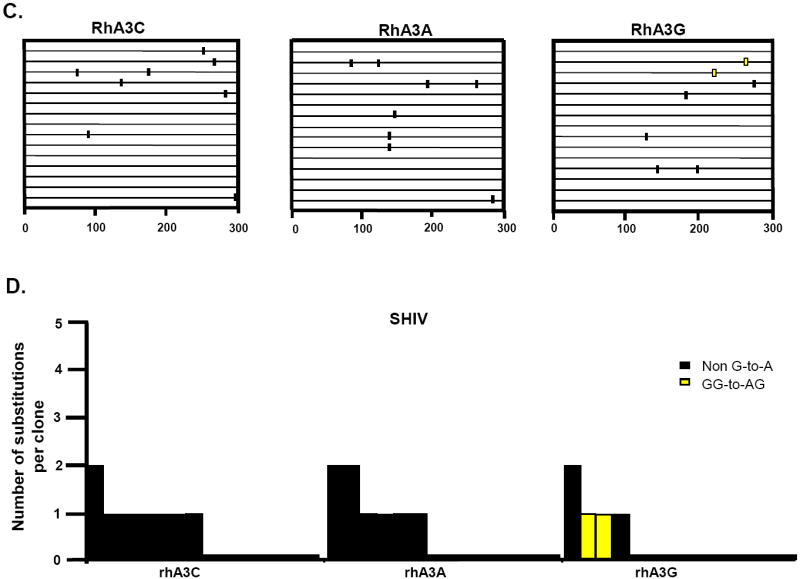
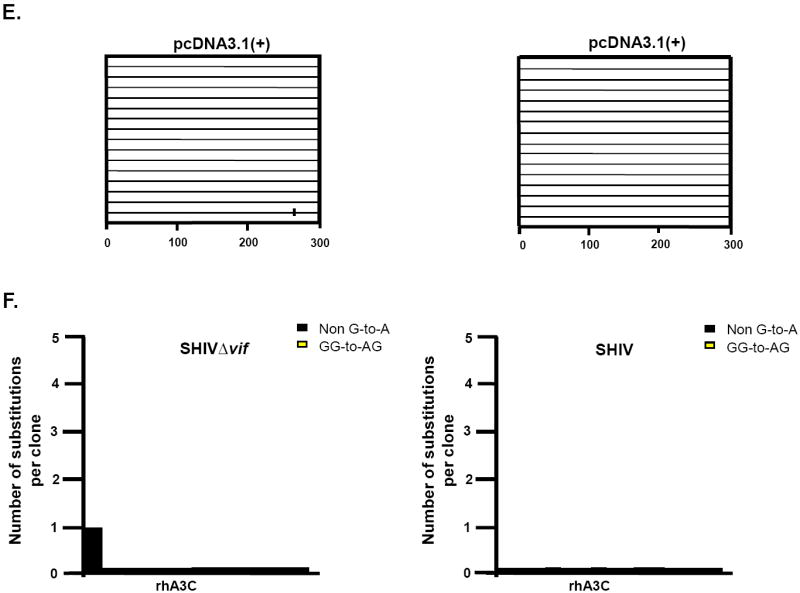
RhA3A induces low frequencies of G-to-A mutations in SHIVΔvif nascent reverse transcripts. TZM-Bl cells were infected with DNAse-I treated SHIVΔvif or SHIV virions and DNA was extracted after 24 h. A 300-bp segment of nef was amplified, cloned and 15 independent clones were sequenced. Panel A. Moderate G-to-A mutations induced by rhA3A on SHIVΔvif. Panel B. Graph depicting the cumulative number of mutations from the 15 clones. RhA3A induced 5-fold lower G-to-A mutations than rhA3G against SHIVΔVif. Panel C. Lack of G-to-A substitutions in wild-type SHIV in the presence of either rhA3A or rhA3G. Panel D. Graph of cumulative mutations in wild-type SHIV. In panels A and C, each mutation is denoted by a vertical line that is color coded with respect to the dinucleotide context: GA (red), GG (yellow), or non-G-to-A (black). In panels B and D, Each bar is shaded according to the proportion of G-to-A-substitutions that occurred in the context of GG (yellow) or non-G-to-A (black).
Rhesus A3A does not inhibit adeno-associated virus 2 (AAV-2)
Previous studies have shown that hA3A can inhibit the replication of AAV-2 and autonomously replicating parvoviruses (Chen et al., 2006). We determined if rhA3A could inhibit the replication of AAV-2. 293 cells were transfected with vectors expressing hA3A, rhA3G, rhA3A or the empty vector and plasmids expressing AAV-2 and a helper plasmid. At 48 h, the cells were harvested, the extrachromosomal DNA isolated by Hirt extraction and analyzed using Southern blots for the presence of replicating AAV-2 DNA. The results, shown in Fig. 10A, indicate that hA3A, but not rhA3G, inhibited AAV-2 replication, as previously reported (Narvaiza et al., 2009; Chen et al., 2006). In contrast, rhA3A did not significantly inhibit AAV-2 replication. A Western blot from an aliquot of the 293 lysates showed that the A3 proteins were similarly expressed (Fig. 10B).
Fig. 10.
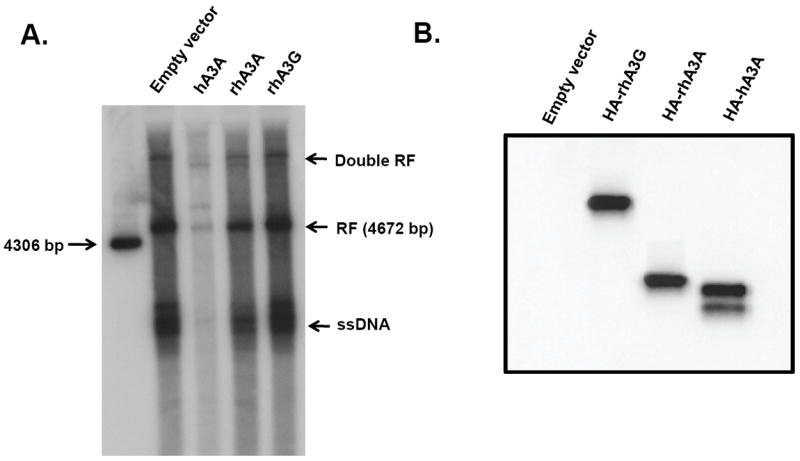
RhA3A does not inhibit AAV2 replication. Panel A. Low molecular weight DNA extracted from 293 cells transfected with recombinant AAV-2 plasmids and either empty vector (293), hA3A, rhA3A, or rhA3G A 1% southern blot was run and hybridized with a 32P-labeled probe from SSV9 as previously described (Qiu et al., 2002). The following AAV replication products are shown as dRF (double recombinant form), RF (recombinant form), and ssDNA. These experiments were performed two times with the same result. Panel B. A Western blot was performed on an aliquot of the lysate to show the expression of each A3 protein was expressed.
RhA3A does not inhibit LINE-1 retrotransposition
L1 elements are autonomous non-LTR retrotransposons that constitute about 17% of the human genome (Babushok and Kazazian,2007). L1 elements, through the ORF2 gene product, also facilitate the retrotransposition of Alu elements, which are present in an additional 11% of the genome. It has been estimated that for every person, 80-100. These in vitro retrotransposition assays require the co-transfection of hA3A with L1 plasmids(Rangwala and Kazazian, 2009) and assaying after 3-4 days, a timepoint where significant hA3A inhibition of plasmid DNA may be observed (Stenglein et al., 2010).
To determine whether rhA3A could inhibit L1 retrotransposition, we used an L1-eGFP plasmid construct that contains a self-splicing intron in the opposite orientation within eGFP (pLRE3-EF1-mEGFPI) (Wissing et al., submitted). As a positive control, an intronless isogenic construct that encoded an intact eGFP (pLRE3-EF1-mEGFP) was used. L1 (1 μg) and A3A (200 ng) plasmids were co-transfected into 293T cells and the cells were harvested at 2 and 4 days. Similar to our earlier results (Fig. 5A), at 2 days post-transfection, we observed minimal (22%) inhibition of the eGFP control plasmid, while rhA3A had no effect (data not shown). However, the L1 signals we obtained at 2 days were still very low, which is line with previous reported data (Kroutter et al., 2009). By 4 days post-transfection, we observed detectable L1 retrotransposition (Fig. 11A). As expected, hA3A potently inhibited L1 retrotransposition. In contrast, rhA3A, as well as the rhA3ACSVR mutant, had no effect (Fig. 11A). The effect of hA3A on L1 retrotransposition could be partially attributed to foreign DNA restriction, since at 4 days post-transfection, we observed a 2-fold reduction in eGFP signal from the control plasmid (Fig. 11B). However, it should be noted that substantial eGFP signals were still detected, in contrast to near-complete inhibition of L1 by hA3A (Fig. 11A). Importantly, under conditions where we detected significant foreign DNA restriction by hA3A, no such activity was observed for rhA3A and rhA3ACSVR. Thus, we conclude that unlike hA3A, rhA3A does not restrict L1 retrotransposons and/or foreign DNA.
