

Introduction
Liposomal formulations offer several potential advantages for the intravenous delivery of antitumor agents due to their ability to increase drug solubility, reduce drug toxicity, and prolong drug release [1]. Pegylated liposomes have the added benefit of longer systemic circulation due to reduced clearance by the mononuclear phagocytic system [1-4]. This prolonged circulation time when combined with an appropriate particle size provides preferential delivery of liposomes to solid tumors, a result of the well-known enhanced permeability and retention of nanoparticles in tumors [1, 3, 5]. These properties have led to the FDA-approved liposomal formulation of doxorubicin (DOXIL®) as well as other drug products, including several currently in clinical trials [6-13].
When investigated systematically, the antitumor efficacy of drug-loaded liposomal formulations has been closely linked to the drug release rate [14-17] Such investigations imply that the ability to tailor liposomal drug release rates could enable clinicians to optimize efficacy for a specific tumor by selecting the delivery system that produces the optimal tumor concentration profile. Protracted or metronomic dosing regimens have shown such promise with enhanced antitumor agent efficacy [18-20], but these approaches are unable to take advantage of the localized intratumoral drug release afforded by liposomes and other nanoparticulate systems. Liposomal systems that provide predictable drug release rates would reduce the frequent visits and monitoring currently necessary due to the narrow therapeutic window and rapid clearance of many chemotherapeutics.
While many models have been developed to describe drug loading [21-24], few have considered release kinetics [25-27] and even fewer have been validated experimentally [21, 26, 27]. Mechanistic models that incorporate physicochemical properties of the drug in solution including the drug species present as a function of intraliposomal pH, their interactions with the lipid bilayer, and their membrane permeabilities would be essential to release rate design and optimization.
Liposomal formulations of topotecan (TPT) serve as a prime example of the need for mechanistic models to reliably predict drug release rates under a variety of loading and release conditions. TPT is a topoisomerase I inhibitor currently approved to treat cervical, ovarian, and small cell lung cancers as an injectable solution and in multiple clinical trials as the sole medication or in conjunction with other medications and/or radiation [28-32]. Like other weakly basic drugs, TPT exhibits high encapsulation efficiency in liposomal formulations utilizing active loading strategies [14, 33-38]. Previous work with liposomal TPT has mainly focused on encapsulation strategies, while the emphasis on controlled or extended release has been limited [17, 35, 36, 39, 40].
Studies that systematically examine various formulation and releasing-media parameters are necessary to develop a model capable of understanding and controlling release. Since the generation of a lowintravesicular pH is a prerequisite for active loading of weak bases while drug release occurs under physiological conditions near a neutral pH [34, 37, 39], evaluating the sensitivity of TPT release to both the intra- and extravesicular pH is critical to the development of a mechanistic model having practical utility. Over this pH range, TPT is assumed to exist in solution as one of four major species as illustrated in Scheme 1.
Scheme 1.

The major species of TPT in solution in the low to neutral pH range. Two lactone forms are present (Lp and Ln) differing in the state of ionization of the phenol on ring A. Reversible hydrolysis of the ring may transform these species to their lactone E-ring may transform these species to their carboxylate counterparts (Cn and Ca respectively).
The aim of the present work was to determine the pH sensitivity of TPT release from unilamellar liposomes and develop a mechanism-based mathematical model to account for the observed transport rates. To completely account for the pH-permeability profiles obtained experimentally, the mathematical model had to include the effects of TPT speciation via ionization, membrane-binding equilibria, drug species’ permeability coefficients, and the kinetics and pH dependence of TPT lactone ring-opening/closing.
Materials and Methods
Materials
Powders of 1,2-Distearoyl-sn-glycero-3-phosphatidylcholine (DSPC, >99% purity) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethyleneglycol)-2000] (m-PEG DSPE, MW = 2806, >99% purity) were purchased from Avanti Polar Lipids (Alabaster, AL). Topotecan hydrochloride was purchased from AK Scientific (Union City, CA). Millipore ultrafiltration cartridges (Amicon® Ultra 0.5 mL centrifugal filter device with 30,000 MWCO Ultracel® membrane), Nuclepore polycarbonate membranes (0.1 µm), solvents, and buffer salts were purchased from Fisher Scientific (Florence, KY). All solvents were HPLC grade.
Preparation and characterization of DSPC/m-PEG DSPE liposomes for lactone-carboxylate interconversion and release studies
Large unilamellar vesicles were formed using the film hydration and extrusion process described in several previous reports with slight modifications [26, 40]. Briefly, DSPC and m-PEG DSPE (95:5 mol:mol) lipids were weighed into borosilicate vials, then dissolved in chloroform. The chloroform was subsequently evaporated under a stream of N2 and the residue was vacuum-dried at 40°C for 6 hours to form a thin lipid film. Films were hydrated and passed 10 times through two stacked Nuclepore polycarbonate membranes (100 nm pore) using a Liposofast® extrusion device at 60°C to obtain unilamellar vesicles. All solutions for film hydration were made with a buffer concentration of 50 mM and adjusted to an ionic strength of 0.3 with NaCl. The reported pH was measured at 37 °C. Liposomes used in lactone-carboxylate interconversion studies were composed of lipid films hydrated with pH 6.33 (2-(N-morpholino)ethanesulfonic acid (MES)) and pH 7.67 (Tris) to achieve a final lipid concentration of 50 mg/mL. Liposomes for release studies were hydrated with solutions of pH 3.35 (chloroacetate), 4.01 (formate), 5.01 (acetate), 5.92 and 6.33 (MES), 7.04 and 7.39 (phosphate), and 7.67 (Tris) containing 50 μM TPT, yielding a lipid concentration of 40 mg/mL.
For the calculation of vesicle volume parameters, particle size and lipid content were determined. Liposome particle size was analyzed using a Beckman Delsa™ Nano C Particle Sizer with a 70 second accumulation time. Particle size before and after release studies was determined using Cumulants analysis. Liposome suspensions were diluted by a factor of 10 before analysis to obtain intensity readings within the detection range of the instrument. To avoid interference from dust and other artifacts during size analysis, the buffers used in liposome hydration and subsequent release studies were filtered with a 0.22 μm nitrocellulose filter. Samples were stored at 4°C until analysis. Lipid content was determined using HPLC and is described in further detail in the analyses section.
Fluorescence measurements of aqueous TPT solutions
The acid dissociation constant of the TPT A-ring phenol was determined using changes in fluorescence excitation spectra with pH. Solutions of 500 nM TPT were prepared at various pH with buffers of formate (pH 3.50), acetate (4.50 and 5.50), MES (pH 6.00 and 6.27) and phosphate (pH 6.50, 6.80, 7.20, and 7.50) at concentrations of 50 mM while ionic strength was kept at 0.3 by adjustment with sodium chloride. Solutions were scanned with a FluoroMax-3 (Jobin Yvon Inc., Edison, NJ). Excitation scans were made over a range of 300 – 470 nm using an emission wavelength of 560 nm. The temperature of the sample chamber was maintained at 37 °C, and fluorescence intensity was recorded using a 0.5 second integration time and a 3 nm band pass width.
