

Highlights
-
•
A novel brain-specific metabolic model was reconstructed, covering 570 genes.
-
•
The model, iMS570, correctly predicts major metabolic fluxes.
-
•
It is used as a scaffold to map transcriptome data for neurodegenerative diseases.
-
•
Identified reporter metabolites are potential biomarkers for the disease pathology.
-
•
Pathway-specific effect of the diseases on brain metabolism is revealed.
Abbreviations: AD, Alzheimer’s disease; ALS, amyotrophic lateral sclerosis; FBA, flux balance analysis; GABA, gamma-aminobutyric acid; HD, Huntington’s disease; KIV, ketoisovalerate; KLF, Krüppel-like factor; KMV, alpha-keto-beta-methylvalerate; MS, multiple sclerosis; PCA, principal component analysis; PD, Parkinson’s disease; RMA, reporter metabolite analysis; RPA, reporter pathway analysis; SCHZ, schizophrenia; TCA, tricarboxylic acid; USF, upstream stimulatory factor
Keywords: Neurometabolism, Brain metabolic network, Reporter metabolite, Computational systems biology, Transcriptome, Neurodegenerative diseases
Abstract
Network-oriented analysis is essential to identify those parts of a cell affected by a given perturbation. The effect of neurodegenerative perturbations in the form of diseases of brain metabolism was investigated by using a newly reconstructed brain-specific metabolic network. The developed stoichiometric model correctly represents healthy brain metabolism, and includes 630 metabolic reactions in and between astrocytes and neurons, which are controlled by 570 genes. The integration of transcriptome data of six neurodegenerative diseases (Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, multiple sclerosis, schizophrenia) with the model was performed to identify reporter features specific and common for these diseases, which revealed metabolites and pathways around which the most significant changes occur. The identified metabolites are potential biomarkers for the pathology of the related diseases. Our model indicated perturbations in oxidative stress, energy metabolism including TCA cycle and lipid metabolism as well as several amino acid related pathways, in agreement with the role of these pathways in the studied diseases. The computational prediction of transcription factors that commonly regulate the reporter metabolites was achieved through binding-site analysis. Literature support for the identified transcription factors such as USF1, SP1 and those from FOX families are known from the literature to have regulatory roles in the identified reporter metabolic pathways as well as in the neurodegenerative diseases. In essence, the reconstructed brain model enables the elucidation of effects of a perturbation on brain metabolism and the illumination of possible machineries in which a specific metabolite or pathway acts as a regulatory spot for cellular reorganization.
1. Introduction
Neurodegenerative diseases are disorders associated with functional loss of brain cells, especially with advancing age. Reported increase in the average life expectancy throughout the world means a large increase in the number of people with these diseases in the coming decades [1–3], making it essential to understand how these diseases develop. A full understanding of the mechanisms of neurodegenerative diseases will lead to improvement of successive treatment techniques.
Metabolism has an essential role in the cell, therefore, it is of substantial importance to perform a systematic investigation of the effect of neurodegenerative diseases on brain metabolism. Such an analysis can also reveal the common pathways perturbed by neurodegenerative diseases as well as disease-specific metabolic pathways. Limited research has been performed on this, however, a systematic and comparative analysis is still missing [4,5]. Such an analysis necessitates a detailed and curated representation of metabolic reactions occurring in the brain. Consequently, brain-specific metabolic stoichiometric models have recently been reconstructed [6–8]. These models can be used as scaffolds for computational analyses to draw hypotheses, as done for other metabolic systems in literature [9,10].
In this study, we have developed a novel brain-specific stoichiometric metabolic model which covers a large number of metabolic pathways in and between astrocytes and neurons, the two major cell types in the brain. We then used this model as a scaffold to analyze transcriptional changes associated with six common neurodegenerative diseases: Alzheimer’s disease (AD), Parkinson’s disease (PD), Huntington’s disease (HD), multiple sclerosis (MS), amyotrophic lateral sclerosis (ALS), and schizophrenia (SCH). The changes induced by these diseases on brain metabolism at the transcriptional level were systematically and comparatively analyzed by integrating corresponding transcriptome data of the diseases with the here-developed metabolic network of brain. This enabled a comprehensive analysis of the effect of neurodegeneration on metabolism from the perspective of different related diseases.
2. Results
2.1. Validation of the reconstructed model
We have reconstructed a novel brain metabolic model based on a previous study [6] by (i) transforming the lumped reactions in the original model into elementary reactions and (ii) adding new literature-supported pathways, as described in detail in Methods section. The new brain model comprises 630 reactions (571 internal, 59 exchange) and 524 metabolites (465 internal, 59 external). This means a 3-fold increase in the number of reactions compared to the original model [6]. 253 of the reactions occur in neurons while 299 of them are astrocytic. 59 reactions are for the exchange of metabolites, and there are 19 reactions depicting the transfer of metabolites between the two cell types. The model is available both in excel format and in SBML format as a supplement (Supplementary Files 1 and 2). 460 out of 571 internal reactions have gene information obtained from HumanCyc [11]. Due to the high coverage (81%) of gene-associated reactions in the model, it is possible to use this brain model for the integrated computational analysis of brain metabolism and transcriptome data. Since the total number of genes in the model is 570, it was named iMS570. Some of the reactions are controlled by one or more genes whereas some of the reactions do not have gene information. The number of the matched genes is 496 (87%) covering 447 of 571 internal reactions for GEO: GSE26927 dataset used in this study.
Note that the present model, iMS570, was developed purely in a brain-specific manner. Its gene coverage is much higher than the other gene-associated brain metabolic model (iNL403) reported in literature so far (570 vs. 403) [7], which was derived from a generic human metabolic reconstruction. The curation of the present model was achieved in extensive iterative steps such that it can be used in flux-balance based simulations. Flux balance analysis [12] of resting healthy brain metabolism performed with the newly reconstructed model iMS570 revealed a meaningful flux distribution consistent with experimental major flux splits in central metabolism (Table 1). Similar to the original model [6], the resting-state fluxes were calculated using two consecutive simulations: (i) linear programming with the objective function of maximization of the sum of these fluxes through glutamate/glutamine/GABA cycles (primary objective), (ii) after fixing the value of the sum of these three cycle fluxes to the optimum value, quadratic programming with the objective function of minimization of the Euclidean norm of fluxes (secondary objective). The primary objective provides a tight coupling between the two cell types, and it was shown to outperform several other alternative objective functions [6]. On the other hand, linear objective functions usually result in non-unique flux distributions, known as the problem of alternate optima [13]. Our secondary objective function eliminates the alternate optima, resulting in a unique flux distribution [6]. Both of the objective functions have physical meanings. The first one ensures a tight coupling of astrocytes and neurons through the well-known intercellular cycles [14,15]. The second one ensures channeling of fluxes through all pathways with minimal use of enzymes [16,17]. The FBA-based prediction of metabolic fluxes at resting state when combined with a proper objective function is one of the superiority of the iMS570 model over the other comprehensive brain model available in literature [7]. Besides, the FBA results render a verification of the reconstructed model.
Table 1.
Predicted flux results in this study, in comparison to the experimental results in resting (healthy) state. Predictions by the original model [6] are also given for comparison.
| % Flux ratio | This study | Original model | Experimental |
|---|---|---|---|
| Lactate release flux (r11) with respect to CMRglc | 7.2 | 4.5 | 3–9 |
| [132–135] | |||
| Glutamate/glutamine cycle flux (r95) with respect to CMRglc | 73.1 | 68.0 | 40–80 |
| [14,136,137] | |||
| Relative oxidative metabolism of astrocytes (rTCA,A/rTCA,total, r25/(r25 + r69)) | 33.9 | 35.0 | 30 |
| [136,138,139] | |||
| Total lipid synthesis with respect to CMRglc | 3.1 | 2.8 | 2 |
| [140] | |||
| Total PPP flux with respect to CMRglc | 5.5 | 5.6 | 3–6 |
| [141,142] | |||
| Pyruvate carboxylase flux (r12) with respect to CMRglc | 11.1 | 11.7 | 10 |
| [136,138,143] |
The model was reconstructed in such a way that pathways which are of major importance to brain metabolism are included. Although there are still reactions/pathways not covered by iMS570, we have performed an additional analysis to show that missing reactions will not affect current results and conclusions. To this aim, we removed 72 reactions from iMS570 belonging to the purine and pyrimidine nucleoside metabolisms, and repeated the reporter metabolite and reporter metabolism calculations for PD. Among about 40 associated reporter metabolites (Table 2), only three (astrocytic ADP, ATP and GTP) were not captured in this highly reduced model. Additionally, only methionine metabolism (Table 3) was not any more a reporter metabolism in addition to the excluded pathways. This clearly shows that pathways which may not be included in the current reconstruction do not have any practical effect on the conclusions made.