Fig. 11.
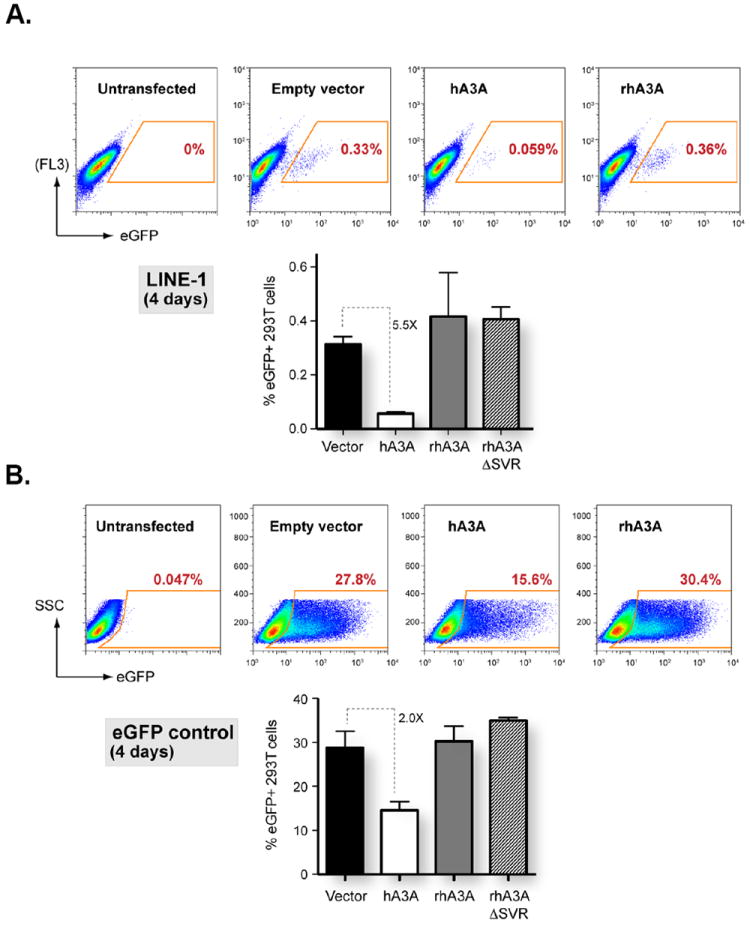
RhA3A does not inhibit retrotransposition. One μg of L1 constructs containing eGFP with or without the γ-globin intron in the opposite orientation and 200 ng of empty vector, hA3A, rhA3A or rhA3AΔSVR plasmid were co-transfected into 293T cells using Fugene 6 (Roche). At 4 days post-transfection, cells were harvested and analyzed for eGFP expression using a FACSCalibur machine (BD Biosciences) with 150,000 events. Panel A. Results of co-transfection of LINE 1 plasmid (with intron) with either the empty vector, hA3A or rhA3A plasmids. To minimize autofluorescence background, the percentage of eGFP+ cells from the untransfected control were gated against an empty fluorescence channel (FL3). Panel B. Results of co-transfection of eGFP control plasmid (no introns) with either the empty vector, hA3A or rhA3A plasmids.
DISCUSSION
SIV and SHIV infections of rhesus macaques have been used extensively as models for studying HIV pathogenesis and in vaccine development. The fact that the dose, timing and route of infection can be controlled also makes it ideal for investigating the earliest events following pathogenic lentivirus infection, including innate immune responses. Recent discoveries have shown that specific lentivirus genes could antagonize the effects of host innate restriction factors. Thus, the macaque/SIV or SHIV models provides a compelling system to explore how these restriction factors are regulated in vivo following lentivirus infection.
Identification of rhA3A as a novel lentivral restriction factor
The APOBEC3 family of deoxycytidine deaminases function as potent antiretroviral factors. Incorporation of select A3 proteins can inhibit reverse transcription and/or induce G-to-A hypermutation in nascent reverse transcripts in the next target cell (Bishop et al., 2004; Celico et al., 2006; Russell et al, 2009; Sova et al., 1993; von Schwedler et al., 1993; Zheng et al., 2005). Seven A3 genes are encoded in the human genome, but only a subset, hA3G, and to a lesser extent, hA3F and hA3C, are expressed in CD4+ T cells, inhibit HIV-1 and are counteracted by the HIV-1 encoded Vif protein (Koning et al., 2009; Refsland et al., 2010). There is uncertainty whether a similar complement of A3 genes are present in rhesus macaques, particularly since no rhA3A gene has yet to be identified. In this study, we cloned the rhA3A gene. This result is consistent with a recent study that revealed evolutionary conservation of A3A among hominids, Old World and New World monkeys (Bulliard et al., 2011). In addition, analysis of the rhesus macaque genome provided evidence of seven encoded A3 members that based on phylogenetic analyses are orthologous to the corresponding hA3 genes. These findings suggest that the seven rhA3 genes have been maintained after the evolutionary split between monkeys and humans. Thus, the vif genes of various SIV strains naturally infecting Old World monkey species have likely been co-evolving with the same set of A3 genes for millions of years.
Rhesus macaque A3G is a potent A3 deaminase against SIV, SHIV and HIV-1 (Schmitt et al., 2010; Virgen et al., 2007; Zennou et al., 2006). We therefore determined the expression of the other rhesus macaque A3 genes in primary, activated CD4+ T cells, the main cellular targets of SIV and SHIV in vivo, in relation to rhA3G. Our results indicate that rhA3B mRNA is not expressed and that rhA3D mRNA is expressed at levels that are likely not biologically relevant in vivo. RhA3F is expressed at 2-fold lower levels than rhA3G, but rhA3F appears to be incorporated promiscuously into virions and is not as functional as rhA3G in side-by-side co-transfection studies (Schmitt et al., 2010). Only rhA3A, rhA3C and rhA3H are expressed at similar or higher levels than rhA3G. RhA3H has been previously shown to have potent antiretroviral activity against HIV-1 (OhAinle et al., 2008) and we found significant activity against SHIV (unpublished data). We also found no significant antiviral activity of rhA3C against SHIV. Surprisingly, rhA3A exhibited potent antiretroviral activity. Based on this data, we conclude that rhA3A, rhA3G, and rhA3H most likely are the dominant rhesus macaque APOBEC3 deaminases that could restrict SHIV or SIV in vivo.
Insights on rhA3A restriction of lentiviruses
Rhesus macaque A3G inhibits SHIVΔvif and HIV-1Δvif virion infectivity through a mechanism that involves virion incorporation. Virion incorporated rhA3G blocks reverse transcription and/or induces G-to-A hypermutation in the next target cell. Consistent with this mechanism, we readily detected rhA3G in SHIVΔvif and HIV-1Δvif virions, and observed substantial G-to-A mutations in reverse transcripts newly formed in target cells. Given these consistent phenotypes, we used rhA3G as a positive control for evaluating the antiretroviral activity of rhA3A. In this study, we found that rhA3A is as potent as rhA3G in restricting virus infectivity. Both proteins inhibited SHIVΔvif infectivity by approximately 20-fold. In addition, rhA3A modestly inhibited HIVΔvif infectivity by about 3-fold. We found that in contrast to rhA3G, rhA3A was incorporated at low levels into SHIVΔvif virions and was undetectable in HIVΔvif virions (Fig. 6B and D). The results may be due to the sensitivity of the Western blots and that rhA3A is incorporated into virions at very low levels compared to rhA3G. This would also suggest that rhA3A enzymatic activity may be higher than rhA3G on a per-molar basis.