TPT interconversion studies
Kinetic studies of the reversible and pH dependent ring-opening/closing of TPT were conducted with 0.4-0.6 μM TPT solutions at pH 5.92, 6.33, 7.04, 7.39, and 7.67 using the same buffers used to hydrate liposomes. At pH 5.92 and 6.33, solutions were spiked with a 50 μM stock solution of the ring-opened TPT carboxylate dissolved in 0.1 N NaOH. Studies at higher pH used a 50 μM stock of lactone TPT in DMSO. To determine the effect (if any) of TPT binding to the bilayer, interconversion studies were also conducted in 50 mg/mL liposome suspensions at pH 6.33 and 7.67 and compared with profiles obtained in aqueous solution.
At various times, 150 μL samples were withdrawn and interconversion was quenched using 300 μL of a chilled (−20 °C) 2:1 (v:v) acetonitrile: methanol solution. Samples were immediately injected and analyzed by HPLC for both ring-opened carboxylate and lactone content. All studies were conducted in a water-jacketed incubator maintained at 37 °C and stirred at 200 rpm with a 10 × 5 mm Teflon stir bar using a Thermo Cimerac iPoly 15 multipoint stirrer.
Release of TPT from DSPC/DSPE-PEG-2K liposomes
Release studies were conducted in a similar manner as reported previously [41]. Unencapsulated TPT present in passively loaded liposome suspensions was removed by passing suspensions through a Sephadex G-25 column equilibrated with the same buffer as the liposome suspension. Aliquots of liposome suspension (0.2–0.5 mL) were passed through the column and the drug-loaded liposome fraction eluting between 2.5–5 mL was collected, yielding liposome suspensions for release studies having lipid concentrations of 1.0-4.5 mg/mL. Liposome suspensions were transferred to glass vials capped with a rubber stopper and stirred at the same conditions used for interconversion studies within a water-jacketed incubator maintained at 37 °C. Suspension temperature was monitored daily using a digital thermometer over the time span of release studies.
Encapsulated drug was monitored by ultrafiltration of 50–150 μL aliquots of liposome suspension taken at various time points. Each aliquot was diluted with chilled (4 °C) buffer to 425 μL and ultrafiltered using an Amicon® Ultra 0.5 mL centrifugal filter device with 30,000 MWCO Ultracel® membrane. Cartridges were then centrifuged at 14,000 rpm for 10 minutes in an Eppendorf 5417R maintained at 4 °C. The concentrated suspension (50 μL) was recovered by inverting the cartridge and centrifuging at 2000 rpm for another 2 minutes. Recovered concentrate was resuspended in another 400 μL of chilled buffer and the process was repeated. The final concentrate was dissolved in acidified methanol and diluted within the calibration range for HPLC analysis.
HPLC analyses
Samples from interconversion studies were analyzed for TPT concentration by HPLC [42]. A Waters Alliance 2695 separation system coupled to a Waters fluorescence detector (M474) was employed with excitation and emission wavelengths at 380 and 560 nm, respectively. Interconversion studies measured both lactone and carboxylate forms of TPT using a Supelcosil™ ABZ+ column (250 × 4.6 mm, 5 μm) and guard column (20 × 4.0 mm, 5 μm) with a mobile phase (14% acetonitrile: 86% (v/v) of 5% (pH = 5.5) triethylamine acetate, 50 mM tetrabutylammonium hydrogen sulfate (TBAHS) buffer) flow rate of 1.5 mL/min. Lactone TPT standards were prepared in chilled, acidified methanol (−20 °C) and carboxylate standards were prepared in 10 mM sodium carbonate buffer (pH 10.1) at 20-200 nM concentrations. Lactone and carboxylate retention times were 5.5 and 2.1 min, respectively.
For release studies, samples were diluted in chilled, acidified methanol (0.001 N HCl for studies at pH ≤ 5.01 and 0.02 N HCl for studies conducted at higher pH) to convert all TPT to its lactone form. Samples were stored at −20 °C until analysis. TPT lactone was then analyzed using a Waters Symmetry® C18 column (4.6×150 mm, 5 μm) and guard column (3.9 × 20 mm) with a mobile phase (16% acetonitrile: 84% (v/v) of 5% (pH = 5.5) triethylamine acetate buffer) flow rate of 1 mL/min. Sample compartment and column were kept at ambient temperature. The retention time for TPT lactone was 4.5 min and response was linear between 20 and 200 nM.
Lipid analysis was performed with HPLC using an evaporative light scattering detector (ELSD, Sedere, Inc., Lawrenceville, NJ) based on a previously described method using an Allsphere (Alltech Associates, Inc., Deerfield, IL) silica column (4 × 150 mm, 5 μm) and guard column (20 × 4.0 mm, 5 μm). The elution method employed a linear gradient composed of 100% mobile phase A (80% chloroform:19.5% methanol:0.5%(v/v) NH4OH) changing to 80% mobile phase A and 20% mobile phase B (80% methanol:19.5% water:0.5% (v/v) NH4OH) at 3 min which was maintained until 7 min, and returned to 100% mobile phase A at 14 min at a flow rate of 1 mL/min [26]. Samples (100 μL) were dried at room temperature under N2, then dissolved in chilled mobile phase A before analysis.
Mechanism-based mathematical model development
Scheme 2 depicts the equilibria and rate constants that influence the rate of liposomal release of TPT as a function of pH. While the mechanistic approach is similar to those employed previously for other compounds [21, 26, 27], the species present and parameter values will obviously differ. Using this scheme, a mathematical model that accounted for the various species of TPT as a function of pH was developed as briefly described below (see Supplemental Data for the full derivation).
Scheme 2.

A schematic of the associated equilibria and kinetics governing liposomal release of TPT. The liposome depicted with radius, r, highlights the volume compartments described in the mathematical model. The different volume compartments are color coded with blue highlighting the inner aqueous volume, , while the green and violet sections refer to the inner, , and outer, , membrane volumes, respectively. The transport pathways and binding/ionization equilibria for all species illustrated in Scheme 1 are also depicted and described in the accompanying table (right).
Total drug release within a liposome suspension may be described by the total rate of change for both intra- and extravesicular concentrations of TPT ( and , respectively). This total rate of change is the sum of the rates of change for both the lactone, , and carboxylate, , forms of TPT in the intra- and extravesicular compartments. This is expressed in Eqs. 1a and b.
| (1a) |
| (1b) |
The driving force governing liposomal drug release is the concentration gradient between the unbound, intra- and extra-vesicular concentrations of the permeable species. In the case of TPT, pH-dependent ring-closure may become the rate-limiting step for drug release under certain conditions. Thus, the rates of change of the intravesicular lactone and carboxylate concentrations are determined by both diffusive and chemical kinetic contributions as depicted in Eq. 2.
| (2) |
These terms can be explicitly written for the rates of both the lactone and carboxylate forms of TPT as shown by Eq. 3a and b for the intravesicular compartment.
| (3a) |
| (3b) |
The colors highlighting the various terms in Eqs. 3a and b correspond to those in Eq. 2. In both equations, the rates of change of intravesicular lactone and carboxylate (highlighted green) are composed of diffusive (yellow) and chemical kinetic components (blue) describing interconversion of TPT between its lactone and carboxylate forms.