Table 2.
Reporter metabolite analysis results in different neurodegenerative diseases. “+” sign states significant changes with p-values less than 0.05 (reporter metabolites), and “−” sign represents no significant change (A: astrocytes, N: neurons).
| Metabolite | AD | ALS | HD | MS | PD | SCH |
|---|---|---|---|---|---|---|
| ADP/ATP (A) | + | − | + | + | + | − |
| ADP/ATP (N) | + | + | + | + | + | + |
| AMP (A) | − | − | + | − | − | + |
| AMP (N) | − | − | − | − | − | + |
| GTP (A,N) | − | − | − | − | + | + |
| NADmit/NADHmit (A) | − | − | + | − | − | − |
| NADmit/NADHmit (N) | − | − | − | − | + | − |
| NADP/NADPH (A) | − | − | − | + | − | − |
| NADPmit/NADPHmit (N) | − | − | − | − | − | + |
| CytCox (A,N) | + | + | + | + | + | − |
| CytCred (A,N) | + | + | + | + | + | − |
| Ubiquinol (A,N) | + | + | − | − | + | − |
| Ubiquinone (A,N) | + | + | − | − | + | − |
| Hc (A,N) | + | + | + | + | + | + |
| Pyruvate (A) | − | − | − | − | + | − |
| Pyruvate (N) | − | − | + | − | + | + |
| Fumarate (N) | − | − | − | − | − | + |
| Malate (A,N) | − | − | − | − | + | − |
| Succinate (A,N) | + | − | − | − | − | − |
| Phosphoenol–pyruvate (A,N) | − | − | − | − | − | + |
| 2-Phosphoglycerate (A,N) | + | − | − | − | − | + |
| Bicarbonate (A) | − | + | − | − | − | − |
| Bicarbonate (N) | + | + | − | − | − | − |
| Serine (A) | − | + | − | − | + | − |
| Serine (N) | − | − | − | − | + | − |
| KIV (A) | − | − | − | − | + | − |
| KMV (A) | − | − | − | − | + | − |
| 3-Phosphohydroxypyruvate (A) | − | − | + | − | − | − |
| Acetamidobutanal (A) | − | − | − | + | − | − |
| S-3-hydroxy-isobutyrate (A) | − | + | − | − | − | − |
| Propionyl-CoA (A) | − | + | − | − | − | − |
| Cholesterol (A) | − | − | − | + | − | − |
| Desmosterol (A) | − | − | − | + | − | − |
| Dimethylallyl_diphosphate (A) | + | − | − | − | − | − |
| Lanosterol (A) | − | − | − | + | − | − |
| Lathosterol (A) | − | − | − | + | − | − |
| 24-25-Dihydrolanosterol (A) | − | − | − | + | − | − |
| 5Alpha-cholesta-7-24-dien-3beta-ol (A) | − | − | − | + | − | − |
| Fatty acid (A,N) | + | − | − | − | − | + |
| Palmitoyl-CoA (A,N) | − | − | − | − | + | − |
| 1-Acyl-sn-glycerol-3-phosphate (A,N) | + | − | − | − | − | + |
| Myo-inositol-(1-3-4)-trisphosphate (A,N) | − | − | − | − | + | − |
| Myo-inositol-(1-3-4-5)-tetrakisphosphate (A,N) | − | − | + | + | + | − |
| Myo-inositol-(1-4)-bisphosphate (A,N) | − | − | − | − | + | − |
| Myo-inositol-(1-4-5)-trisphosphate (A,N) | − | − | + | + | + | − |
| Myo-inositol-(4)-monophosphate (A,N) | − | − | − | − | + | − |
| Phosphatidyl-1D-myo-inositol-4-5-bisphosphate (A,N) | + | − | − | − | + | − |
| Oxidized glutathione (A,N) | − | − | − | − | − | + |
| Reduced glutathione (A,N) | − | − | − | − | − | + |
| 5-Amino-levulinate (A,N) | + | − | − | − | − | − |
Table 3.
Reporter pathway analysis results in different neurodegenerative diseases. “+” sign states significant changes with p-values less than 0.05 (reporter pathways), and “−” sign represents no significant change.
| Pathway | PD | AD | ALS | HD | MS | SCH |
|---|---|---|---|---|---|---|
| Glycolysis | + | + | + | + | − | + |
| Pentose phosphate pathway | − | − | + | − | − | − |
| TCA cycle | + | + | − | + | + | + |
| Oxidative phosphorylation and ATPase | + | + | + | + | + | + |
| Glutamate–glutamine cycle | + | − | − | + | + | + |
| Ketone body metabolism | + | − | + | + | − | + |
| Creatine metabolism | − | + | + | − | + | + |
| Purine nucleoside metabolism | + | + | + | + | + | + |
| Pyrimidine nucleoside metabolism | + | + | − | − | + | − |
| Gaba cycle | + | + | + | + | − | − |
| Aspartate metabolism | − | − | − | − | + | − |
| Asparagine metabolism | − | + | − | + | + | + |
| Alanine metabolism | + | − | + | + | − | + |
| Glycine–serine metabolism | + | − | + | − | − | − |
| Leucine metabolism | + | − | + | + | − | + |
| Valine metabolism | + | − | + | + | + | − |
| Isoleucine metabolism | + | − | + | + | + | − |
| Methionine metabolism | + | − | − | − | − | − |
| Fatty acid synthesis | − | + | − | + | − | − |
| CDP-diacylglycerol biosynthesis | − | + | − | − | − | − |
| Cholesterol synthesis | − | − | − | − | + | − |
| Phosphatidylethanolamine metabolism | − | − | − | + | − | − |
| Phosphatidylcholine metabolism | − | + | − | + | + | − |
| Sphingomyelin metabolism | + | − | − | − | − | − |
| Inositol metabolism | + | + | + | + | + | − |
| Reactive oxygen species pathway | − | + | − | + | + | + |
2.2. Reporter metabolite analysis (RMA)
The developed brain model was integrated with the transcriptome data (GEO: GSE26927) [18] for a comprehensive investigation of neurodegenerative diseases of AD, PD, HD, ALS, MS and schizophrenia. RMA was applied to the data as described in Methods section, and the reporter metabolites specific to each disease were tabulated comparatively (Table 2). ATP and ADP in neurons are found to be reporter metabolites in all investigated neurological diseases together with Hc, the proton in oxidative phosphorylation. When the shared reporter metabolites for Parkinson’s disease, Alzheimer’s disease, and amyotrophic lateral sclerosis are closely compared (Table 2), most of them are found in oxidative phosphorylation metabolism (ubiquinol, ubiquinone, cytochrome C). Several inositol derivatives are reporter metabolites in Parkinson’s disease.
As for disease-specific reporter metabolites, ketoisovalerate (KIV), alpha-keto-beta-methylvalerate (KMV), palmitoyl-CoA, and malate are found to be reporter metabolites only in PD. Significant transcriptional changes around succinate, 5-amino-levulinate and dimethylallyl-diphosphate are specific only to AD. Succinate plays a role in the citric acid cycle, an energy-yielding process and the latter two metabolites are known to take part in the biosynthesis of secondary metabolites. S-3-hydroxy-isobutyrate, bicarbonate and propionyl-CoA satisfy the reporter metabolites criteria only in ALS. The reporter metabolite specific to HD is 3-phosphohydroxypyruvate, which is involved in serine biosynthesis. The transcriptional changes significant only in multiple sclerosis are around lanosterol, lathosterol, 5alpha-cholesta-7-24-dien-3beta-ol, desmosterol, cholesterol, 24-25-dihydrolanosterol, and acetamidobutanal. Fumarate, oxidized and reduced glutathione, and phosphoenol-pyruvate are reporter metabolites solely in schizophrenia. The disease-specific reporter metabolites are potential candidates as biomarkers for the identification of the corresponding diseases.