As a first step in gaining mechanistic insights on rhA3A-mediated restriction of SHIV and HIV-1, we evaluated whether two rhA3A domains are critical for restriction. Comparison of the amino acid sequences of human and rhesus macaque A3A reveals a monkey-specific insertion at position 27-29 (SVR) that is critical for SHIVΔvif and HIV-1ΔVif inhibition by rhA3A that was lost in the hominid lineage (Fig. 1A). In a recent modeling study of hA3A, it was shown that amino acids 29 and 30 likely form part of a polynucleotide accommodating groove near the active-site pocket (Bulliard et al., 2011). We observed that the rhA3A ΔSVR mutant is totally inactive, while remaining sensitive to SHIV and HIV-1 Vif-dependent degradation. In addition, we made just the opposite mutation, the addition of the SVR motif to hA3A. We found that this mutant gained partial activity against HIV-1Δvif but was not as active as rhA3A, suggesting that other amino acid substitutions are likely involved in this function. We also determined if the canonical cytidine deaminase Zn+2 binding domain (C/H-X-E-X23-28-P-C-X2-4-C) was critical for rhA3A anti-lentiviral activity. Mutation of the histidine at position 70 to glutamic acid resulted in no restriction of SHIVΔvif (unpublished data). Thus, the monkey-specific residues SVR (27-29) and the canonical Zn+2 binding domain are critical for rhA3A-mediated Δvif lentivirus restriction.
Human A3A is largely inactive against HIV-1CVif, but when targeted to the viral nucleocapsid, it has potent activity against HIV-1CVif (Goila-Gaur et al., 2007). Since hA3A and the C-terminal region of hA3G are derived from a common ancestor, the N-terminal domain of hA3G was fused to hA3A (hA3G-A3A). This hA3G-A3A fusion protein localized to the cytoplasm, incorporated into the viral nucleoprotein complexes, and inhibited HIV-1 replication (Goila-Gaur et al., 2007). Similarly, fusion of hA3A to the Vpr protein resulted in its incorporation into viral nucleoprotein complexes and inhibition of HIV-1 and SIV replication (Aguiar et al., 2008). These results suggested that the reason that hA3A could not restrict lentiviruses is that it could not be targeted to the virion core. While hA3A had no substantial activity against HIV-1CVif, hA3A surprisingly decreased SHIVCVif virion infectivity by approximately 10-fold. Although we did not detect hA3A in virions, it is possible that like rhA3A, very little is incorporated into nascent virions and is below the limits of detection by Western blot analysis.
Evidence for recent evolutionary gain of function of DNA restriction in hominids
Previous studies have shown that while hA3A is inactive against HIV-1, it can block endogenous retroviral elements such as LTR-retrotransposons or Alu elements (Chen et al., 2006; Bogerd et al., 2006b). Human A3A has antiviral activity against autonomous (minute virus of mice; MVM) and non-autonomous (AAV-2) replicating parvoviruses and has editing activity against the human papillomavirus genome (Chen et al., 2006; Narvaiza et al., 2009; Vartanian et al., 2008). Finally, hA3A has been shown to counteract foreign DNA (Stenglein et al., 2010) and promote DNA damage (Landry et al., 2011). These studies suggest that hA3A biology is intimately linked to the restriction of double-stranded DNA elements. Surprisingly, we found no evidence for rhA3A restricting AAV-2, L1 elements and foreign DNA. While it can be argued that rhA3A may have activity against rhesus macaque AAV, no infectious macaque AAVs have been isolated to date. Thus, the DNA restriction properties of A3A may have been a recent evolutionary development in the hominid lineage.
The mammalian A3 locus encodes genes that have among the highest detectable evolutionary pressures known (Sawyer et al., 2004). This is thought to reflect a long-standing host-pathogen genetic conflict that eventually helps dictate current trends in cross-species transmission and viral host range. Our findings now enter A3A into this select group of genes, given the divergent virus specificities of the human and monkey homologues. RhA3A inhibits the SHIVΔvif and HIV-1 CVif, but not the DNA virus AAV-2 and L1 retroelements, while hA3A inhibits SHIVΔvif, AAV-2 and L1 elements but not HIV-1 Δvif. Thus, it would appear that rhA3A could inhibit a broader range of lentiviruses at the cost of having no activity against DNA viruses. On the other hand, hA3A appears to have acquired activity against DNA viruses, at the expense of losing broad potency against lentiviruses. The rhA3A mutant with amino acids 27-29 deleted (rhA3AΔSVR) was incapable of restricting SHIVΔvif and HIV-1Δvif. Thus, the divergent viral specificities of hA3A and rhA3A may be dictated, at least in part, by an evolutionary switch that involved alterations in the polynucleotide-binding groove of A3A. The 81% identity between hA3A and rhA3A at the amino acid level should enable structure-based domain-swap experiments to determine which residues account for differential virus restriction. These types of studies should provide a biologically relevant system to interrogate which changes in the molecular determinants of nucleic acid specificity of the A3A proteins dictates the antiviral activity of A3A against lentiviruses, parvoviruses and L1 retroelements.
MATERIALS AND METHODS
Cells, viruses, and plasmids
HeLa and 293 cells were used for transfections of vectors expressing various APOBEC3 proteins, full-length SHIV or HIV-1 (NL4-3). The TZM-bl cell line was used as an indicator cell line to measure the infectivity of viruses (Derdeyn et al., 2000; Platt et al., 2009; Takeuchi et al., 2008; Wei et al., 2002). Both cell lines was maintained in Dulbecco’s minimal essential medium (DMEM) with 10% fetal bovine serum (R10FBS), 10 mM Hepes buffer, pH 7.3, and 100 U/μg penicillin-streptomycin and 5 μg gentamicin. Rhesus macaque PBMCs were obtained from uninfected animals and isolated on Ficoll/Hypaque gradients. The derivation of SHIVKU-2MC4 (a pathogenic molecular clone; referred to in the text as SHIV) and SHIVVifSTOP (referred to in the text as SHIVΔvif) has been previously described (Liu et al., 1999; Schmitt et al., 2010). A plasmid with the genome of HIV-1 strain NL4-3 (referred to in the text as HIV-1) was used to construct a Δvif version (referred to in text as HIV-1Δvif) (pNL4-3; NIH AIDS Research and Reference Reagent Program). This plasmid was digested with PflM1, phenol: chloroform extracted, and ethanol precipitated. The 5’-protruding ends were filled using DNA Polymerase I Large Fragment (Klenow; Promega). The resulting Klenow fragment reaction produced blunt ended fragments that were purified and re-ligated. The resulting plasmid was sequenced and found to contain a deletion in pNL4-3 vif between base pairs 5301 to 5308. Therefore, only the first 79 amino acids of pNL4-3 Vif could be expressed. The plasmids pcDNA3.1(+)-HA-rhA3G and pcDNA3.1(+)-HA-hA3A were kindly provided by Nathaniel Landau (New York University School of Medicine). A plasmid expressing HA-rhA3C was constructed in this laboratory and cloned into the pCruz-HA vector (Santa Cruz Biotechnology) using restriction sites Sca I and Kpn I. A plasmid expressing pcDNA3.1(+)-HA-hA3G used in these studies was provided by the NIH AIDS Reference and Reagent Program.
Molecular cloning of rhA3A
Total RNA from mitogen-activated rhesus macaque PBMCs was isolated using the RNAEasy Kit (QIAGEN; Valencia, CA). Three hundred ng of sample was subjected to random hexamer priming using the RT2 EZ first strand kit (SA Biosciences), and 4 μl of this cDNA was subjected to a 40 μl PCR reaction consisting of 1x Sweet PCR Mix (SA Biosciences), 12.5 pmol of rhA3A.F (5’-GACAAGCACATGGACGGCAG) and rhA3A.R (5’-CATCCTTCAGTTTCCCTGAT) primers, designed based on the first and last exons of human A3A (hA3A). Amplification conditions included 95°C for 15 min hot-start, followed by 40 cycles at 94°C 30 s, 50°C 30 s and 72°C 3 min in a PE 9700 machine (Perkin Elmer). A dominant PCR amplicon of 600 bp was subcloned into the TOPO-TA vector (Invitrogen) and sequenced. The sequence for rhesus macaque A3A (rhA3A) was submitted to GenBank with Accession number JF831054. An expression construct containing rhA3A linked to an N-terminal HA-tag was synthesized (GenScript). The HA-rhA3A gene was subcloned into pcDNA3.1(+) using restriction sites Kpn I and Eco RV sites of pcDNA3.1(+) and sequence-confirmed.