In the diffusion term, and are effective transport rate constants for the lactone and carboxylate species, respectively. These constants are pH dependent, as determined by the various ionization states of the lactone and carboxylate species (Scheme 1) and their permeability coefficients. The superscript, u, indicates that only lactone and carboxylate species not bound to the membrane contribute to the diffusive driving force governing release. The fraction of unbound drug can be solved for mathematically (see Supplementary Data). The interconversion term accounts for the kinetics of lactone-carboxylate interconversion.
Using this mechanistic model to obtain release rate constants for each TPT species from transport studies also required separate experiments to generate parameters governing the ionization state of the TPT A-ring phenol and the lactone-carboxylate interconversion kinetics. These determinations are described in the upcoming sections.
Spectrometric determination of the TPT A-ring phenol (pKa1)
The ionization state of drugs and small molecules has been shown to alter release kinetics, typically due to the likelihood that the neutral form is more permeable than charged species [26, 43, 44]. In the case of TPT, the phenolic -OH group ortho to the dimethyl-aminomethyl substituent on the A ring may ionize and alter the charge of TPT as depicted in Scheme 3. The value of pKa1 was determined by fitting the fluorescence intensities generated from excitation spectra of TPT at varying pH (3.5 – 7.5). Details of these analyses are provided in Supplementary Data.
Scheme 3.

Phenol ionization on the A ring of TPT is governed by the acid dissociation constant, Ka1.
TPT lactone-carboxylate interconversion kinetics in solution and in the presence of liposomes
The lactone E-ring in TPT can undergo reversible, base-catalyzed hydrolysis to form its ring-opened, carboxylate form via deprotonation of the carboxylic acid intermediate, CCOOH (Scheme 4). This process is pH-dependent, similar to other camptothecin analogues, with the lactone form, L, dominating at low pH. As pH is increased ionization of the ring-opened carboxylic acid to form CCOO- shifts the equilibrium toward the ring-opened species [42, 45]. Note, however, that both the ring-opening and closing reactions described in Scheme 4 are base-catalyzed in accordance with the principle of microscopic reversibility.
Scheme 4.
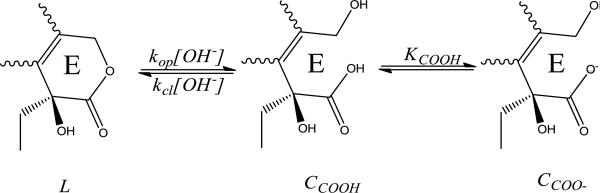
The proposed mechanism for reversible, pH dependent ring opening of TPT from its lactone, L, to carboxylate, CCOO-, form. Because ring opening proceeds through the carboxylic acid species, CCOOH, ring opening increases as more carboxylate is formed at higher pH, as governed by the acid dissociation constant for the E-ring carboxylic acid, KCOOH.
The rate constants governing the base-catalyzed ring opening/closing kinetics were determined by monitoring the lactone and carboxylate concentrations at various pH (6.33 – 7.67) and fitting the data to an interconversion model based on Scheme 4 (see Supplementary Data).
Results
Validation of analytical methods and liposome characterization
TPT concentrations were analyzed by HPLC with fluorescence detection using excitation and emission wavelengths of 380 nm and 550 nm, respectively. Peak areas varied linearly with concentration between 20 and 200 nM for both lactone and carboxylate standards. Release studies monitored total TPT concentration by converting all drug to its lactone form by diluting samples with chilled acidified methanol into the concentration range of standards. Coefficients of variation for response factors of standards were ±1.4% intraday and ±2.6% interday. Initial TPT concentrations ranged from 0.2-1.0 μM for these experiments. Studies monitoring TPT interconversion kinetics were performed with initial concentrations of 0.4-0.6 μM and coefficients of variation for response factors were ±2.0% intraday and ±7.1% interday for carboxylate standards and ±1.5% intraday and ±2.1% interday for lactone standards. Phospholipid content was determined by a previously validated HPLC method with slight modifications [26, 27]. ELSD, necessary due to the lack of chromophore/fluorophore in the lipid molecules, provided linear log-log plots of the peak areas versus DSPC concentration between 0.025 – 0.3 mg/mL and a peak retention time of 7.9 minutes, similar to that previously reported [26].
To validate the ultrafiltration procedure as a reliable technique to analyze drug release kinetics, the recovery efficiency of TPT and phospholipid after ultrafiltration was determined. Sephadex separation of liposomally entrapped TPT from unencapsulated drug was used as previously reported [41] to determine this recovery value. By analyzing TPT concentrations in liposomal fractions collected immediately after Sephadex purification, the percentage of TPT recovered after the ultrafiltration procedure was determined to be 88.0 ± 2.4 %; however, this value may be an underestimation if trace amounts of unentrapped TPT were present after Sephadex separation. To further explore this as a possible source of error, phospholipid content was also determined in liposome samples after ultrafiltration. Phospholipid recovery was determined to be 94.0 ± 3.9 %. The slightly higher recovery of phospholipid in comparison to TPT after ultrafiltration of freshly purified drug-loaded liposomes provides evidence for the presence of a small percentage of unentrapped drug. Consequently, phospholipid and TPT recovery were compared at the start of a release experiment to estimate the initial amount of TPT in the extra-vesicular solution. During these experiments, the initial extra-vesicular TPT was never more than 2.5 % of the initial intra-vesicular TPT. As a final validation, suspensions of liposomes containing no drug were spiked with TPT and then ultrafiltered. Ultrafiltration of these spiked solutions showed only trace amounts of TPT present well, below the limit of quantitation (less than 0.3 % of initial TPT). Because passive loading was used to conduct these experiments, encapsulation efficiency was not determined as it was expected to be low (< 3%) and not germane to the goals of this study.
Volume parameters (Table 1) used in model fitting were calculated from particle sizes and phospholipid contents determined during release studies. Particle size was determined at each pH (8 measurements) for the suspensions before and after the conclusion of release studies. The average particle size before release studies (with 95 % confidence interval) was 96 ± 2 nm and 98 ± 3 nm after release studies were finished, indicating no statistically significant change in particle size during the experiments. Because of the narrow particle size distribution, average particle size was used to determine the aqueous/total entrapped volume ratio (a) and membrane/total entrapped volume ratio (b), parameters that were necessary for the analysis of the transport data (see Supplementary Data). Extra-vesicular volume ratios (c and d) analogous to a and b were also required for the determination of membrane binding constants and subsequently release rate constants because these studies were not performed under sink conditions. The entrapped/unentrapped volume ratio (fv) calculated from the lipid content present in suspension was varied in release studies by altering the lipid concentration to avoid possible systematic errors in estimation of membrane binding constants from release profiles. Thus, the lipid content determined for each release study was used rather than the average of values for all experiments. The values in Table 1 reflect the parameter ranges explored. [43, 46, 47].