The number of common reporter metabolites for these 6 diseases is shown in Fig. 1, where the diagonal cells represent a total number of reporter metabolites for the corresponding neurological diseases. Parkinson’s disease has the highest number of reporter metabolites (40) compared to the other neurodegenerative diseases. The lowest number of the reporter metabolites is found in amyotrophic lateral sclerosis, indicating a relatively unaffected metabolism in this disease.
Fig. 1.
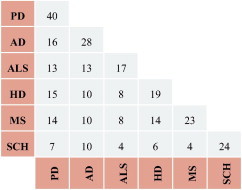
Distributions of the common reporter metabolites for neurodegenerative diseases.
The highest number of common reporter metabolites is 16 in the investigated diseases and found to be between Parkinson’s disease and Alzheimer’s disease. In general, schizophrenia has the lowest number of shared reporter metabolites with the other diseases. This indicates that transcriptional changes taking place in schizophrenia are quite different from the others. In fact, that result is expected as schizophrenia is also known to be a psychiatric disorder. The comparison of the common reporter metabolites in different neurodegenerative diseases (Table 2, Fig. 1) elucidates the effect of the transcriptional changes on brain metabolism due to different kind of perturbations (diseases).
Fig. 2 compares the diseases (observations) and the metabolites (variables) in a PCA bi-plot. PCA bi-plots are useful in visualizing the relationship between observations and variables. The figure demonstrates that ALS, SCHZ and AD exhibit relatively similar phenotypes in terms of reporter metabolites whereas MS and PD exhibit distinct profiles. Additionally, metabolites which are scattered around a disease indicate a specific role for these metabolites for the related disease. For example, lathosterol, cholesterol, and desmosterol populate around MS. These metabolites are specifically significantly affected from the perturbation caused by MS, as also revealed in Table 2. On the other hand, these metabolites do not have any reporter role in diseases such as AD, ALS and SCHZ since they are located on the opposite side of the graph.
Fig. 2.
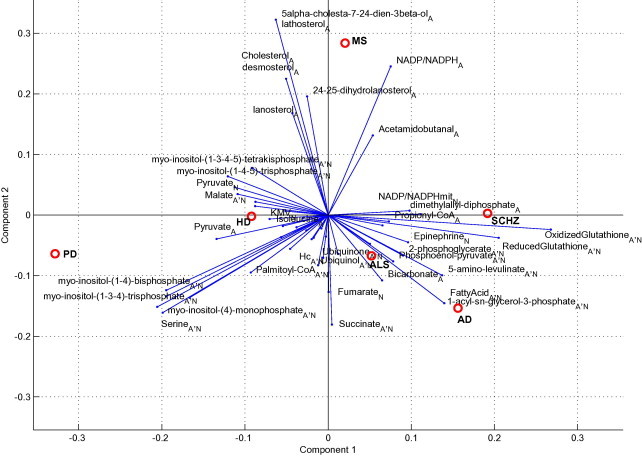
PCA bi-plot comparing all investigated diseases in this study. The plot is based on the Z-scores of metabolites. The relationship between the diseases and the effect of metabolites on the diseases are visualized.
2.3. Reporter pathway analysis (RPA)
Reporter pathway analysis was performed based on the results of RMA to explicate the pathways significantly affected in the perturbed (disease) state. The pathways having a p-value of less than 0.05 are defined as reporter pathways and they involve significant change due to disease condition. A comparison of the reporter pathways in Table 3 indicates that fatty acid synthesis is regulated in both Alzheimer’s disease and Huntington’s disease significantly. Phosphatidylcholine metabolism is affected in Alzheimer’s disease, Huntington’s disease, and multiple sclerosis. Creatine metabolism is significantly perturbed in Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and schizophrenia (Table 3). Disease-specific reporter pathways are as follows: aspartate and cholesterol metabolisms for MS, phosphatidylethanolamine metabolism for HD, sphingomyelin metabolism for PD, CDP-diacylglycerol biosynthesis for AD and pentose phosphate pathway for ALS. There is no unique reporter pathway for schizophrenia.
Fig. 3 compares common reporter pathways for the investigated neurological diseases, offering a more systematic demonstration of their effect on cell metabolism. Huntington’s disease has the highest number of reporter pathways (17) whereas schizophrenia has the lowest number of the reporter pathways (11). The highest number of common reporter pathways (12) is found between Parkinson’s disease and Huntington’s disease. Although the number of the reporter metabolites in Parkinson’s disease is considerably higher than that in Huntington’s disease, total number of reporter pathways in Huntington’s disease is higher than that in Parkinson’s disease. Similarly, despite having the highest number of common reporter metabolites, Parkinson’s disease and Alzheimer’s disease have the lowest number of common reporter pathways. These cases show that the averaged effect of metabolites over pathways is much more significant than the individual effects.
Fig. 3.
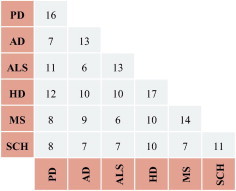
Distributions of the common reporter pathways for neurodegenerative diseases.
2.4. Computational binding site analysis
Potential transcription factors for the studied diseases were determined based on binding sites of genes neighboring the reporter metabolites, as described in Methods. Identified over-represented motifs for each reporter metabolite for the up- and down-regulated genes are given in detail in Supplementary File 3, together with a lumped version of all identified motifs irrespective of corresponding reporter metabolite for each neurodegenerative disease. According to the results, a number of transcription factors common in more than one disease was pinpointed, among which are paired box (Pax), forkhead box (FOX) and Sox protein families as well as proteins such as Krüppel-like factor (KLF) 4, Upstream stimulatory factor (USF1) 1, and SP1.
3. Discussion
3.1. Energy metabolism
According to the RMA and RPA results, energy-related metabolisms including glycolysis, TCA cycle, oxidative phosphorylation and ATPase, ketone body, and creatine metabolisms are significantly regulated in the neurodegenerative diseases.
Experimental studies on Parkinson’s disease showed that oxidative phosphorylation does not function properly due to the reduced activity of NADH-ubiquinone reductase (Complex I) and NADH cytochrome c reductase [19–21]. Indeed NADH was detected to be a reporter metabolite for PD in our analysis (Table 2). Mitochondrial studies of multiple sclerosis similarly revealed decreased activities of respiratory chain complexes I and III [22]. Succinate, the AD-specific reporter metabolite (Table 2), was reported as a potential biomarker for this disease [23]. The alterations in TCA cycle observed by our computational analysis for PD and AD (Tables 2 and 3) are in agreement with the studies showing a decrease in the α-ketoglutarate dehydrogenase complex enzyme due to Parkinson’s disease [24,25], as well as with the findings reporting changes in a number of mitochondrial enzymes that function in TCA cycle on Alzheimer’s disease [26–28]. Significant perturbations pinpointed by RPA in schizophrenia are supported by studies demonstrating dysregulation of several TCA cycle enzymes and energy metabolism [4,29,30]. Our results on HD are in agreement with the fact that energy metabolism in Huntington’s disease was altered because of dysfunction of oxidative phosphorylation pathway and TCA cycle [31,32]. The reported change in lactate-to-pyruvate ratio [33] and reduced pyruvate dehydrogenase activity [31] in HD are also compatible with the fact that neuronal pyruvate is a reporter metabolite for this disease. Regulation of energy metabolism through altered glycolysis, as predicted for all the diseases except MS (Table 3), can be explained by the role reported for Paired box (Pax) 6 protein, since its mutation leads to abnormal glucose metabolism [34]. Indeed, this transcription factor was predicted to have a role in HD, PD and schizophrenia in our analysis (Supplementary File 3). The potential role of pax family in neurodegenerative diseases is also stated in literature [35].
Another pathway related to neurodegenerative diseases is reactive oxygen species, which is an indicator of oxidative stress in the cell. This pathway was identified to be reporter in AD, HD, MS, and schizophrenia. It is widely known that neurodegenerative diseases are associated with oxidative stress [36,37]. Interestingly, PD and ALS seem to be not regulated at the transcriptional level in terms of this pathway according to our results although the opposite was reported in literature. Our binding-site analysis, however, revealed FOXO3 as a regulating factor for PD and AD (Supplementary File 3), which is in accordance with the role of this protein in oxidative stress [38,39]. Therefore, we can state that the interrelation of PD with oxidative stress was not on the pathway level but through the controlling transcription factors. Indeed, a mechanism was suggested in PD for the link between FOXO3 and FOXF2, another identified motif in our analysis, and neurogenesis [40]. A number of sox family proteins were identified as potential regulators in PD (Supplementary File 3). Among those, the dysregulation of Sox2, which was also predicted as a regulator in schizophrenia, led to oxidative stress and, hence, neurodegeneration [41,42]. Krüppel-like factor (KLF) 4, which is identified in our analysis in all the investigated diseases except Schizophrenia (Supplementary File 3), have recently been assigned a role in Parkinson’s disease through oxidative stress [43].