Positioning of rhA3 genes in the rhesus macaque genome
Analysis of the Apobec3 loci was performed using NCBI Build 1.2 version of the macaque chromosome 10 sequence. RhA3C (Accession # EU381233), rhA3G (XM_001094452), rhA3F (DQ514917) and rhA3H (DQ507277) cDNA sequences were subjected to BLAST analysis against the macaque genome, and the corresponding coordinates were obtained. To characterize the intronic sequence flanked by exons 1 and 2 of rhA3A, exon 1 (5’-AGAAGAGACAAGCACATGGAC) and exon 2 (5’-GCTCCACCTCGTAGCACAA) specific primers were used to directly amplify rhesus macaque genomic DNA. The resulting PCR amplicon was directly sequenced (Figure 2; see text for details). RhA3B and rhA3DE sequences were predicted based on the genome sequences upstream of rhA3C and between rhA3C and rhA3F, respectively, using the geneid program (http://genome.crg.es/geneid.html).
Phylogenetic analysis of rhApobec3A
The A3A amino acid sequences from various primate species were obtained from (Bulliard et al. 2011). Sequences were aligned using ClustalX using the Gonnet series, and neighbor-joining trees were constructed with 1000 subreplicates, correcting for multiple substitutions and excluding gaps. The C-terminal half of rhA3G was used as outgroup. A3 amino acid sequences from human, mouse, rat and rhesus macaque were likewise aligned using Clustal X, after splitting the sequences of rodent A3 and primate A3B, A3DE, A3F and A3G into N-terminal and C-terminal halves. Neighbor-joining trees were performed unrooted, with 1000 subreplicates and excluding gaps.
Expression of rhA3A in rhesus macaque tissues
A rhesus macaque that euthanized and perfused with 2 liters of Ringer’s saline to remove contaminating blood was used to examine the expression of rhA3A mRNA in different tissues (visceral organs and brain). RhA3A was amplified from 30 μg of each visceral and CNS tissue using the RNAEasy kit (Qiagen; Valencia, CA). RNA was digested with 1 U/μl DNase-I (Fermentas) for 30 min. RT-PCR was performed using the Titan One RT-PCR kit (Roche). Each reaction used 100 ng of total RNA and was amplified using oligonucleotide primers specific for rhA3A. The oligonucleotides used for the reverse transcriptase reaction and first round PCR were 5’-GGACGGCAGCCCAGCATCCAGGCCCAG-3’ (sense) and 5’-CGTAGGTCATGATGGAGACTTGGGC -’3 (antisense), which are complementary to bases 3-29 and 451-475, respectively. The nested oligonucleotides used were 5’-CCAGGCCCAGACACTTGATGGATCC-3’ (sense) and 5’-CCGCAGCGTTCGCAGTGCCTCCTGATACAGG-3’ (antisense), which are complementary to bases 20-44 and 411-441, respectively. As a control, GAPDH was amplified to verify the integrity the RNA using oligonucleotides 5’-GCCATCACTGCCACCCAG-3’ (sense) and 5’-GCCACATACCAGGAAATGAGC-3’ (antisense). The nested oligonucleotides used were 5’-CCTCCGGGAAACTGTGGC-3’ (sense) and 5’-CGTTGAGGGCAATGCCAG-3’ (antisense). The reactions were performed in an ABI 2720 Thermal Cycler using the following thermal profile: 42°C 30 min for 1 cycle; 94°C 2 min for 1 cycle, 94°C 30 s, 55°C 30 s, and 68°C 45s for 10 cycles; 94°C for 30 s, 55°C 30 s, and 68°C 2 min for 25 cycles. One μl of the initial reaction mixture used for nested PCR using rTaq (Takara) and performed with the following thermal cycle profile: 95°C for 1 min, 48°C 2 min, and 72°C 3 min for 35 cycles. The 450 bp amplicon was visualized in a 1.5% agarose gel, excised and gel purified (Qiagen; Valencia, CA) and directly sequenced.
Expression of rhA3 genes in activated CD4+ T cells
Rhesus PBMCs were obtained from three donors by Ficoll separation. 5 × 106 cells were seeded into each well of a 6-well plate containing 3 ml RPMI medium (Mediatech) with 10% Fetal Bovine Serum (Gemini Biosciences). Half of the cultured PBMCs were activated with 2.5 μg/ml Staphylococcal Enterotoxin B (SEB; Toxin Technology) overnight. The following day, the PBMCs were stained with CD3-V450 (clone SP34-2), CD4-APC-H7 (L200) and CD25-PerCP-Cy5.5 (M-A251) (BD Biosciences) and analyzed in an LSR-II flow cytometer (BD Biosciences). Histogram plots to highlight CD25 expression in CD4+ T cells were constructed using the FlowJo software (Treestar). CD4+ T cells were purified from activated PBMCs by negative selection with magnetic beads (Miltenyi Biotec). Total RNA was extracted from CD4+ T cells using the RNAEasy kit, and cDNA was synthesized by random hexamer priming using the RT2 EZ first strand kit (SA Biosciences). The cDNA was used for triplicate real-time PCR evaluations (25 μl) with optimized A3-specific primers and a housekeeping gene, GAPDH, in a BioRad real-time PCR machine. Briefly, 5 μl of cDNA was mixed with 12.5 μl of 1× Universal Master Mix (Applied Biosystems), 10 pmol of primers and 5 pmol of probe and subjected to 40 cycles of denaturation and annealing/elongation. The A3-specific primers, probes and cycling conditions are listed in Table 1. Cloned cDNA sequences were used as standards and for evaluating cross-reactivity (Table 2). The A3-specific assays had a limit of detection of 5 copies per reaction, and standard curves had r2 values >98%.
Subcellular localization of rhA3A
For intracellular localization of rhA3A, 293 cells were plated onto 13 mm cover slips in a 6 well plate and transiently co-transfected with vectors expressing HA-tagged hA3A, rhA3G or rhA3A and one expressing a eGFP tagged nuclear marker using a polyethylenimine (PEI) transfection reagent (ExGen™ 500, MBI Fermentas) according to the manufacturer’s instructions. Cultures were maintained for 36-48 h before being were fixed in 100% cold methanol (-20C) for 10 min and washed twice for 5 min in 1x PBS (pH7.4). Cover slips were incubated with a rabbit polyclonal anti-HA antibody (HA-probe, Santa Cruz) for 1 h at ambient temperature in 1x PBS plus 1% BSA. Cover slips were washed twice for 5 min in PBS and reacted with a Cy-5 conjugated secondary antibody (Abcam) 1x PBS plus 1% BSA for 30 min. The cover slips were washed twice for 5 min in 1x PBS and mounted in glycerol containing mounting media (Slowfade antifade solution A, Invitrogen). A Nikon A1 confocal microscope was used to collect 100x images with a 2x digital zoom, using EZ-C1 software. The pinhole was set to large for all wavelengths used with Cy5 and eGFP excited using a 638 nm and 488 nm diode lasers, respectively. Images were collected using a 670 nm filter for Cy5 and 525/25 nm fiter for eGFP.
Foreign DNA restriction by rhA3A
293 cells were seeded into 6-well tissue culture plates 24 h prior to transfection. Cells were first transfected with 3 μg of either HA-rhA3A, HA-hA3A, HA-rhA3G HA-rhA3AΔSVR or pcDNA3.1(+) vector using a polyethylenimine transfection reagent (ExGen™ 500, MBI Fermentas) according to the manufacturer’s instructions. After 24 h, the cells were re-transfected with 3 μg of a vector expressing eGFP. Cells were incubated at 37°C in 5% CO2 atmosphere for either 2 or 5 days. The cells were removed from the plate using Ca2+/Mg2+-free PBS containing 10 mM EDTA. Cells were then fixed in 2% paraformaldehyde, for 5 min. The cells were washed twice in 1x PBS plus and analyzed using an LSRII flow cytometer. The mean fluorescence intensity (MFI) for eGFP positive cells was calculated. The MFI ratio and percentage of eGFP positive cells was calculated for each sample. Normalized ratios from three separate experiments were averaged and the standard error calculated. All groups were compared to pcDNA3.1(+) plus eGFP control using a Student’s t-test with p<0.05 considered significant.