Table 1.
Volume parameters used in TPT release studies.
| Parameter | Average | Range |
|---|---|---|
| a | 0.855 | NA |
| b | 0.145 | NA |
| c | 0.9997 | 0.9995 – 0.9999 |
| d | 0.0003 | 0.0001 – 0.0005 |
| fv | 0.0067 | 0.0024 – 0.0110 |
Phospholipids undergo acid-catalyzed ester hydrolysis [48-50] that could lead to hydrolysis-induced changes in bilayer integrity over longer periods of time. This possibility was examined by monitoring lipid content in solution using HPLC with an ELSD. No loss of lipid was detected in release studies at pH 4.01 over 72 hr but >10% lipid loss was found after 48 hr at pH 3.35 (Supplementary Data, Fig. S1) which is qualitatively consistent with literature data [51]. Previous literature reports have shown compromised bilayer integrity when 15% or more phospholipid has degraded [49]. Consequently, only samples taken before 36 hours from the pH 3.35 release study were used in data fitting.
Spectrometric determination of the TPT A-ring phenol (pKa1)
Because of the high dependency of bilayer permeability on permeant charge [1, 27, 37, 43, 44], the pKa of the phenolic -OH on the A ring was determined at 37 °C from changes in the TPT fluorescence excitation spectra with pH as seen in Fig. 1. Spectral changes as a function of pH (Supplementary Data, Fig. S2) were used to determine pKa1 to be 6.56 ± 0.12. This value is similar to those previously reported at lower temperatures [45, 52-54].
Fig 1.

TPT excitation spectra at varying pH (3.50, 4.50, 5.50, 6.00, 6.27, 6.50, 6.80, 7.10, and 7.50) obtained at an emission wavelength of 560 nm. As pH is increased, a red shift occurs with maximum excitation shifting from 380 nm to 410 nm.
TPT lactone-carboxylate interconversion kinetics
The kinetics of interconversion of TPT between its lactone and carboxylate forms was monitored as a function of pH by following both carboxylate and lactone species (Fig. 2). A kinetic model based on a ring opening/closing mechanism previously described in the literature, [45] was able to account for the pH-dependence (Supplementary Data), resulting in a base-catalyzed ring-closing rate constant (kc,OH) of 7.4 ± 0.3 × 108 mol−1hr−1 and a carboxylic acid/lactone equilibrium constant (K0) of 1.98 ± 0.07 × 10−3. Combining the pKa of the ring-opened carboxylic assumed to be 3.9 (based on values previously assumed for camptothecin and its analogues and a pKa of 3.86 for the α-hydroxy acid glycolic acid) with K0 gave an effective pKa for the ring-opening/ionization reaction (pKa2) of 6.60. This value is similar to that reported for camptothecin and other analogues [42, 45].
Fig 2.

Ring opening/closing kinetics of TPT as a function of pH. The plots display the fractions of total TPT in the lactone form (closed symbols) or carboxylate form (open symbols) versus time at the same pH of 5.92 ( ,
,  ), 6.33(
), 6.33( ,
,  ), 7.04 (
), 7.04 ( ,
,  ), 7.39 (
), 7.39 ( ,
,  ), or 7.67 (x, X). The curves of matching color represent simultaneous fits of the kinetic interconversion model to the lactone (solid lines) and carboxylate (dashed lines) data. Most interconversion occurred within the first three hours while the inset shows that equilibrium was achieved.
), or 7.67 (x, X). The curves of matching color represent simultaneous fits of the kinetic interconversion model to the lactone (solid lines) and carboxylate (dashed lines) data. Most interconversion occurred within the first three hours while the inset shows that equilibrium was achieved.
Interconversion studies conducted in liposomal suspensions at high lipid content revealed no significant changes in the kinetic parameters or value of K0 from those obtained at the same pH in aqueous solutions (Supplementary Data, Fig. S3). This supports the negligible effect of membrane binding on interconversion kinetics and the use of kcl and K0 to describe inter-conversion kinetics of total intravesicular TPT.
pH Sensitive release of TPT
Transport experiments were performed at varying pH and at a recorded average temperature of 38.7 ± 0.1 °C during the time period of the studies. The fractions of TPT retained in DSPC/DSPEPEG2K liposomes versus time at varying pH are shown in Fig. 3. The curves displayed in Fig. 3 represent simultaneous fits to the mechanism-based mathematical model for TPT release developed in the Supplementary Data using the equilibria and chemical kinetic constants determined from spectrometric and interconversion studies. From these data, transport rate constants and partition coefficients were obtained for the various ionization states of the lactone and carboxylate species (see Scheme 1). Transport rate constants of 0.51 ± 0.07 hr−1 and 33.9 ± 4.6 hr−1 were found for the cationic lactone species (Lp) and the zwitterionic lactone (Ln), respectively, while the ring-opened carboxylate zwitterion (Cn) had a rate constant of 5.7 ± 0.5 hr−1 and its anionic form (Ca) was found to be impermeable.
Fig 3.

Fraction of TPT retained in DSPC/DSPE-PEG2K liposomes vs. time at varying pH (right panel displays only the first 6 hrs). Release studies were conducted at pH 3.35  , 4.10
, 4.10  , 5.10 +, 5.93
, 5.10 +, 5.93  , 6.33
, 6.33  , 7.04
, 7.04  , 7.39
, 7.39  , and 7.67 x. The solid curves of the same color represent the simultaneous fit of the mechanism-based mathematical model developed in this paper to the entire data set.
, and 7.67 x. The solid curves of the same color represent the simultaneous fit of the mechanism-based mathematical model developed in this paper to the entire data set.
The same analysis also indicated that the zwitterionic lactone (Ln) and anionic carboxylate form Ca exhibited negligible binding to the phospholipid bilayer. Both of these species have in common a phenolate moiety that evidently disfavors interaction with the bilayer. This and the similar partition coefficients of 61.7 ± 5.9 and 42.1 ± 6.4 for the lactone and carboxylate species in which the phenol is unionized (Lp and Cn respectively) suggest that binding likely occurs with preferential orientation of the TPT A-ring toward the hydrophobic region of the phospholipid bilayer. Opening of the lactone ring had a negligible effect on membrane binding.
Discussion
Effect of TPT ring-opening on pH sensitive release kinetics
The significantly greater permeability of the zwitterionic lactone species (Ln) than any of the other TPT species present in the pH range explored (Scheme 1) raises the possibility that, under certain pH conditions, this species might be depleted from the intravesicular compartment due to its slow regeneration from the ring-opened form. Simulated profiles of the concentrations of carboxylate and lactone species at pH 6.33 (where the lactone fraction is greater) and pH 7.04 (where the carboxylate fraction is greater) are shown in Fig. 4 along with the profile of total drug released in Fig. 4. Because interconversion is not instantaneous, the more permeable lactone zwitterion was depleted at a faster rate than its carboxylate counterpart, resulting in biphasic release profiles at certain pH values such as within the first 30 min of release at pH 7.04 (Fig. 4B, see also Fig. 5B).