Creatine helps to supply energy to all cells in the body, and deficiencies in the creatine biosynthetic pathway lead to the malfunction of brain and various severe neurological defects [44]. Our reporter pathway analysis points out alterations in creatine metabolism in different neurodegenerative diseases (AD, ALS, schizophrenia), in agreement with several literature reports [45–48]. There are some studies pointing to creatine supplementation as a potential therapeutic in neurological diseases for energy homeostasis [49–51]. Similar to creatine, ketone bodies, metabolites from another reporter pathway for some of the investigated diseases, are also used in therapeutic strategies to provide energy [52].
3.2. Amino acid metabolism
Reporter pathway analysis indicates that amino acid metabolism is affected in the case of neurodegenerative diseases. Methionine metabolism is regulated only in Parkinson disease. In line with this finding, plasma analysis of L-Dopa treated Parkinson’s patients demonstrates a decreased level of methionine and an increased level of homocysteine in methionine metabolism [53]. Significant perturbation in glutamate–glutamine cycle was found by RPA for PD, HD, MS and schizophrenia. The experimental study performed in cerebrospinal fluid stated that glutamate is declined and glutamine level is raised notably in PD [54]. RPA identified alterations in alanine, valine, leucine, and isoleucine metabolisms due to Parkinson’s disease in agreement with cerebrospinal fluid studies which also revealed changes in these amino acids, among others [55]. Similarly, in the case of Huntington’s disease, results of our reporter pathway and reporter metabolite analyses are in accord with the literature findings indicating increased levels of alanine, isoleucine, and glutamate [56,57]. Alteration in valine metabolism in multiple sclerosis observed by RPA is also supported by another experimental study [58]. This study also reports a change in tryptophan and some others, but these are not captured by our analysis. In schizophrenia, significant perturbations observed in leucine and glutamate metabolisms by RPA can be linked to the literature studies reporting alterations in the levels of these amino acids, among others [57,59].
All the above mentioned findings indicate that amino acids, the precursors of neurotransmitters, play key roles in brain. There are abundant evidences in literature such that complex interactions among various amino acids and metabolites in brain support proper functioning of metabolism, leading to a healthy state. Disturbances in the levels and balance among them cause cognitive impairments associated with many psychiatric disorders, such as schizophrenia, and with neurological disorders, such as Parkinson’s, Alzheimer’s disease and many others.
3.3. Lipid and related metabolisms
The studied neurodegenerative disorders were found to cause significant perturbations in lipid metabolism (Tables 2 and 3). Having an important role in nourishing brain cells, inositol metabolism is predicted to be affected in PD, AD, HD, ALS and MS, except schizophrenia, whereas sphingomyelin metabolism is only affected in Parkinson disease. In PD, sphingolipid structures are altered due to interactions with alpha-synuclein protein [60]. In Alzheimer’s disease, fatty acid synthesis, phosphatidylcholine metabolism, CDP-diacylglycerol biosynthesis, and inositol metabolism are determined as reporter pathways by RPA. Inositol metabolites are known to play a major role in transducing signals in the nervous system [61,62]. Identification of inositol metabolism as a reporter pathway in AD is in line with literature finding stating a changed level of inositol-related compounds [63,64]. In amyotrophic lateral sclerosis, inositol metabolism is the only reporter pathway among the other lipid-related metabolisms. This result is in accordance with experimental study showing perturbed composition of inositol due to this disease [65]. Reported link between cholesterol metabolism and AD and PD in literature could not be captured by our analysis. Cholesterol synthesis is influenced only in multiple sclerosis according to the RPA results. This finding is in agreement with a study, which demonstrates a decline in cholesterol, lathosterol, desmosterol and lanosterol in multiple sclerosis [66]. The identification of cholesterol precursors as MS-specific reporter metabolites points to their potential as biomarkers for MS pathology, as suggested by a recent experimental study [67]. Although there are similarities, the effects of the different neurodegenerative diseases are different in pathway level in the brain.
Many of the reactions in lipid metabolism are associated with ATP, and cofactors, NADH and NADPH. ATP is a reporter metabolite in all investigated diseases, and the cofactor NADH is a reporter in HD and PD while NADPH is a reporter in MS and schizophrenia (Table 3). The identification of hubs (high connection nodes) in the metabolic network as reporters was also stated before [10]. The binding site analysis of neighbor genes of these hub reporters predicted many common putative regulators, some of which were discussed in previous sections. SP1 and USF1, which are known to regulate the genes of lipid metabolism, are also among them (Supplementary File 3). USF1 is a ubiquitously expressed upstream transcription factor with multiple roles including the regulation of apolipoprotein E [68], an AD-risk gene and known to transport cholesterol from astrocytes to neurons [69]. SP1 belongs to zinc-finger protein family with widespread functions, among which is a reported essential role in the regulation of cholesterol metabolism [70]. The transcriptional activity of SP1 is known to change in Huntington’s disease with neuroprotective role [71,72], in Alzheimer’s disease, [73,74] and in multiple sclerosis [75]. Similarly, literature findings associate the upstream transcription factor 1 (USF1) with AD [68], and MS [76].
4. Concluding remarks
A comprehensive stoichiometric model of brain metabolism covering interactions in and between astrocytes and neurons was reconstructed via an extensive literature survey. The model, termed iMS570, has the associated gene information for each included reaction whenever possible, with much higher gene coverage compared to the only other similar brain-specific model. After it was shown that the model correctly represents healthy resting brain metabolism in terms of the distribution of major fluxes, we used it as a scaffold for a detailed and systematic analysis of the metabolic effects of six neurodegenerative diseases comparatively. The identified reporter metabolites revealed parts of brain metabolic network perturbed by these diseases at transcriptional level. Such metabolites respond to the perturbations by a change in the expression levels of the surrounding genes, either to adjust their level into a new level or to keep their level constant. Therefore, they are potential biomarkers for the pathology of the related diseases, and they can be used as potential drug targets. Major effects around energy metabolism, reactive oxygen species and lipid metabolism were explained by common transcription factors regulating the genes neighboring the reporter metabolites. Furthermore, the identified transcription factors had roles on neurodegeneration, strengthening the potential functioning of the reporter metabolites as regulatory hot spots. The results, which are presented in the form of reporter metabolites, reporter pathways, and putative transcription factors, present a detailed catalogue of similarities and distinctive properties of the studied diseases at the level of metabolism, and can therefore be used as basis to develop and test several hypotheses. A close clinical inspection is needed to further elaborate on the differences in the affected metabolic pathways observed for these neurodegenerative diseases.
5. Methods
5.1. Transcriptome data
Transcriptome data belonging to six neurodegenerative diseases (AD, PD, HD, ALS, MS and Schizophrenia) were downloaded from Gene Expression Omnibus (Dataset series number: GEO: GSE26927). The dataset was originally generated by Prof. Richard Reynolds and co-workers [18] by using Illumina BeadChip platform for the analysis of common neuroinflammatory pathways in these diseases. Disease samples are from different regions of the brain since the most affected part of the brain is different for each disease. The downloaded dataset including 118 samples was subjected to outlier analysis by using Sammon mapping [77,78] within MATLAB 2012b. Sammon mapping is a dimension reduction method used for high dimensional data and it helps the identification of outliers. In Sammon mapping, distance matrix between the arrays is used in place of original data matrix. A new representation of the arrays in a lower-dimensional space is aimed by minimizing the total stress with reference to the original one. This method is performed on MATLAB with the commands of ‘pdist’ giving Euclidean distances between each pair of observations in the data matrix, and ‘mdscale’ performing non-metric multidimensional scaling on the Euclidean distances. The results of Sammon mapping are plotted to visualize samples among replicates which show a distinct behavior from the rest. In this way, outlier samples are determined. The identified outlier samples were discarded from the dataset before further analysis. One such outlier sample for HD and MS disease sets were identified (GSE: GSM663064 for HD, GSE: GSM663079, respectively), and two outlier samples for schizophrenia healthy set were identified (GSE: GSM663106 and GSE: GSM663108).