Production of SHIVΔVif virions in the presence of A3A
293 cells were seeded into a 12-well tissue culture plate 24 h prior to transfection. Cells were first transfected with 1 μg of HA-tagged rhA3A, hA3A, rhA3G or pcDNA3.1(+) vector using PEI (Fermentas). After 24 h, cells were transfected with 1 μg of SHIVΔVif or HIV-1Δvif plasmid. Cells were incubated at 37°C in 5% CO2 atmosphere for 48 h. Supernatants were collected and cellular debris removed by low speed centrifugation. The cells were lysed in 500 μl of RIPA (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 0.5% deoxycholate, 0.2% SDS, 10 mM EDTA) and the nuclei were removed through high speed centrifugation. The amount of Gag p27 or p24 present in the viral supernatants was measured using a commercially available p27 and p24 ELISA kits (Zeptometrix). The experiment was run at least three separate times, normalized to the empty vector control samples. Differences between mean percentages were calculated using a two-tailed Student’s t-test with p<0.05 considered significant.
Inhibition of SHIV and HIV-1 virion infectivity by A3A
SHIV, SHIVΔVif, HIV-1 or HIVΔVif infectious molecular clones (3 μg) were co-transfected (1.5 μg) with plasmids expressing rhA3A, rhA3G, rhA3C, hA3A, or hA3G using PEI (Fermentas) in a 6-well plate. At 48 h post-transfection, the culture medium was harvested and clarified by low speed centrifugation. Equivalent amounts of p27 were serially diluted using 10-fold dilutions from 101 to 106 and used to inoculate TZM-bl cells. At 48 h post-inoculation, the media was removed, cells washed with PBS and the monolayer fixed using 1% formaldehyde-0.2% glutaraldehyde in PBS. The cells were washed and incubated in a solution for 2 h at 37°C containing 4 mM potassium ferrocyanide, 4 mM potassium ferricyanide, 4 mM magnesium chloride, and 0.4 mg X-gal per ml. The reaction was stopped and the infectious units (IU) per ml were calculated (Derdeyn et al., 2000; Wei et al., 2002).
Site-directed mutagenesis
Mutations introduced into all plasmids were accomplished using a QuikChange site-directed mutagenesis kit (Stratagene) according to the manufacturer’s protocol. All plasmid inserts were sequenced to ensure the validity of the mutations and that no other mutations were introduced during the cloning process.
Virion incorporation assays
SHIV, SHIVΔVif, HIV-1 or HIVΔVif infectious molecular clones (3 μg) were co-transfected with 1.5 μg of plasmids expressing either rhA3A, rhA3G, hA3G or hA3A proteins into 293 cells using PEI reagent (Ex-Gen™ 500, Fermentas) in each well of a 6-well plate. At 48 h, virus supernatants were harvested and clarified by low speed centrifugation. The clarified supernatant was ultracentrifuged to pellet virions (SW41 rotor, 247,000×g, 1 h, 4°C). The pellet was resuspended in PBS and layered on a 20%/60% sucrose step gradient and again subjected to ultracentrifugation (SW55Ti, 247,000×g, 1 h) at 4°C. The interface (containing virions) was harvested, pelleted again by ultracentrifugation as described above, and resuspended in 150 μl of PBS. An aliquot was saved to determine the p27 or p24 content by antigen capture assay (Zeptometrix). The remaining sample was boiled in 2× sample reducing buffer. Equivalent amounts of p27 or p24 were loaded on a 12% SDS-PAGE gel. A3 proteins were detected by Western blotting using an anti-HA antibody (HA-probe; Santa Cruz). Blots were placed in stripping buffer (25 mM glycine, pH 2.0 and 1% SDS) and reprobed using either rabbit polyclonal antibody specific for p27 or a mouse monoclonal antibody to p24 (NIH AIDS Research and Reference Reagent Program).
Stability of rhA3A in the presence of SHIV and HIV-1 genomes
Infectious molecular clones of HIV-1, HIV-1Δvif or SHIV and SHIVΔvif were co-transfected in a 2:1 ratio with rhA3A, rhA3G, hA3G, or hA3A using PEI transfection reagent (Ex-Gen500; Fermentas) into a 12-well plate. At 24 h post-transfection, the supernatant was removed, the cells were harvested and lysed using RIPA buffer. Following lysis, the nuclei were removed by centrifugation at 14,000 rpm for 15 min in a microfuge at 4°C. The protein was precipitated using methanol, resuspended in 2x sample reducing buffer, and boiled for 5 min. Proteins were separated on a 12% SDS-PAGE gel and probed using commercially available rabbit polyclonal HA antibody (HA-probe, Santa Cruz). All samples were normalized to the same amount of β-actin protein using a mouse monoclonal antibody specific for β-actin (AC15; Novous Biologicals).
Hypermutation assays in the presence of rhA3A
To determine whether virion-incorporated rhA3A can induce G-to-A mutations in nascent reverse transcripts, 293 cells were co-transfected with vectors containing the genomes SHIV or SHIVΔVif and vectors expressing rhA3G (positive control), rhA3C (negative control) or rhA3A using PEI (Fermentas). At 24 h post-transfection, cells were washed and fresh DMEM was added. At 48 h, the supernatant containing virus was subjected to ultracentrifugation through a 20% sucrose cushion. The resulting supernatant was DNase-I-treated (Fermentas) at 37°C for 30 min to minimize plasmid carry-over. Infectious titers in the supernatants were measured on TZM-bl cells. In addition, at 24 h post-infection of TZM-bl cells, total cellular DNA was harvested and extracted using the DNeasy kit (Qiagen). The DNA was used in a nested DNA PCR to amplify a 300 base pair fragment of nef. The PCR reaction was carried out using rTaq and the manufacturer’s instructions (Takara). The oligonucleotides employed during in the first round PCR to amplify SIV nef were 5’-GGTGGAGCTATTTCCATGAGG-3’ (sense) and 5’-GTCTTCTTGGACTGTAAT AAATCCC-3’ (anti-sense). One μl of the first PCR product was added to a nested reaction. The oligonucleotides used during the nested PCR reaction were 5’-CCATGAGGCGGTCCAGGCAGTCTAGAG-3’ (sense) and 5’-CCTCCCAGTCCCCCCT TTTC-3’ (anti-sense). The PCR reactions were performed using an ABI 2720 Thermal Cycler with the following thermal profile: 95°C for 2 min, 1 cycle; 95°C for 30 s, 55° for 30 s, 65°C for 2 min, for 35 cycles; 65°C for 7 min. The PCR products were separated by electrophoresis, isolated, purified, sequenced and sub-cloned into pGEM-TEasy (Promega). Fifteen independent clones were sequenced and assessed for each mutant SHIV as described above for each condition.
Adeno-associated virus-2 (AAV-2) replication assay
The ability of rhA3A to inhibit the replication of AAV2 was assessed. 293 cells were transfected using LipoD 293 transfection reagent (SignaGen Labs;, Rockville, MD) into 60 mm dishes in duplicate with 1 μg SSV9 (an AAV2 infectious clone, psub201) (Qiu et al., 2002), 2μg pHelper (Stratagene, La Jolla, CA) (Xiao et al., 1998), and 1 μg of an A3 expression vector, rhA3G, hA3A, rhA3A, or as a negative control pBluescript SK (+) (Stratagene, La Jolla, CA). Forty-eight h post-transfection, the cells were harvested and resuspended in PBS. The supernatant was discarded and Hirt DNA was extracted using Hirt solution (10 mM Tris, 10 mM EDTA, pH 7.5, 0.6% SDS) at room temperature for 10 min followed by 5 M NaCl at 4°C overnight. The mixture was centrifuged at 14,000 rpm at 4°C for 20 min. Following centrifugation, the pellet was discarded and the supernatant was treated with Proteinase K (50 μg/ml) at 37°C for 1 h. The low molecular weight (LMW) DNA was extracted twice with saturated phenol and once with chloroform/isoamyl alcohol. The resulting DNA was ethanol precipitated and column purified using the Qiagen gel extraction kit (Qiagen). The LMW DNA was run on a 1% agarose gel, transferred to nitrocellulose and analyzed by Southern hybridization using a 32P-labeled probe consisting of a fragment representing nucleotides 184-4490 (Xba I digestion) from SSV9 as previously described (Qiu et al., 2002). To ensure that all samples were expressing the desired A3 proteins, aliquots of harvested cells utilized in the Southern blot were lysed in RIPA buffer. The nuclei were removed and the protein was precipitated using methanol, resuspended in 2X sample reducing buffer, and boiled for 5 min. Proteins were separated on a 12% SDS-PAGE gel and probed using a rabbit polyclonal anti-HA antibody (HA-probe, Santa Cruz).