Fig 4.

Liposomal TPT release profiles at pH 6.33 (A) and 7.04 (B). The observed fraction of total TPT retained ( ) and the resulting fit to the model described within this paper (
) and the resulting fit to the model described within this paper ( ) are shown. The simulated profiles of the lactone (
) are shown. The simulated profiles of the lactone ( ) and ring-opened (
) and ring-opened ( ) forms are also displayed to illustrate the rapid depletion of the lactone. The lactone to carboxylate ratio, R (
) forms are also displayed to illustrate the rapid depletion of the lactone. The lactone to carboxylate ratio, R ( ), is also shown to highlight the role of slow carboxylate → lactone conversion during TPT release.
), is also shown to highlight the role of slow carboxylate → lactone conversion during TPT release.
Fig 5.

(A) Comparison of the experimental pH profile of TPT release half-lives ( ) to model fits that account for the kinetics of lactone-carboxylate interconversion (
) to model fits that account for the kinetics of lactone-carboxylate interconversion ( ) or assume lactone-carboxylate equilibrium (
) or assume lactone-carboxylate equilibrium ( ). The equilibrium model was unable to account for the steep changes in half-life in the neutral pH region. The blue section highlights the pH region in which release was slowed by greater than 25% due to rate-limiting ring-closure. (B) The biphasic release profile observed at pH 7.04 and the fits of models that either include interconversion kinetics (
). The equilibrium model was unable to account for the steep changes in half-life in the neutral pH region. The blue section highlights the pH region in which release was slowed by greater than 25% due to rate-limiting ring-closure. (B) The biphasic release profile observed at pH 7.04 and the fits of models that either include interconversion kinetics ( ) or assume interconversion equilibrium (
) or assume interconversion equilibrium ( ).
).
The ratio of lactone to carboxylate species, R, during release is also depicted in Fig. 4. The profile of R initially shows a rapid decrease as the more permeable lactone species are depleted during the early phase of release. During the later phase of release, R approaches its initial equilibrium value as the less permeable carboxylate specie continues to release. Similar trends of R are evident for release studies conducted in the neutral pH range (Supplementary Data, Fig. S4)
Comparison of kinetic and equilibrium models of lactone ring-opening/closing
In previous pH-dependent release studies with the camptothecin analogue AR-67, ring-opening kinetics could be assumed to be instantaneous because the high membrane-binding constant for the lactone form reduced the driving force for release and provided a reservoir of the lactone species [26, 27, 55]. Both of these factors minimized the depletion of lactone. In contrast, the low membrane-partitioning observed for TPT required such an assumption to be tested. To compare the release model that incorporated the kinetics of ring-opening with one that assumed ring-opening/closing equilibrium (see Supplementary Data), the pH profiles for the release half-lives (t1/2) generated by the two models as a function of pH were compared in Fig. 5 along with the experimentally-observed half-lives.
Because the equilibrium model does not account for interconversion kinetics, it tries to compensate for the steep change in half-life (resulting from rate-limited ring-closing) seen at neutral pH (6.8 – 8.0) by overestimating release in the acidic region and underestimating release at higher pH. Fig. 5 also demonstrates the inadequate fit of the equilibrium model to a single release profile. Here, the equilibrium model underestimated the initial phase of drug release as it could not account for biphasic kinetics. In contrast, the model incorporating interconversion kinetics is able to account for the rapid initial phase of drug release that leads to lactone depletion followed by subsequent slower release limited by intravesicular regeneration of lactone from the carboxylate.
Determination of species permeability coefficients
The apparent permeability coefficient, , is related to the apparent transport rate constant at a given pH and the radius of the particle, r, as expressed by Eq. 4.
| (4) |
Using the release model that included the kinetics of ring-opening/closing, three TPT species were determined to contribute to the liposomal transport. From these release rate constants, permeability coefficients could be calculated for these species.
Based on the 100 nm liposomes used in these studies, the cationic and zwitterionic lactone permeability coefficients were 2.4 × 10–10 cm/s and 1.5 × 10–8 cm/s while TPT carboxylate zwitterion permeability coefficient was 3.0 × 10–9 cm/s. Generally, neutral, non-ionized species are orders-of-magnitude more permeable than charged species; however, few zwitterions have been explored [26, 43, 44]. The permeability coefficient for the lactone zwitterion determined from this work is similar to one obtained for the neutral, unionized lactone species of another camptothecin analogue [26]. This is likely due to an intramolecular interaction between the A-ring phenolate and dimethyl-ammonium substituents. Because of their close proximity and orientation, the free energy required for bilayer partitioning of this specie may be lower due to an electrostatic interaction or an intramolecular hydrogen bond between the heteroatoms which would partially shield the charge of each ion. This is supported by the large pKa shifts observed in aqueous solution for the phenolic –OH (i.e., from 10.0 in phenol to 6.56 in TPT) and dimethyl-aminomethyl substituent (from 8.93 in benzyldimethylamine to 10.5 in TPT), respectively. These shifts in pKa are a direct consequence of the stabilization of the zwitterionic form in Scheme 3. A similar effect has also been reported for other compounds with this same feature [45, 56], and recent studies of TPT fluorescence lifetimes in aqueous solution have distinguished intramolecular and bulk solution contributions to phenol deprotonation [54]. These interactions may help explain the small but significant permeability of the cationic species, Lp, seen in this study. Partial shielding of the cationic charge of Lp may be through hydrogen bonding with the phenol which may be further stabilized by resonance forms that delocalize the charge throughout TPT's conjugated ring structure.
Unlike the lactone zwitterion, the carboxylate form, Cn, does not offer the possibility of such intramolecular interactions between adjacent charged residues. Without this feature, the observed permeability may reflect the minor fraction of unionized neutral species, C0, as depicted in Scheme 5.
Scheme 5.
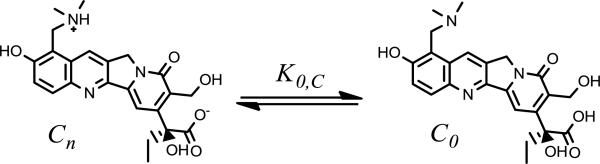
The equilibrium between TPT's carboxylate zwitterion and neutral, unionized form is governed by K0,C.
To ascertain whether the observed permeability for the carboxylate could be attributable to C0, the equilibrium constant between and Cn, K0,C, must be determined. This may be written in terms of the fraction of the zwitterion, fz,C, and non-ionized, f0,C, species as expressed in Eq. 5.
| (5) |
Using this ratio, a maximum permeability for C0 may be determined if one assumes C0 is the sole specie contributing to the observed permeability of the carboxylate. Using this assumption and a pKa3 of 10.5 for the dimethyl-amino group [45], the maximum permeability coefficient is estimated to be 9.3 × 10–3 cm/s for C0.