5.2. Model development
A novel brain metabolic model is developed based on a previous study [6] by (i) expanding the lumped reactions in the original model and (ii) adding new reactions that existed in the literature. Transforming lumped reactions into individual reactions led to a better representation of metabolic pathways. For example, the number of reactions in lipid metabolism is increased about fivefold after unlumping. It is aimed to integrate transcriptome data with the brain reaction model, and gene information controlling each reaction is obtained from HumanCyc, (www.humancyc.org) [11]. New reactions were appended to the model in four steps: (i) Addition of reactions missing in the original model following a detailed investigation of central carbon metabolism, (ii) Addition of pathways for essential and non-essential amino-acids that were not covered in the original model, (iii) Addition of pathways which are reported to have highly expressed controlling genes based on the transcriptomic data of healthy brain, (iv) Addition of other pathways which are reported to be important for brain metabolism. The ultimate goal is to construct a metabolic model of brain with a correct representation of healthy metabolism. The steps followed from model development stage to the final analyses are summarized in a flowchart in Fig. 4. Supplementary File 1, which lists all reactions in iMS570, also provides a complete overview of the model expansion by giving the reference for each reaction in the model.
Fig. 4.
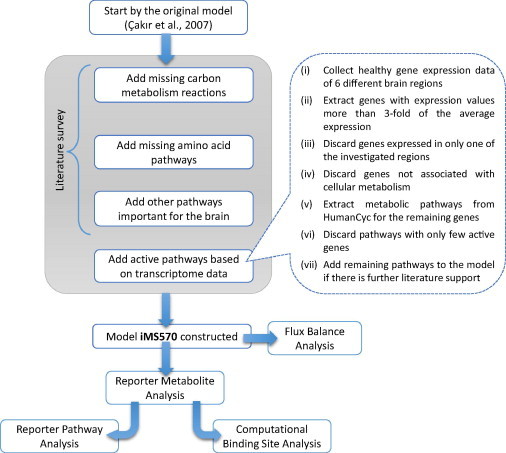
Flowchart depicting the model development steps for iMS570 and further analyses.
5.2.1. Central carbon metabolism
A literature survey yielded few reactions related to the central carbon metabolism which were not accounted for in the original model [6], but included in the current model. In glycolysis, 3-phosphoglycerate is also produced from 1-3-biphosphoglycerate via 2-3-Disphospho-D-glycerate without yielding ATP in both astrocytes and neurons (r15, r16, r59, r60) [79]. In addition to the already considered NADPH dependence, NADH is also used in the production of glutamate from alpha–ketoglutarate in astrocytes and neurons (r90, r93) [80]. Phosphoenolpyruvate carboxykinase, transforming oxaloacetate to phosphoenol pyruvate, is expressed in both astrocytes and neurons (r14, r58) [81,82]. In addition to isocitrate biosynthesis, citrate is also converted to oxaloacetate and acetyl-CoA in an ATP-dependent manner in neurons as well as astrocytes enzymatically (r28, r72) [83]. The conversion of citrate to oxaloacetate and acetate is taken into account in astrocytes part of the present brain model (r29) because citrate lyase subunit beta-like protein, responsible for this reaction, was reported to be an astrocyte-specific enzyme [84].
5.2.2. Amino-acid pathways
Pathways for asparagine, histidine, methionine, threonine, arginine, and proline amino acids were added into the present model following an extensive literature survey. All other amino acids were already included in the original model [6].
Asparagine is a non-essential amino acid, and its biosynthesis is taken into account only in neurons, since asparagine synthetase, responsible for enzymatic biosynthesis of glutamate and asparagine from glutamine and aspartate is found to be neuron-specific (r106) [84]. Histidine, an essential amino acid, is taken up by the brain [85] and converted into neurotransmitter histamine in neurons by histidine decarboxylase (r107) [86,87]. Neurons are also capable of synthesizing histamine N-methyltransferase, producing S-adenosyl-L-homocysteine and methylhistamine from histamine and S-adenosyl-L-methionine (r108) [88]. Methionine, an essential amino acid, is taken up by the brain [85]. It is converted into cysteine via the transsulfuration pathway in astrocytes and neurons (r181–r186, r189–r194) [89,90]. In this pathway, homocysteine is formed by three reactions in series and reacts with serine to produce cystathionine which is transformed into cysteine, alpha-ketobutyrate (2-oxobutanoate), and ammonia. Homocysteine is also recycled to methionine due to the activity of 5-methyltetrahydrofolate-homocysteine methyltransferase enzyme in the brain (r187, r195) [91]. The conversion of alpha-ketobutyrate to propionyl-CoA is thought to be active in astrocytes due to the control of this reaction by astrocyte-specific branched-chain α-keto acid dehydrogenase complex (r188) [84]. Threonine is an essential amino acid and can cross the blood-brain barrier with a slow rate compared to other amino acids [85]. Its metabolism contains only two enzymatic reactions in the brain cells (r196–r201) [92]. First, threonine is converted to 2-amino-3-ketobutyrate by threonine 3-dehydrogenase. Then, 2-amino-3-ketobutyrate is catabolized to glycine and acetyl-CoA via 2-amino-3-ketobutyrate ligase. Arginine is taken up by the astrocytes (r607) [93]. It is used to produce agmatine (r446) [93,94] and ornithine [95] (r447) in astrocytes. Arginine is also transferred to neurons by astrocytes to synthesize agmatine (r453) [96], citrulline (r455) [97], and ornithine (r458) [95]. In addition to its uptake, arginine is also produced in neurons from aspartate and citrulline (r456–r457) [95,98,99]. Ornithine is taken up by both cell types, besides its production in astrocytes and neurons [100]. In the cytosol of astrocytes, pyrroline-5-carboxylate, an intermediate metabolite in glutamate synthesis from ornithine, and NADPH are degraded into proline and NADP+ (r179). In astrocytic mitochondria, proline and FAD are converted to pyrroline-5-carboxylate and FADH2 (r180). These two proline producing and consuming reactions are considered in only astrocytes in the brain model, because Pyrroline-5-carboxylate reductase-like protein and Proline dehydrogenase were stated to be astrocyte-specific [84].
5.2.3. Active pathways based on transcriptome data
Pathways controlled by the highly expressed genes were identified based on the transcriptome data of a healthy brain. To this aim, gene expression data of different brain regions for healthy individuals were used (control samples of GEO: GSE26927 dataset [18], storing the transcriptome data for 6 common neurodegenerative diseases). Since the aim of the present study is to develop a general model for the whole brain, the brain model constructed here is not region-specific. The identification of metabolic genes with high expression was achieved in 5 steps: (i) Genes with expression values higher than 3-fold of the average expression values of all genes in the same transcriptome data were extracted. (ii) Among these genes, those which are expressed in only one of the investigated regions were discarded to minimize false positives. (iii) Of the remaining genes, the ones associated with cellular metabolism were identified by matching with reaction-controlling genes from HumanCyc. (iv) Metabolic pathways corresponding to these active genes were extracted using the information given by HumanCyc when available. If a pathway includes many reactions, but genes controlling only few of its reactions are found to be active in this pathway according to transcriptome data analysis, the related pathway was disregarded since such genes are active probably because of different functions other than metabolic control. (v) The candidate pathways were added into the present brain model if further support from literature was found as to the activity of the pathway in the brain. This procedure led to the involvement of following pathways into the brain model: inositol, creatine, polyamine, heme, pyrimidine and purine nucleosides.