L1 retrotransposition assay
The engineered L1 enhanced green fluorescent protein (EGFP) reporter (Ostertag et al., 2000) was cloned into the pLRE3-EF1-mEGFPI, which contains a retrotransposition competent L1 element (LRE3) (Brouha et al., 2003) under the control of an EF-1α promoter in addition to the internal 5’ UTR promoter. The enhanced GFP (eGFP) retrotransposition indicator cassette is under the control of an ubiquitin promoter (UBC), and the SV40 late polyadenylation signal. The construct was cloned into pBSKS-II+ (Stratagene). The positive control pLRE3-EF1-mEGFP(Δintron) is similar to the pLRE3-EF1-mEGFPI but lacks the intron in the mEGFPI indicator cassette and therefore serves as a positive control for transfection efficiency. One μg of L1 construct and 200 ng of empty vector, hA3A, rhA3A or rhA3ACSVR plasmid was co-transfected into 293T cells using Fugene 6 (Roche). All transfections were performed in triplicate. Cells were harvested at 2 and 4 days and analyzed for eGFP expression using a FACSCalibur machine (BD Biosciences), collecting 150,000 events.
Supplementary Material
Acknowledgments
The work reported here is supported by a grants NIH AI090795 to M.L.S. and NIH AI51981 to E.B.S. We thank members of the KUMC Biotechnology Support Facility and Northwestern University for their assistance with the sequence analysis and oligonucleotide synthesis. The following reagents were obtained through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH: TZM-bl from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc.; pNL4-3 from Dr. Malcolm Martin; and p24 antibody from Dr. Michael H. Malim.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe H, Ochi H, Maekawa T, Hatakeyama T, Tsuge M, Kitamura S, Kimura T, Miki D, Mitsui F, Hiraga N, Imamura M, Fujimoto Y, Takahashi S, Nakamura Y, Kumada H, Chayama K. Effects of structural variations of APOBEC3A and APOBEC3B genes in chronic hepatitis B virus infection. Hepatol Res. 2009;39:1159–1168. doi: 10.1111/j.1872-034X.2009.00566.x. [DOI] [PubMed] [Google Scholar]
- Aguiar RS, Lovsin N, Tanuri A, Peterlin BM. Vpr.A3A chimera inhibits HIV replication. J Biol Chem. 2008;283:2518–2525. doi: 10.1074/jbc.M706436200. [DOI] [PubMed] [Google Scholar]
- Babushok DV, Kazazian HH., Jr Progress in understanding the biology of the human mutagen LINE-1. Hum Mut. 2007;28:527–539. doi: 10.1002/humu.20486. [DOI] [PubMed] [Google Scholar]
- Baumert TF, Rosler C, Malim MH, von Weizsäcker F. Hepatitis B virus DNA is subject to extensive editing by the human deaminase APOBEC3C. Hepatology. 2007;46:682–689. doi: 10.1002/hep.21733. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Holmes RK, Sheehy AM, Davidson NO, Cho SJ, Malim MH. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr Biol. 2004;14:1392–1406. doi: 10.1016/j.cub.2004.06.057. [DOI] [PubMed] [Google Scholar]
- Bishop KN, Verma M, Kim EY, Wolinsky SM, Malim MH. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008;4:e1000231. doi: 10.1371/journal.ppat.1000231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Doehle BP, Lueders KK, Cullen BR. APOBEC3A and APOBEC3B are potent inhibitors of LTR-retrotransposon function in human cells. Nucleic Acids Res. 2006a;34:89–95. doi: 10.1093/nar/gkj416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd HP, Wiegand HL, Hulme AE, Garcia-Perez JL, O’Shea KS, Moran JV, Cullen BR. Cellular inhibitors of long interspersed element 1 and Alu retrotransposition. Proc Natl Acad Sci USA. 2006b;103:8780–8785. doi: 10.1073/pnas.0603313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonvin M, Achemann F, Greeve I, Stroka D, Keogh A, Inderbitzin D, Candinas D, Sommer P, Wain-Hobson S, Vartanian JP, Greeve J. Interferon-inducible expression of APOBEC3C editing enzymes in human hepatocytes and inhibition of hepatitis B virus replication. Hepatology. 2006;43:1364–1374. doi: 10.1002/hep.21187. [DOI] [PubMed] [Google Scholar]
- Bonvin M, Greeve J. Effects of point mutations in the cytidine deaminase domains of APOBEC3B on replication and hypermutation of hepatitis B virus in vitro. J Gen Virol. 2007;88:3270–3274. doi: 10.1099/vir.0.83149-0. [DOI] [PubMed] [Google Scholar]
- Brouha B, Schustak J, Badge RM, Lutz-Prigge S, Farley AH, Moran JV, Kazazian HH., Jr Hot L1s account for the bulk of retrotransposition in the human population. Proc Natl Acad Sci U S A. 2003;100:5280–5285. doi: 10.1073/pnas.0831042100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulliard T, Narvaiza I, Bertero A, Peddi S, Röhrig UF, Oritz M, Zoete V, Castro-Diaz N, Turelli P, Telenti A, Michielin O, Weitzman MD, Trono D. Structure-function analyses point to a polynucleotide-accommodating groove essential for APOBEC3A restriction activities. J Virol. 2011;85:1765–76. doi: 10.1128/JVI.01651-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celico L, Pham P, Calabrese P, Goodman MF. APOBEC3G DNA deaminase acts processively 3’-5’ on single stranded DNA. Nat Struct Mol Bio. 2006;13:392–399. doi: 10.1038/nsmb1086. [DOI] [PubMed] [Google Scholar]
- Chen H, Lilley CE, Yu Q, Lee DV, Chou J, Narvaiza I, Landau NR, Weitzman MD. APOBEC3A is a potent inhibitor of adeno-associated virus and retrotransposons. Curr Biol. 2006;16:480–485. doi: 10.1016/j.cub.2006.01.031. [DOI] [PubMed] [Google Scholar]
- Chiu YL, Greene WC. APOBEC3G: an intracellular centurion. Philos Trans R Soc Lond B Biol Sci. 2009;364:689–703. doi: 10.1098/rstb.2008.0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Thomas CJ, Petersen-Mahrt SK, Neuberger MS. Evolution of the AID/APOBEC family of polynucleotide (deoxy)cytidine deaminases. Biol Evol. 2005;22:367–377. doi: 10.1093/molbev/msi026. [DOI] [PubMed] [Google Scholar]
- Dang Y, Wang X, Esselman WJ, Zheng YH. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J Virol. 2006;80:10522–10533. doi: 10.1128/JVI.01123-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derdeyn CA, Decker JM, Sfakianos JN, Wu X, O’Brien WA, Ratner L, Kappes JC, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J Virol. 2000;74:8358–8367. doi: 10.1128/jvi.74.18.8358-8367.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehle BP, Schafer A, Cullen BR. Human APOBEC3B is a potent inhibitor of HIV-1 infectivity and is resistant to HIV-1 Vif. Virology. 2005;339:281–288. doi: 10.1016/j.virol.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Khan MA, Miyagi E, Kao S, Strebel K. Targeting APOBEC3A to the viral nucleoprotein complex confers antiviral activity. Retrovirology. 2007;4:61. doi: 10.1186/1742-4690-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goila-Gaur R, Strebel K. HIV-1 Vif, APOBEC, and intrinsic immunity. Retrovirology. 2008;5:51. doi: 10.1186/1742-4690-5-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Saadatmand J, Kleiman L. Inhibition of formula-primed reverse transcription by human APOBEC3G during human immunodeficiency virus type 1 replication. J Virol. 2006;80:11710–11722. doi: 10.1128/JVI.01038-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Cen S, Niu M, Yang Y, Gorelick RJ, Kleiman L. The interaction of APOBEC3G with human immunodeficiency virus type 1 nucleocapsid inhibits tRNA3Lys annealing to viral RNA. J Virol. 2007;81:11322–11331. doi: 10.1128/JVI.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harari A, Ooms M, Mulder LC, Simon V. Polymorphisms and splice variants influence the antiretroviral activity of human APOBEC3H. J Virol. 2009;83:295–303. doi: 10.1128/JVI.01665-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry M, Guètard D, Suspène R, Rusniok C, Wain-Hobson S, Vartanian JP. Genetic editing of HBV DNA by monodomain human APOBEC3 cytidine deaminases and the recombinant nature of APOBEC3G. PLoS One. 2009;4:e4277. doi: 10.1371/journal.pone.0004277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes RK, Malim MH, Bishop KN. APOBEC-mediated viral restriction: not simply editing? Trends Biochem Sci. 2007;32:118–128. doi: 10.1016/j.tibs.2007.01.004. [DOI] [PubMed] [Google Scholar]