To assess the significance of this permeability coefficient, comparison to the theoretical maximum permeability, Pmax, is needed. Pmax can be described by diffusion-limited transport through the boundary layer of a spherical particle. This is given by Eq. 6.
| (6) |
Here, the diffusivity of TPT, DTPT, was determined by the Stokes-Einstein equation for diffusivity in water at 37 °C and a molecular volume of 366.8 Å3 (ACD labs). Based on this information, DTPT was calculated to be 7.52 × 10–6 cm2/s and Pmax was estimated to be 1.50 cm/s. While the estimated permeability coefficient of necessary to account for the experimental data is below Pmax, it is still several orders of magnitude higher than that of TPT's lactone zwitterion and another neutral, nonionized camptothecin of similar size [26]. This analysis suggests transport of the neutral, unionized ring-opened species is unlikely to fully account for the transport observed. Other mechanisms that may stabilize the ring-opened carboxylate zwitterion as it traverses the bilayer include: long-range intramolecular substituent effects on membrane partitioning, formation of water bridges through the bilayer, or ion-pairing within the barrier domain during TPT transport [57-61].
Conclusions
The pH dependent release of TPT from DSPC/m-PEG DSPE liposomes was characterized and the contribution of the kinetics of the pH-dependent ring-closure reaction to this process was assessed. These factors were incorporated into a mechanism-based mathematical model to describe TPT release. Based on this model, three TPT species were determined to be permeable to the membrane with the A-ring zwitterion form being the most permeable species. Within a defined pH region lactone depletion resulted in ring-closure of the ring-opened carboxylate form becoming at least partially rate-determining. A mathematical model that assumed equilibrium between the lactone and ring-opened species was inadequate in accounting for the complete profile for the dependence of t1/2 on pH and the biphasic release kinetics observed at certain pH values. The mechanism-based model developed in these studies will provide a basis for understanding the loading and release kinetics of actively-loaded formulations of TPT.
Supplementary Material
Acknowledgements
The project described was supported by Grant Number R25CA153954 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The authors would also like to thank Laura Cheng for her aid in data collection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Drummond DC, Noble CO, Hayes ME, Park JW, Kirpotin DB. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J. Pharm. Sci. 2008;97:4696–4740. doi: 10.1002/jps.21358. [DOI] [PubMed] [Google Scholar]
- 2.Charrois GJR, Allen TM. Rate of biodistribution of STEALTH® liposomes to tumor and skin: influence of liposome diameter and implications for toxicity and therapeutic activity. Biochim. Biophys. Acta. 2003;1609:102–108. doi: 10.1016/s0005-2736(02)00661-2. [DOI] [PubMed] [Google Scholar]
- 3.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Mirovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54:3352–3356. [PubMed] [Google Scholar]
- 4.Chonn A, Semple SC, Cullis PR. Association of blood proteins with large unilamellar liposomes in vivo. Relation to circulation lifetimes. J. Biol. Chem. 1992;267:18759–18765. [PubMed] [Google Scholar]
- 5.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul. 2001;41:189–207. doi: 10.1016/s0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 6.Gabizon AA. Liposomal drug carrier systems in cancer chemotherapy: current status and future prospects. J. Drug Target. 2002;10:535–538. doi: 10.1080/1061186021000043061. [DOI] [PubMed] [Google Scholar]
- 7.O'Brien S, Schiller G, Lister J, Damon L, Goldberg S, Aulitzky W, Ben-Yehuda D, Stock W, Coutre S, Douer D, Heffner LT, Larson M, Seiter K, Smith S, Assouline S, Kuriakose P, Maness L, Nagler A, Rowe J, Schaich M, Shpilberg O, Yee K, Schmieder G, Silverman JA, Thomas D, Deitcher SR, Kantarjian H. High-dose vincristine sulfate liposome injection for advanced, relapsed, and refractory adult philadelphia chromosome–negative acute lymphoblastic leukemia. J. Clin. Oncol. 2013;31:676–683. doi: 10.1200/JCO.2012.46.2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Batist G, Gelmon KA, Chi KN, Miller WH, Chia SKL, Mayer LD, Swenson CE, Janoff AS, Louie AC. Safety, pharmacokinetics, and efficacy of CPX-1 liposome injection in patients with advanced solid tumors. Clin. Cancer Res. 2009;15:692–700. doi: 10.1158/1078-0432.CCR-08-0515. [DOI] [PubMed] [Google Scholar]
- 9.Montanari M, Fabbri F, Rondini E, Frassineti GL, Mattioli R, Carloni S, Scarpi E, Zoli W, Amadori D, Cruciani G. Phase II trial of non-pegylated liposomal doxorubicin and low-dose prednisone in second-line chemotherapy for hormone-refractory prostate cancer. Tumori. 2012;98:696–701. doi: 10.1177/030089161209800604. [DOI] [PubMed] [Google Scholar]
- 10.Koudelka S, Turanek J. Liposomal paclitaxel formulations. J. Control. Release. 2012;163:322–334. doi: 10.1016/j.jconrel.2012.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Deeken JF, Slack R, Weiss GJ, Ramanathan RK, Pishvaian MJ, Hwang J, Lewandowski K, Subramaniam D, He AR, Cotarla I, Rahman A, Marshall JL. A phase I study of liposomal-encapsulated docetaxel (LE-DT) in patients with advanced solid tumor malignancies. Cancer Chemother. Pharmacol. 2013;71:627–633. doi: 10.1007/s00280-012-2048-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaspers GJ, Zimmermann M, Reinhardt D, Gibson BE, Tamminga RY, Aleinikova O, Armendariz H, Dworzak M, Ha SY, Hasle H, Hovi L, Maschan A, Bertrand Y, Leverger GG, Razzouk BI, Rizzari C, Smisek P, Smith O, Stark B, Creutzig U. Improved outcome in pediatric relapsed acute myeloid leukemia: results of a randomized trial on liposomal daunorubicin by the International BFM Study Group. J. Clin. Oncol. 2013;31:599–607. doi: 10.1200/JCO.2012.43.7384. [DOI] [PubMed] [Google Scholar]
- 13.Mamot C, Ritschard R, Wicki A, Stehle G, Dieterle T, Bubendorf L, Hilker C, Deuster S, Herrmann R, Rochlitz C. Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: a phase 1 dose-escalation study. Lancet Oncol. 2012;13:1234–1241. doi: 10.1016/S1470-2045(12)70476-X. [DOI] [PubMed] [Google Scholar]