Inositol is taken up by the brain [101]. It is used to produce diacylglycerol, complex inositol phosphate, and phosphatidylinositol (r388–r415) [102,103]. Another important metabolism consuming arginine and functioning in both neurons and astrocytes is creatine metabolism (r474–r479) consisting of ornithine and guanidinoacetate production from glycine and arginine [104]. Guanidinoacetate is then transformed to creatine to be used in creatine phosphate synthesis (r475–r1476, r478–r479) [105]. Agmatine synthesized in neurons from arginine is converted into putrescine (r454) [106,107] to be utilized both in polyamine metabolism including interconversion of precursor putrescine, spermidine, and spermine in neurons (r467–r473) [108] and in GABA synthesis from putrescine in astrocytes (r461–r464) [109,110]. The production of putrescine from ornithine is neuron-specific (r459) [111] and glutamate production from ornithine and alpha-ketoglutarate is astrocyte-specific (r448–r450) [112]. The metabolism of heme, an iron containing chemical compound, is included in the present study in both neurons and astrocytes since it has active genes satisfying our criteria and supported by literature [113,114]. Heme is synthesized from glycine and succinyl-CoA by eight reactions in series and degraded to bilirubin via biliverdin intermediate enzymatically (r480–r499) [115]. Adenosine, guanosine, uridine, and cytidine are nucleosides encountered in brain metabolism and they are mainly synthesized in liver and transported into the brain through the blood-brain barrier [116]. These nucleosides, in addition to a five-carbon sugar, contain the bases adenine, guanine, uracil and cytosine, respectively. These nucleosides are transformed into related nucleotides in the brain, which are utilized in metabolic processes or recycled to their nucleosides (r500–r571) [116].
5.2.4. Addition of other pathways
Pathways for ketone body metabolism, cardiolipin and sphingomyelin were included in the present model due to their importance for brain metabolism.
Acetoacetate and 3-hydroxybutyrate are the most important ketone bodies in human brain energy metabolism. They are produced by liver and transported by blood to brain where they can be used in ATP production as an alternative to glucose [117]. The uptake rates of ketone bodies from blood-brain barrier into brain are very small in comparison with glucose in resting condition; however, they significantly change in case of starvation in which uptake rates of ketone bodies increase with a decline in glucose uptake rate [118]. Acetoacetate and 3-hydroxybutyrate are converted into acetyl-CoA (r438–r444, r127–r128, r164) which participates in TCA cycle reactions or other metabolic reactions both in astrocytes and neurons [119]. Cardiolipin is a phospholipid and found in the mitochondrial membrane [120]. It is synthesized from CDP-diacylglycerol and Glycerol-3-phosphate by three enzymatic reactions in series in the brain (r366–r371) [121]. Sphingomyelin, a type of sphingolipid, is localized in the brain [120,122]. Precursors of sphingomyelin synthesis are serine and palmitoyl-CoA. Astrocytes use its own serine; however, neurons utilize serine transported from astrocytes in sphingomyelin synthesis since there is no serine synthesis in neurons (r372–r376, r378–r382) [123–125]. Sphingomyelin biosynthesis includes five metabolic reactions with 3-dehydrosphinganine, sphinganine, dihydroceramide, and ceramide as the intermediate metabolites. Sphingomyelin is also converted to ceramide and phosphoryl-choline due to the availability of sphingomyelinase in the brain (r377, r383) [126].
5.3. Flux balance analysis
The model was validated by applying flux balance analysis [12] for the prediction of major flux splits in the resting (healthy) state. Simulations were performed in MATLAB 2012b environment. Optimization (linear programming and quadratic programming) was performed using MATLAB bindings of CPLEX ILOG optimization algorithms provided by IBM’s Academic Initiative Program. The healthy-case FBA simulation was performed using the exact constraints as reported for the original model [6], as well as few other following constraints. The uptake rates for newly added amino acids were constrained as 0.0025 [85], 0.0017 [85], 0.0008 [85], 0.0020 [127], and 0.0031 [85] in units of μmole/g tissue/min for histidine, methionine, threonine, arginine and ornithine, respectively. Uptake rates of ketone bodies acetoacetate and 3-hydroxybutryrate were constrained to be zero since this pathway is known to be inactive in resting state [118].
5.4. Reporter metabolite analysis
Reporter metabolites are defined as metabolites around which the most significant transcriptional changes occur in response to a genetic or environmental perturbation, including diseases [10,128]. In this way, alterations in a metabolic network are elucidated at the transcriptional level. For this, genes are assigned p-values based on the differential transcriptome data using t-test. p-value of each gene (pi), representing the significance of differential gene data, is transformed into Z score (Zi) by using the inverse normal cumulative distribution. Then, each metabolite in the metabolic network in question is scored based on the aggregated Z-scores of the genes controlling the reactions in which the metabolite is involved, i.e., each metabolite is assessed by the Z-scores of these k neighbor genes.
| (1) |
In the case where a reaction is associated with more than one gene, the one with most significant change is taken into account [10]. Then, Z scores are corrected by using mean (μk) and standard deviation (σk) of the aggregated Z scores of many sets of randomly selected k genes from the metabolic network.
| (2) |
In the last step, corrected Z scores are converted to p-values, and metabolites having p-values less than the selected cut-off are named as reporter metabolites. For reporter metabolite analysis (RMA), p-value of 0.05 was selected as cut-off. Those metabolites with only a single neighbor gene were not considered as reporter metabolites in evaluating the results. RMA calculation was performed using the online BioMet Toolbox [129] ttest2 command of Statistics Toolbox of MATLAB was used to calculate p-values based on two-sample t-test.
5.6. Reporter pathway analysis
A novel analysis, termed reporter pathway analysis (RPA), was performed to predict significantly affected metabolic pathways due to the investigated diseases with the primary goal of making it easier to analyze RMA results. Here, each pathway is scored based on the p-values of its metabolites, calculated through RMA. The scoring is based on Eqs. (1) and (2), leading to pathway p-values. The pathways having p-values less than the selected cut-off (0.05) are defined as reporter pathways, i.e., pathways significantly affected due to the perturbation. Calculations were performed using BioMet Toolbox [129]. Note that we hereby opted for a global network-centered effect of the diseases on pathways by using metabolite p-values to calculate pathway scores since metabolite p-values include information on the interactions with other pathways.
5.7. Computational binding site analysis
To investigate whether genes associated with a reporter metabolite are controlled by common transcription factors, computational binding site analysis was performed. For each reporter metabolite, its neighboring genes were grouped into two sets with respect to up- and down-regulation [10]. For the sets with at least 4 gene members, DNA sequence regions with −950 to +50 base pairs were scanned from the JASPAR database [130] for the significantly over-represented motifs of the stored transcription factors (p-value: 0.05). The analysis was performed using the internet-based software tool, Pscan [131].
5.8. Principal component analysis (PCA)
Metabolites which did not show a significant change in any of the studied diseases (p-value cut-off: 0.05) were discarded from the set. Z-scores of the remaining metabolites were used in the PCA analysis. The analysis was performed using pca function of Statistical Toolbox of MATLAB.
Acknowledgements
The financial supports by TUBITAK, The Scientific and Technological Research Council of Turkey, through a career grant (Project Code: 110M464) and by Gebze Institute of Technology Research Project (Project Code: BAP 2011-A-27) are gratefully acknowledged. Emrah Özcan (Gebze Institute of Technology) is acknowledged for his help in reorganizing the reconstructed model.
Appendix A. Supplementary data
The reconstructed brain metabolic network (iMS570) in Excel format.
The reconstructed brain metabolic network (iMS570) in SBML format.
Detailed results of computational binding site analysis.