- Holmes RK, Koning FA, Bishop KN, Malim MH. APOBEC3F can inhibit the accumulation of HIV-1 reverse transcription products in the absence of hypermutation. Comparisons with APOBEC3G. J Biol Chem. 2007;282:2587–2595. doi: 10.1074/jbc.M607298200. [DOI] [PubMed] [Google Scholar]
- Huthoff H, Malim MH. Cytidine deamination and resistance to retroviral infection: towards a structural understanding of the APOBEC proteins. Virology. 2005;334:147–153. doi: 10.1016/j.virol.2005.01.038. [DOI] [PubMed] [Google Scholar]
- Iwatani Y, Chan DS, Wang F, Maynard KS, Sugiura W, Gronenborn AM, Rouzina I, Williams MC, Musier-Forsyth K, Levin JG. Deaminase-independent inhibition of HIV-1 reverse transcription by APOBEC3G. Nucleic Acids Res. 2007;35:7096–7108. doi: 10.1093/nar/gkm750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarmuz A, Chester A, Bayliss J, Gisbourne J, Dunham I, Scott J, Navaratnam N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics. 2002;79:285–296. doi: 10.1006/geno.2002.6718. [DOI] [PubMed] [Google Scholar]
- Kakimoto WM, Gettie A, Smith S, Donahoe SM, Jin X, Marx P, Connor R, Nixon DF. Comparison of restimulation methods to elicit SIV specific cytotoxic T-lymphocytes (CTL) in vitro: Staphylococcal enterotoxin B (SEB) provides a novel method for the quantification of SIV specific CTL precursors. Immunol Lett. 1999;66:35–140. doi: 10.1016/s0165-2478(98)00173-4. [DOI] [PubMed] [Google Scholar]
- Kao S, Khan MA, Miyagi E, Plishka R, Buckler-White A, Strebel K. The human immunodeficiency virus type 1 Vif protein reduces intracellular expression and inhibits packaging of APOBEC3G (CEM15), a cellular inhibitor of virus infectivity. J Virol. 2003;77:11398–11407. doi: 10.1128/JVI.77.21.11398-11407.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köck J, Blum HE. Hypermutation of hepatitis B virus genomes by APOBEC3G, APOBEC3C, and APOBEC3H. J Gen Virol. 2008;89:1184–1191. doi: 10.1099/vir.0.83507-0. [DOI] [PubMed] [Google Scholar]
- Koning FA, Newman EN, Kim EY, Kunstman KJ, Wolinsky SM, Malim MH. Defining APOBEC3 expression patterns in human tissues and hematopoietic cell subsets. J Virol. 2009;83:9474–9485. doi: 10.1128/JVI.01089-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroutter EN, Belancio VP, Wagstaff BJ, Roy-Engel AM. The RNA polymerase dictates ORF1 requirement and timing of LINE and SINE retrotransposition. PLoS Genet. 2009;5:e1000458. doi: 10.1371/journal.pgen.1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landry S, Narvaiza I, Linfesty DC, Weitzman MD. APOBEC3A can activate the DNA damage response and cause cell-cycle arrest. EMBO Rep. 2011 doi: 10.1038/embor.2011.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois MA, Beale RC, Conticello SG, Neuberger MS. Mutational comparison of the single-domained APOBEC3C and double-domained APOBEC3F/G anti-retroviral cytidine deaminases provides insight into their DNA target site specificities. Nucleic Acids Res. 2005;33:1913–1923. doi: 10.1093/nar/gki343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRue RS, Jónsson SR, Silverstein KA, Lajoie M, Bertrand D, El-Mabrouk N, Hötzel I, Andrésdóttir V, Smith TP, Harris RS. The artidactyl APOBEC3 innate immune repertoire shows evidence for a multi-functional domain organization that existed in the ancestor of placental mammals. BMC Mol Biol. 2008;18:104. doi: 10.1186/1471-2199-9-104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MM, Wu LI, Emerman M. The range of human APOBEC3H sensitivity to lentiviral Vif proteins. J Virol. 2010;84:88–95. doi: 10.1128/JVI.01344-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XY, Guo F, Zhang L, Kleiman L, Cen S. APOBEC3G inhibits DNA strand transfer during HIV-1 reverse transcription. J Biol Chem. 2007;282:32065–32074. doi: 10.1074/jbc.M703423200. [DOI] [PubMed] [Google Scholar]
- Liddament MT, Brown WL, Schumacher AJ, Harris RS. APOBEC3F properties and hypermutation preferences indicate activity against HIV-1 in vivo. Curr Biol. 2004;14:1385–1391. doi: 10.1016/j.cub.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Liu B, Yu X, Luo K, Yu Y, Yu XF. Influence of primate lentiviral Vif and proteasome inhibitors on human immunodeficiency virus type 1 virion packaging of APOBEC3G. J Virol. 2004;78:2072–2081. doi: 10.1128/JVI.78.4.2072-2081.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu ZQ, Muhkerjee S, Sahni M, McCormick-Davis C, Leung K, Li Z, Gattone VH, 2nd, Tian C, Doms RW, Hoffman TL, Raghavan R, Narayan O, Stephens EB. Derivation and biological characterization of a molecular clone of SHIV(KU-2) that causes AIDS, neurological disease, and renal disease in rhesus macaques. Virology. 1999;260:295–307. doi: 10.1006/viro.1999.9812. [DOI] [PubMed] [Google Scholar]
- Luo K, Wang T, Liu B, Tian C, Xiao Z, Kappes J, Yu XF. Cytidine deaminases APOBEC3G and APOBEC3F interact with human immunodeficiency virus type 1 integrase and inhibit proviral DNA formation. J Virol. 2007;81:7238–7248. doi: 10.1128/JVI.02584-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahieux R, Suspène R, Delebecque F, Henry M, Schwartz O, Wain-Hobson S, Vartanian JP. Extensive editing of a small fraction of human T-cell leukemia virus type 1 genomes by four APOBEC3 cytidine deaminases. J Gen Virol. 2005;86:2489–2494. doi: 10.1099/vir.0.80973-0. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Muckenfuss H, Hamdorf M, Held U, Perkovic M, Lower J, Cichutek K, Flory E, Schumann GG, Munk C. APOBEC3 proteins inhibit human LINE-1 retrotransposition. J Biol Chem. 2006;281:22161–22172. doi: 10.1074/jbc.M601716200. [DOI] [PubMed] [Google Scholar]
- Narvaiza I, Linfesty DC, Greener BN, Hakata Y, Pintel DJ, Logue E, Landau NR, Weitzman MD. Deaminase-independent inhibition of parvoviruses by the APOBEC3A cytidine deaminase. PLoS Pathog. 2009;5:e1000439. doi: 10.1371/journal.ppat.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman EN, Holmes RK, Craig HM, Klein KC, Lingappa JR, Malim MH, Sheehy AM. Antiviral function of APOBEC3G can be dissociated from cytidine deaminase activity. Curr Biol. 2005;15:166–70. doi: 10.1016/j.cub.2004.12.068. [DOI] [PubMed] [Google Scholar]
- Noguchi C, Hiraga N, Mori N, Tsuge M, Imamura M, Takahashi S, Fujimoto Y, Ochi H, Abe H, Maekawa T, Yatsuji H, Shirakawa K, Takaori-Kondo A, Chayama K. Dual effect of APOBEC3G on hepatitis B virus. J Gen Virol. 2007;88:432–440. doi: 10.1099/vir.0.82319-0. [DOI] [PubMed] [Google Scholar]