- 14.Kirpotin DB, Drummond DC, Shao Y, Shalaby MR, Hong K, Nielsen UB, Marks JD, Benz CC, Park JW. Development of a highly active nanoliposomal irinotecan using a novel intraliposomal stabilization strategy. Cancer Res. 2006;66:6732. doi: 10.1158/0008-5472.CAN-05-4007. [DOI] [PubMed] [Google Scholar]
- 15.Noble CO, Krauze MT, Drummond DC, Yamashita Y, Saito R, Berger MS, Kirpotin DB, Bankiewicz KS, Park JW. Novel nanoliposomal CPT-11 infused by convection-enhanced delivery in intracranial tumors: pharmacology and efficacy. Cancer Res. 2006;66:2801–2806. doi: 10.1158/0008-5472.CAN-05-3535. [DOI] [PubMed] [Google Scholar]
- 16.Patankar N, Waterhouse D, Strutt D, Anantha M, Bally M. Topophore C: A liposomal nanoparticle formulation of topotecan for treatment of ovarian cancer. Invest. New Drugs. 2013;31:46–58. doi: 10.1007/s10637-012-9832-8. [DOI] [PubMed] [Google Scholar]
- 17.Ramsay EC, Anantha M, Zastre J, Meijs M, Zonderhuis J, Strutt D, Webb MS, Waterhouse D, Bally MB, Irinophore C. A liposome formulation of irinotecan with substantially improved therapeutic efficacy against a panel of human xenograft tumors. Clin. Cancer Res. 2008;14:1208–1217. doi: 10.1158/1078-0432.CCR-07-0780. [DOI] [PubMed] [Google Scholar]
- 18.Romiti A, Cox MC, Sarcina I, Rocco R, D'Antonio C, Barucca V, Marchetti P. Metronomic chemotherapy for cancer treatment: a decade of clinical studies. Cancer Chemother. Pharmacol. 2013 doi: 10.1007/s00280-013-2125-x. [DOI] [PubMed] [Google Scholar]
- 19.Kehrer DFS, Soepenberg O, Loos WJ, Verweij J, Sparreboom A. Modulation of camptothecin analogs in the treatment of cancer: a review. Anti-Cancer Drugs. 2001;12:89–105. doi: 10.1097/00001813-200102000-00002. [DOI] [PubMed] [Google Scholar]
- 20.Santana VM, Zamboni WC, Kirstein MN, Tan M, Liu T, Gajjar A, Houghton PJ, Stewart CF. A pilot study of protracted topotecan dosing using a pharmacokinetically guided dosing approach in children with solid tumors. Clin. Cancer Res. 2003;9:633–640. [PubMed] [Google Scholar]
- 21.Modi S, Xiang T-X, Anderson BD. Enhanced active liposomal loading of a poorly soluble ionizable drug using supersaturated drug solutions. J. Control. Release. 2012;162:330–339. doi: 10.1016/j.jconrel.2012.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Clerc S, Barenholz Y. A quantitative model for using acridine orange as a transmembrane pH gradient probe. Anal. Biochem. 1998;259:104–111. doi: 10.1006/abio.1998.2639. [DOI] [PubMed] [Google Scholar]
- 23.Zucker D, Marcus D, Barenholz Y, Goldblum A. Liposome drugs' loading efficiency: A working model based on loading conditions and drug's physicochemical properties. J. Control. Release. 2009;139:73–80. doi: 10.1016/j.jconrel.2009.05.036. [DOI] [PubMed] [Google Scholar]
- 24.Ceh B, Lasic DD. A rigorous theory of remote loading of drugs into liposomes. Langmuir. 1995;11:3356–3368. doi: 10.1006/jcis.1996.4555. [DOI] [PubMed] [Google Scholar]
- 25.Čeh B, Lasic DD. A rigorous theory of remote loading of drugs into liposomes: transmembrane potential and induced pH-gradient loading and leakage of liposomes. J. Colloid Interface Sci. 1997;185:9–18. doi: 10.1006/jcis.1996.4555. [DOI] [PubMed] [Google Scholar]
- 26.Vijay Joguparthi T-XX, Anderson Bradley D. Liposome transport of hydrophobic drugs: Gel phase lipid bilayer permeability and partitioning of the lactone form of a hydrophobic camptothecin, DB-67. J. Pharm. Sci. 2008;97:400–420. doi: 10.1002/jps.21125. [DOI] [PubMed] [Google Scholar]
- 27.Vijay Joguparthi BDA. Liposomal delivery of hydrophobic weak acids: Enhancement of drug retention using a high intraliposomal pH. J. Pharm. Sci. 2008;97:433–454. doi: 10.1002/jps.21135. [DOI] [PubMed] [Google Scholar]
- 28.Bruce JNMD, Fine RLMD, Canoll PMDP, Yun JMD, Kennedy BCMD, Rosenfeld SSMDP, Sands SAP, Surapaneni KMD, Lai RMD, Yanes CLRN, Bagiella EP, DeLaPaz RLMD. Regression of recurrent malignant gliomas with convection-enhanced delivery of topotecan. Neurosurgery. 2011;69:1272–1280. doi: 10.1227/NEU.0b013e3182233e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Potter SL, Berg S, Ingle AM, Krailo M, Adamson PC, Blaney SM. Phase 2 clinical trial of intrathecal topotecan in children with refractory leptomeningeal leukemia: a Children's Oncology Group trial (P9962) Pediatr. Blood Cancer. 2012;58:362–365. doi: 10.1002/pbc.23317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rose PG, Sill MW, McMeekin DS, Ahmed A, Salani R, Yamada SD, Wolfson AH, Fusco N, Fracasso PM. A phase I study of concurrent weekly topotecan and cisplatin chemotherapy with whole pelvic radiation therapy in locally advanced cervical cancer: a gynecologic oncology group study. Gynecol. Oncol. 2012;125:158–162. doi: 10.1016/j.ygyno.2011.12.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGonigle KF, Muntz HG, Vuky J, Paley PJ, Veljovich DS, Greer BE, Goff BA, Gray HJ, Malpass TW. Combined weekly topotecan and biweekly bevacizumab in women with platinum-resistant ovarian, peritoneal, or fallopian tube cancer: results of a phase 2 study. Cancer. 2011;117:3731–3740. doi: 10.1002/cncr.25967. [DOI] [PubMed] [Google Scholar]
- 32.NCI Cancer drug information : Topotecan hydrochloride. 2006 [Google Scholar]
- 33.Mayer LD, Tai LCL, Bally MB, Mitilenes GN, Ginsberg RS, Cullis PR. Characterization of liposomal systems containing doxorubicin entrapped in response to pH gradients. Biochim. Biophys. Acta. 1990;1025:143–151. doi: 10.1016/0005-2736(90)90091-2. [DOI] [PubMed] [Google Scholar]
- 34.Ramsay E, Alnajim J, Anantha M, Taggar A, Thomas A, Edwards K, Karlsson G, Webb M, Bally M. Transition metal-mediated liposomal encapsulation of irinotecan (CPT-11) stabilizes the drug in the therapeutically active lactone conformation. Pharm. Res. 2006;23:2799–2808. doi: 10.1007/s11095-006-9111-5. [DOI] [PubMed] [Google Scholar]