References
- 1.Forman M.S., Trojanowski J.Q., Lee V.M. Neurodegenerative diseases: a decade of discoveries paves the way for therapeutic breakthroughs. Nat. Med. 2004;10:1055–1063. doi: 10.1038/nm1113. [DOI] [PubMed] [Google Scholar]
- 2.Wancata J., Musalek M., Alexandrowicz R., Krautgartner M. Number of dementia sufferers in Europe between the years 2000 and 2050. Eur. Psychiatry. 2003;18(6):306–313. doi: 10.1016/j.eurpsy.2003.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Bach J.-P., Ziegler U., Deuschl G., Dodel R., Doblhammer-Reiter G. Projected numbers of people with movement disorders in the years 2030 and 2050. Mov. Disord. 2011;26:2286–2290. doi: 10.1002/mds.23878. [DOI] [PubMed] [Google Scholar]
- 4.Prabakaran S., Swatton J.E., Ryan M.M., Huffaker S.J., Huang J.T.-J., Griffin J.L., Wayland M., Freeman T., Dudbridge F., Lilley K.S. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol. Psychiatry. 2004;9:684–697. doi: 10.1038/sj.mp.4001511. [DOI] [PubMed] [Google Scholar]
- 5.Brooks W.M., Lynch P.J., Ingle C.C., Hatton A., Emson P.C., Faull R.L.M., Starkey M.P. Gene expression profiles of metabolic enzyme transcripts in Alzheimer’s disease. Brain Res. 2007;1127:127–135. doi: 10.1016/j.brainres.2006.09.106. [DOI] [PubMed] [Google Scholar]
- 6.Çakır T., Alsan S., Saybaşılı H., Akın A., Ülgen K.Ö. Reconstruction and flux analysis of coupling between metabolic pathways of astrocytes and neurons: application to cerebral hypoxia. Theor. Biol. Med. Model. 2007;4:48. doi: 10.1186/1742-4682-4-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lewis N.E., Schramm G., Bordbar A., Schellenberger J., Andersen M.P., Cheng J.K., Patel N., Yee A., Lewis R.A., Eils R. Large-scale in silico modeling of metabolic interactions between cell types in the human brain. Nat. Biotechnol. 2010;28:1279–1285. doi: 10.1038/nbt.1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Occhipinti R., Puchowicz M.A., LaManna J.C., Somersalo E., Calvetti D. Statistical analysis of metabolic pathways of brain metabolism at steady state. Ann. Biomed. Eng. 2007;35:886–902. doi: 10.1007/s10439-007-9270-5. [DOI] [PubMed] [Google Scholar]
- 9.Gille C., Bölling C., Hoppe A., Bulik S., Hoffmann S., Hübner K., Karlstädt A., Ganeshan R., König M., Rother K. HepatoNet1: a comprehensive metabolic reconstruction of the human hepatocyte for the analysis of liver physiology. Mol. Syst. Biol. 2010;6(411) doi: 10.1038/msb.2010.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zelezniak A., Pers T.H., Soares S., Patti M.E., Patil K.R. Metabolic network topology reveals transcriptional regulatory signatures of type 2 diabetes. PLoS Comput. Biol. 2010;6:e1000729. doi: 10.1371/journal.pcbi.1000729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Romero P., Wagg J., Green M.L., Kaiser D., Krummenacker M., Karp P.D. Computational prediction of human metabolic pathways from the complete human genome. Genome Biol. 2004;6:2. doi: 10.1186/gb-2004-6-1-r2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kauffman K.J., Prakash P., Edwards J.S. Advances in flux balance analysis. Curr. Opin. Biotechnol. 2003;14(5):491–496. doi: 10.1016/j.copbio.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 13.Mahadevan R., Schilling C.H. The effects of alternate optimal solutions in constraint-based genome-scale metabolic models. Metab. Eng. 2003;5:264–276. doi: 10.1016/j.ymben.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 14.Shen J., Petersen K.F., Behar K.L., Brown P., Nixon T.W., Mason G.F., Petroff O.A., Shulman G.I., Shulman R.G., Rothman D.L. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc. Natl. Acad. Sci. U.S.A. 1999;96(14):8235–8240. doi: 10.1073/pnas.96.14.8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gruetter R. In vivo 13C NMR studies of compartmentalized cerebral carbohydrate metabolism. Neurochem. Int. 2002;41(2–3):143–154. doi: 10.1016/s0197-0186(02)00034-7. [DOI] [PubMed] [Google Scholar]
- 16.Holzhütter H.-G. The principle of flux minimization and its application to estimate stationary fluxes in metabolic networks. Eur. J. Biochem. 2004;271(14):2905–2922. doi: 10.1111/j.1432-1033.2004.04213.x. [DOI] [PubMed] [Google Scholar]
- 17.Bonarius H.P., Hatzimanikatis V., Meesters K.P., de Gooijer C.D., Schmid G., Tramper J. Metabolic flux analysis of hybridoma cells in different culture media using mass balances. Biotechnol. Bioeng. 1996;50(3):299–318. doi: 10.1002/(SICI)1097-0290(19960505)50:3<299::AID-BIT9>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 18.Durrenberger P.F., Fernando F.S., Magliozzi R., Kashefi S.N., Bonnert T.P., Ferrer I., Seilhean D., Nait-Oumesmar B., Schmitt A., Gebicke-Haerter P.J., Falkai P., Grünblatt E., Palkovits M., Parchi P., Capellari S., Arzberger T., Kretzschmar H., Roncaroli F., Dexter D.T., Reynolds R. Selection of novel reference genes for use in the human central nervous system: a BrainNet Europe Study. Acta Neuropathol. 2012;124(6):893–903. doi: 10.1007/s00401-012-1027-z. [DOI] [PubMed] [Google Scholar]
- 19.Schapira A.H.V., Cooper J.M., Dexter D., Clark J.B., Jenner P., Marsden C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990;54(3):823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 20.Janetzky B., Hauck S., Youdim M.B.H., Riederer P., Jellinger K., Pantucek F., Zöchling R., Boissl K.W., Reichmann H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson’s disease. Neurosci. Lett. 1994;169:126–128. doi: 10.1016/0304-3940(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 21.Tretter L., Sipos I., Adam-Vizi V. Initiation of neuronal damage by complex I deficiency and oxidative stress in Parkinson’s disease. Neurochem. Res. 2004;29:569–577. doi: 10.1023/b:nere.0000014827.94562.4b. [DOI] [PubMed] [Google Scholar]
- 22.Dutta R., McDonough J., Yin X., Peterson J., Chang A., Torres T., Gudz T., Macklin W.B., Lewis D.A., Fox R.J., Rudick R., Mirnics K., Trapp B.D. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Ann. Neurol. 2006;59(3):478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 23.Forster D.M., James M.F., Williams S.R. Effects of Alzheimer’s disease transgenes on neurochemical expression in the mouse brain determined by 1H MRS in vitro. NMR Biomed. 2012;25:52–58. doi: 10.1002/nbm.1712. [DOI] [PubMed] [Google Scholar]
- 24.Mizuno Y., Matuda S., Yoshino H., Mori H., Hattori N., Ikebe S.-I. An immunohistochemical study on α-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann. Neurol. 1994;35:204–210. doi: 10.1002/ana.410350212. [DOI] [PubMed] [Google Scholar]
- 25.Gibson G.E., Kingsbury A.E., Xu H., Lindsay J.G., Daniel S., Foster O.J.F., Lees A.J., Blass J.P. Deficits in a tricarboxylic acid cycle enzyme in brains from patients with Parkinson’s disease. Neurochem. Int. 2003;43:129–135. doi: 10.1016/s0197-0186(02)00225-5. [DOI] [PubMed] [Google Scholar]
- 26.Gibson G.E., Sheu K.-F.R., Blass J.P. Abnormalities of mitochondrial enzymes in Alzheimer disease. J. Neural Transm. 1998;105:855–870. doi: 10.1007/s007020050099. [DOI] [PubMed] [Google Scholar]
- 27.Bubber P., Haroutunian V., Fisch G., Blass J.P., Gibson G.E. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann. Neurol. 2005;57:695–703. doi: 10.1002/ana.20474. [DOI] [PubMed] [Google Scholar]
- 28.Gibson G.E., Blass J.P., Beal M.F., Bunik V. The α-ketoglutarate-dehydrogenase complex. Mol. Neurobiol. 2005;31:43–63. doi: 10.1385/MN:31:1-3:043. [DOI] [PubMed] [Google Scholar]