- OhAinle M, Kerns JA, Li MM, Malik HS, Emerman M. Antiretroelement activity of APOBEC3H was lost twice in recent human evolution. Cell Host Microbe. 2008;11:249–259. doi: 10.1016/j.chom.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms M, Majdak S, Seibert CW, Harari A, Simon V. The localization of APOBEC3H variants in HIV-1 virions determines their antiviral activity. J Virol. 2010;84:7961–7969. doi: 10.1128/JVI.00754-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostertag EM, Prak ET, DeBerardinis RJ, Moran JV, Kazazian HH., Jr Determination of L1 retrotransposition kinetics in cultured cells. Nucl Acids Res. 2000;28:1418–1423. doi: 10.1093/nar/28.6.1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paprotka T, Venkatachari NJ, Chaipan C, Burdick R, Delviks-Frankenberry KA, Hu WS, Pathak VK. Inhibition of xenotropic murine leukemia virus-related virus by APOBEC3 proteins and antiviral drugs. J Virol. 2010;84:5719–5729. doi: 10.1128/JVI.00134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkovic M, Schmidt S, Marino D, Russell RA, Stauch B, Hofmann H, Kopietz F, Kloke BP, Zielonka J, Ströver H, Hermle J, Lindemann D, Pathak VK, Schneider G, Löchelt M, Cichutek K, Münk C. Species-specific inhibition of APOBEC3C by the prototype foamy virus protein bet. J Biol Chem. 2009;284:5819–5826. doi: 10.1074/jbc.M808853200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platt EJ, Bilska M, Kozak SL, Kabat D, Montefiori DC. Evidence that ecotropic murine leukemia virus contamination in TZM-bl cells does not affect the outcome of neutralizing antibody assays with human immunodeficiency virus type 1. J Virol. 2009;83:8289–8292. doi: 10.1128/JVI.00709-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu J, Pintel DJ. The adeno-associated virus type 2 rep protein regulates RNA processing via interaction with the transcription template. Mol Cell Biol. 2002;22:3639–3652. doi: 10.1128/MCB.22.11.3639-3652.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala SH, Kazazian HH., Jr The L1 retrotransposition assay: a retrospective and toolkit. Methods. 2009;49:219–226. doi: 10.1016/j.ymeth.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Refsland EW, Stenglein MD, Shindo K, Albin JS, Brown WL, Harris RS. Quantitative profiling of the full APOBEC3 mRNA repertoire in lymphocytes and tissues: implications for HIV-1 restriction. Nucleic Acids Res. 2010;38:4274–84. doi: 10.1093/nar/gkq174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RA, Moore MD, Hu WS, Pathak VK. APOBEC3G induces a hypermutation gradient: purifying selection at multiple steps during HIV-1 replication results in levels of G-to-A mutations that are high in DNA, intermediate in cellular viral RNA, and low in virion RNA. Retrovirology. 2009;13:6–16. doi: 10.1186/1742-4690-6-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer SL, Emerman M, Malik HS. Ancient adaptive evolution of the primate antiviral DNA-editing enzyme APOBEC3G. PLoS Biol. 2004;2:E275. doi: 10.1371/journal.pbio.0020275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K, Hill MS, Ruiz A, Culley N, Pinson DM, Wong SW, Stephens EB. Mutations in the highly conserved SLQXLA motif of Vif results in a simian-human immunodeficiency virus results in less pathogenic virus and is associated with G to A mutations in the viral genome. Virology. 2009;383:62–72. doi: 10.1016/j.virol.2008.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt K, Liu Q, Hill MS, Ruiz A, Culley N, Pinson DM, Wong SW, Stephens EB. Comparison of the pathogenicity of simian-human immunodeficiency viruses expressing Vif proteins with mutation of the SLQYLA and HCCH domains in macaques. Virology. 2010;404:87–203. doi: 10.1016/j.virol.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröfelbauer B, Senger T, Manning G, Landau NR. Mutational alteration of human immunodeficiency virus type 1 Vif allows for functional interaction with nonhuman primate APOBEC3G. J Virol. 2006;80:5984–5991. doi: 10.1128/JVI.00388-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Smith JL, Pathak VK. Identification of specific determinants of human APOBEC3F, APOBEC3C, and APOBEC3DE and African green monkey APOBEC3F that interact with HIV-1 Vif. J Virol. 2010;84:12599–608. doi: 10.1128/JVI.01437-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sova P, Volsky DJ. Efficiency of viral DNA synthesis during infection of permissive and nonpermissive cells with vif-negative human immunodeficiency virus type-1. J Virol. 1993;67:6322–6326. doi: 10.1128/jvi.67.10.6322-6326.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenglein MD, Burns MB, Li M, Lengyel J, Harris RS. APOBEC3 proteins mediate the clearance of foreign DNA from human cells. Na t Struct Mol Biol. 2010;17:222–229. doi: 10.1038/nsmb.1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strebel K. APOBEC3G & HTLV-1: inhibition without deamination. Retrovirology. 2005;2:37. doi: 10.1186/1742-4690-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Y, Zhu Q, Gagnon S, Dzutsev A, Terabe M, Vaccari M, Venzon D, Klinman D, Strober W, Kelsall B, Franchini G, Belyakov IM, Berzofsky JA. Innate and adaptive immune correlates of vaccine and adjuvant-induced control of mucosal transmission of SIV in macaques. Proc Natl Acad Sci U S A. 2010;107:9843–9848. doi: 10.1073/pnas.0911932107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi Y, McClure MO, Pizzato M. Identification of gammaretroviruses constitutively released from cell lines used for human immunodeficiency virus research. J Virol. 2008;82:12585–12588. doi: 10.1128/JVI.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli P, Mangeat B, Jost S, Vianin S, Trono D. Inhibition of hepatitis B virus replication by APOBEC3G. Science. 2004;303:1829. doi: 10.1126/science.1092066. [DOI] [PubMed] [Google Scholar]
- Vartanian JP, Guétard D, Henry M, Wain-Hobson S. Evidence for editing of human papillomavirus DNA by APOBEC3 in benign and precancerous lesions. Science. 2008;320:230–233. doi: 10.1126/science.1153201. [DOI] [PubMed] [Google Scholar]
- Virgen CA, Hatziioannou T. Antiretroviral activity and vif sensitivity of rhesus macaque APOBEC3 proteins. J Virol. 2007;81:13932–13937. doi: 10.1128/JVI.01760-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler U, Song J, Aiken C, Trono D. Vif is crucial for human immunodeficiency virus type 1 proviral cDNA synthesis in infected cells. J Virol. 1993;67:4945–4955. doi: 10.1128/jvi.67.8.4945-4955.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Abudu A, Son S, Dang Y, Venta PJ, Zheng YH. Analysis of human APOBEC3H haplotypes and anti-human immunodeficiency virus type-1 activity. J Virol. 2011;85:3142–3152. doi: 10.1128/JVI.02049-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby JM, Saag MS, Wu X, Shaw GM, Kappes JC. Emergence of resistant human immunodefici ency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother. 2002;46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegand HL, Doehle BP, Bogerd HP, Cullen BR. A second human antiretroviral factor, APOBEC3F, is suppressed by the HIV-1 and HIV-2 Vif protein. EMBO J. 2004;23:2451–2458. doi: 10.1038/sj.emboj.7600246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Li J, Samulski RJ. Production of high-titer recombinant adeno-associated virus vectors in the absence of helper adenvirus. J Virol. 1998;72:2224–2232. doi: 10.1128/jvi.72.3.2224-2232.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Yu Q, Konig R, Pillai S, Chiles K, Kearney M, Palmer S, Richman D, Coffin JM, Landau NR. Single-strand specificity of APOBEC3G accounts for minus-strand deamination of the HIV genome. Nat Struct Mol Biol. 2004;11:435–442. doi: 10.1038/nsmb758. [DOI] [PubMed] [Google Scholar]
- Zennou V, Bieniasz PD. Comparative analysis of the antiretroviral activity of APOBEC3G and APOBEC3F from primates. Virology. 2006;25:31–40. doi: 10.1016/j.virol.2005.12.035. [DOI] [PubMed] [Google Scholar]
- Zhang W, Zhang X, Tian C, Wang T, Sarkis PT, Fang Y, Zheng S, Yu XF, Xu R. Cytidine deaminase APOEC3B interacts with heterogeneous nuclear ribonucleoprotein K and suppresses hepatitis B virus expression. Cell Microbiol. 2008;10:112–121. doi: 10.1111/j.1462-5822.2007.01020.x. [DOI] [PubMed] [Google Scholar]
- Zheng YH, Irwin D, Krosu T, Tokunaga K, Sata T, Peterlin BM. Human APOBEC3F is another host factor that blocks human immunodeficiency virus type 1 replication. J Virol. 2004;78:6073–6076. doi: 10.1128/JVI.78.11.6073-6076.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng YH, Peterlin BM. Intracellular immunity to HIV-1:newly defined retroviral battles inside infected cells. Retrovirology. 2005;13:2–25. doi: 10.1186/1742-4690-2-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.