- 35.Taggar AS, Alnajim J, Anantha M, Thomas A, Webb M, Ramsay E, Bally MB. Copper–topotecan complexation mediates drug accumulation into liposomes. J. Control. Release. 2006;114:78–88. doi: 10.1016/j.jconrel.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 36.Drummond DC, Noble CO, Guo Z, Hayes ME, Connolly-Ingram C, Gabriel BS, Hann B, Liu B, Park JW, Hong K, Benz CC, Marks JD, Kirpotin DB. Development of a highly stable and targetable nanoliposomal formulation of topotecan. J. Control. Release. 2010;141:13–21. doi: 10.1016/j.jconrel.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta. 1993;1151:201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 38.Maurer-Spurej E, Wong KF, Maurer N, Fenske DB, Cullis PR. Factors influencing uptake and retention of amino-containing drugs in large unilamellar vesicles exhibiting transmembrane pH gradients. Biochim. Biophys. Acta. 1999;1416:1–10. doi: 10.1016/s0005-2736(98)00204-1. [DOI] [PubMed] [Google Scholar]
- 39.Abraham SA, Edwards K, Karlsson G, Hudon N, Mayer LD, Bally MB. An evaluation of transmembrane ion gradient-mediated encapsulation of topotecan within liposomes. J. Control. Release. 2004;96:449–461. doi: 10.1016/j.jconrel.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 40.Liu J-J, Hong R-L, Cheng W-F, Hong K, Chang F-H, Tseng Y-L. Simple and efficient liposomal encapsulation of topotecan by ammonium sulfate gradient: stability, pharmacokinetic and therapeutic evaluation. Anti-Cancer Drugs. 2002;13:709–717. doi: 10.1097/00001813-200208000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Fugit KD, Anderson BD. A dynamic, non-sink method for the simultaneous determination of drug permeability and binding coefficients in liposomes. doi: 10.1021/mp400765n. (In submission) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xiang T.-x., Anderson BD. Stable supersaturated aqueous solutions of silatecan 7-t-butyldimethylsilyl-10-hydroxycamptothecin via chemical conversion in the presence of a chemically modified β-cyclodextrin. Pharm. Res. 2002;19:1215–1222. doi: 10.1023/a:1019862629357. [DOI] [PubMed] [Google Scholar]
- 43.Xiang TX, Anderson BD. Permeability of acetic acid across gel and liquid-crystalline lipid bilayers conforms to free-surface-area theory. Biophys. J. 1997;72:223–237. doi: 10.1016/S0006-3495(97)78661-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiang T-X, Anderson BD. Influence of chain ordering on the selectivity of dipalmitoylphosphatidylcholine bilayer membranes for permeant size and shape. Biophys. J. 1998;75:2658–2671. doi: 10.1016/S0006-3495(98)77711-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fassberg J, Stella VJ. A kinetic and mechanistic study of the hydrolysis of camptothecin and some analogues. J. Pharm. Sci. 1992;81:676–684. doi: 10.1002/jps.2600810718. [DOI] [PubMed] [Google Scholar]
- 46.Huang CH. A 13C and 2H nuclear magnetic resonance study of phosphatidylcholine/cholesterol interactions: characterization of liquid-gel phases. Biochemistry. 1993;32:11. doi: 10.1021/bi00211a041. [DOI] [PubMed] [Google Scholar]
- 47.Kodati VR, Lafleur M. Comparison between orientational and conformational orders in fluid lipid bilayers. Biophys. J. 1993;64:163–170. doi: 10.1016/S0006-3495(93)81351-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grit M, Crommelin DJA. Chemical stability of liposomes: implications for their physical stability. Chem. Phys. Lipids. 1993;64:3–18. doi: 10.1016/0009-3084(93)90053-6. [DOI] [PubMed] [Google Scholar]
- 49.Grit M, Crommelin DJA. The effect of aging on the physical stability of liposome dispersions. Chem. Phys. Lipids. 1992;62:113–122. doi: 10.1016/0009-3084(92)90089-8. [DOI] [PubMed] [Google Scholar]
- 50.Grit M, de Smidt JH, Struijke A, Crommelin DJA. Hydrolysis of phosphatidylcholine in aqueous liposome dispersions. Int. J. Pharm. 1989;50:1–6. [Google Scholar]
- 51.Zuidam NJ, Crommelin DJA. Chemical hydrolysis of phospholipids. J. Pharm. Sci. 1995;84:1113–1119. doi: 10.1002/jps.2600840915. [DOI] [PubMed] [Google Scholar]
- 52.Strel'tsov SA, Grokhovskii SL, Kudelina IA, Oleinikov VA, Zhuze AL. Interaction of topotecan, DNA topoisomerase I inhibitor, with double-stranded polydeoxyribonucleotides. 1. topotecan dimerization in solution. Mol. Biol. 2001;35:365–373. [PubMed] [Google Scholar]
- 53.Kearney AS, Patel K, Palepu NR. Preformulation studies to aid in the development of a ready-to-use injectable solution of the antitumor agent, topotecan. Int. J. Pharm. 1996;127:229–237. [Google Scholar]
- 54.di Nunzio MR, Wang Y, Douhal A. Structural spectroscopy and dynamics of inter- and intramolecular H-bonding interactions of topotecan, a potent anticancer drug, in organic solvents and in aqueous solution. J. Phys. Chem. B. 2012;116:7522–7530. doi: 10.1021/jp302923a. [DOI] [PubMed] [Google Scholar]
- 55.Joguparthi V, Feng S, Anderson BD. Determination of intraliposomal pH and its effect on membrane partitioning and passive loading of a hydrophobic camptothecin, DB-67. Int. J. Pharm. 2008;352:17–28. doi: 10.1016/j.ijpharm.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Epstein J, Michel HO, Rosenblatt DH, Plapinger RE, Stephani RA, Cook E. Reactions of isopropyl methylphosphonofluoridate with substituted phenols. II. J. Am. Chem. Soc. 1964;86:4959–4963. [Google Scholar]
- 57.Bemporad D, Luttmann C, Essex JW. Behaviour of small solutes and large drugs in a lipid bilayer from computer simulations. Biochim. Biophys. Acta. 2005;1718:1–21. doi: 10.1016/j.bbamem.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 58.Heimburg T. Lipid ion channels. Biophys. Chem. 2010;150:2–22. doi: 10.1016/j.bpc.2010.02.018. [DOI] [PubMed] [Google Scholar]
- 59.Tejwani RW, Stouch TR, Anderson BD. Substituent effects on the ionization and partitioning of p-(aminoethyl)phenols and structurally related compounds: Electrostatic effects dependent on conformation. J. Pharm. Sci. 2009;98:4534–4544. doi: 10.1002/jps.21775. [DOI] [PubMed] [Google Scholar]
- 60.Shirai O, Yoshida Y, Kihara S. Voltammetric study on ion transport across a bilayer lipid membrane in the presence of a hydrophobic ion or an ionophore. Anal. Bioanal. Chem. 2006;386:494–505. doi: 10.1007/s00216-006-0435-0. [DOI] [PubMed] [Google Scholar]
- 61.Shirai O, Yoshida Y, Kihara S, Ohnuki T, Uehara A, Yamana H. Ion transport across a bilayer lipid membrane facilitated by gramicidin A - Effect of counter anions on the cation transport. J. Electroanal. Chem. 2006;595:53–59. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.