- 29.Martins-de-Souza D., Gattaz W.F., Schmitt A., Novello J.C., Marangoni S., Turck C.W., Dias-Neto E. Proteome analysis of schizophrenia patients Wernicke’s area reveals an energy metabolism dysregulation. BMC Psychiatry. 2009;9:17. doi: 10.1186/1471-244X-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bubber P., Hartounian V., Gibson G.E., Blass J.P. Abnormalities in the tricarboxylic acid (TCA) cycle in the brains of schizophrenia patients. Eur. Neuropsychopharmacol. 2011;21:254–260. doi: 10.1016/j.euroneuro.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Browne S.E., Ferrante R.J., Beal M.F. Oxidative stress in Huntington’s disease. Brain Pathol. 1999;9:147–163. doi: 10.1111/j.1750-3639.1999.tb00216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins B.G., Koroshetz W.J., Beal M.F., Rosen B.R. Evidence for irnnairment of energy metabofism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 33.Koroshetz W.J., Jenkins B.G., Rosen B.R., Beal M.F. Energy metabolism defects in Huntington’s disease and effects of coenzyme Q10. Ann. Neurol. 1997;41:160–165. doi: 10.1002/ana.410410206. [DOI] [PubMed] [Google Scholar]
- 34.Wen J.H., Chen Y.Y., Song S.J., Ding J., Gao Y., Hu Q.K., Feng R.P., Liu Y.Z., Ren G.C., Zhang C.Y. Paired box 6 (PAX6) regulates glucose metabolism via proinsulin processing mediated by prohormone convertase 1/3 (PC1/3) Diabetologia. 2009;52:504–513. doi: 10.1007/s00125-008-1210-x. [DOI] [PubMed] [Google Scholar]
- 35.Thompson J.A., Ziman M. Pax genes during neural development and their potential role in neuroregeneration. Prog. Neurobiol. 2011;95(3):334–351. doi: 10.1016/j.pneurobio.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 36.Schulz J.B., Lindenau J., Seyfried J., Dichgans J. Glutathione, oxidative stress and neurodegeneration. Eur. J. Biochem. 2000;267(16):4904–4911. doi: 10.1046/j.1432-1327.2000.01595.x. [DOI] [PubMed] [Google Scholar]
- 37.Barnham K.J., Masters C.L., Bush A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- 38.Chong Z.Z., Li F., Maiese K. Oxidative stress in the brain: novel cellular targets that govern survival during neurodegenerative disease. Prog. Neurobiol. 2005;75(3):207–246. doi: 10.1016/j.pneurobio.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 39.Essers M.A., Weijzen S., de Vries-Smits A.M., Saarloos I., de Ruiter N.D., Bos J.L., Burgering B.M. FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. EMBO J. 2004;23:4802–4812. doi: 10.1038/sj.emboj.7600476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Desplats P., Patel P., Kosberg K., Mante M., Patrick C., Rockenstein E., Fujita M., Hashimoto M., Masliah E. Combined exposure to Maneb and Paraquat alters transcriptional regulation of neurogenesis-related genes in mice models of Parkinson’s disease. Mol. Neurodegener. 2012;7:49. doi: 10.1186/1750-1326-7-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mairet-Coello G., Tury A., Fellmann D., Jouvenot M., Griffond B. Expression of SOx-2, a member of the FAD-dependent sulfhydryl oxidase/quiescin Q6 gene family, in rat brain. NeuroReport. 2002;13:2049–2051. doi: 10.1097/00001756-200211150-00012. [DOI] [PubMed] [Google Scholar]
- 42.Ferri A.L., Cavallaro M., Braida D., Di Cristofano A., Canta A., Vezzani A., Ottolenghi S., Pandolfi P.P., Sala M., DeBiasi S. Sox2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development. 2004;131:3805–3819. doi: 10.1242/dev.01204. [DOI] [PubMed] [Google Scholar]
- 43.Chen J., Wang X., Yi X., Wang Y., Liu Q., Ge R. Induction of KLF4 contributes to the neurotoxicity of MPP+ in M17 cells: a new implication in Parkinson’s disease. J. Mol. Neurosci. 2013:1–9. doi: 10.1007/s12031-013-9961-3. [DOI] [PubMed] [Google Scholar]
- 44.Stockler S., Schutz P.W., Salomons G.S. Creatine and Creatine Kinase in Health and Disease. Springer; 2008. Cerebral creatine deficiency syndromes: clinical aspects, treatment and pathophysiology; pp. 149–166. [DOI] [PubMed] [Google Scholar]
- 45.David S., Shoemaker M., Haley B.E. Abnormal properties of creatine kinase in Alzheimer’s disease brain: correlation of reduced enzyme activity and active site photolabeling with aberrant cytosol-membrane partitioning. Mol. Brain Res. 1998;54:276–287. doi: 10.1016/s0169-328x(97)00343-4. [DOI] [PubMed] [Google Scholar]
- 46.Wendt S., Dedeoglu A., Speer O., Wallimann T., Beal M.F., Andreassen O.A. Reduced creatine kinase activity in transgenic amyotrophic lateral sclerosis mice. Free. Radic. Biol. Med. 2002;32:920–926. doi: 10.1016/s0891-5849(02)00784-0. [DOI] [PubMed] [Google Scholar]
- 47.Öngür D., Prescot A.P., Jensen J.E., Cohen B.M., Renshaw P.F. Creatine abnormalities in schizophrenia and bipolar disorder. Psychiatry Res. Neuroimaging. 2009;172:44–48. doi: 10.1016/j.pscychresns.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burbaeva G.S., Savushkina O.K., Boksha I.S. Creatine kinase BB in brain in schizophrenia. World J. Biol. Psychiatry. 2003;4:177–183. doi: 10.1080/15622970310029916. [DOI] [PubMed] [Google Scholar]
- 49.Adhihetty P.J., Beal M.F. Creatine and its potential therapeutic value for targeting cellular energy impairment in neurodegenerative diseases. Neuromolecular Med. 2008;10:275–290. doi: 10.1007/s12017-008-8053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tarnopolsky M.A., Beal M.F. Potential for creatine and other therapies targeting cellular energy dysfunction in neurological disorders. Ann. Neurol. 2001;49:561–574. [PubMed] [Google Scholar]
- 51.Klein M., Ferrante R.J. Creatine and Creatine Kinase in Health and Disease. Springer; 2008. The neuroprotective role of creatine; pp. 205–243. < http://dx.doi.org/10.1007/978-1-4020-6486-9_11>. [Google Scholar]
- 52.Kashiwaya Y., Takeshima T., Mori N., Nakashima K., Clarke K., Veech R.L. D-β-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. 2000;97:5440–5444. doi: 10.1073/pnas.97.10.5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Müller T., Woitalla D., Hauptmann B., Fowler B., Kuhn W. Decrease of methionine and S-adenosylmethionine and increase of homocysteine in treated patients with Parkinson’s disease. Neurosci. Lett. 2001;308(1):54–56. doi: 10.1016/s0304-3940(01)01972-3. [DOI] [PubMed] [Google Scholar]
- 54.Mally J., Szalai G., Stone T.W. Changes in the concentration of amino acids in serum and cerebrospinal fluid of patients with Parkinson’s disease. J. Neurol. Sci. 1997;151:159–162. doi: 10.1016/s0022-510x(97)00119-6. [DOI] [PubMed] [Google Scholar]
- 55.Antonio Molina J., Javier Jiménez-Jiménez F., Gómez P., Vargas C., AntonioNavarro J., Ortí-Pareja M., Gasalla T., BenitoLeón J., Bermejo F., Arenas J. Decreased cerebrospinal fluid levels of neutral and basic amino acids in patients with Parkinson’s disease. J. Neurol. Sci. 1997;150:123–127. doi: 10.1016/s0022-510x(97)00069-5. [DOI] [PubMed] [Google Scholar]
- 56.Reilmann R., Rolf L.H., Lange H.W. Decreased plasma alanine and isoleucine in Huntington’s disease. Acta Neurol. Scand. 1995;91(3):222–224. doi: 10.1111/j.1600-0404.1995.tb00438.x. [DOI] [PubMed] [Google Scholar]
- 57.Kim J.S., Kornhuber H.H., Holzmüller B., Schmid-Burgk W., Mergner T., Krzepinski G. Reduction of cerebrospinal fluid glutamic acid in Huntington’s chorea and in schizophrenic patients. Arch. Psychiatr. Nervenkr. 1980;228:7–10. doi: 10.1007/BF00365738. [DOI] [PubMed] [Google Scholar]
- 58.Monaco F., Fumero S., Mondino A., Mutani R. Plasma and cerebrospinal fluid tryptophan in multiple sclerosis and degenerative diseases. J. Neurol. Neurosurg. Psychiatry. 1979;42:640–641. doi: 10.1136/jnnp.42.7.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjerkenstedt L., Edman G., Hagenfeldt L., Sedvall G., Wiesel F.A. Plasma amino acids in relation to cerebrospinal fluid monoamine metabolites in schizophrenic patients and healthy controls. Br. J. Psychiatry. 1985;147:276–282. doi: 10.1192/bjp.147.3.276. [DOI] [PubMed] [Google Scholar]
- 60.Piccinini M., Scandroglio F., Prioni S., Buccinnà B., Loberto N., Aureli M., Chigorno V., Lupino E., DeMarco G., Lomartire A. Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. Neurobiol. 2010;41:314–340. doi: 10.1007/s12035-009-8096-6. [DOI] [PubMed] [Google Scholar]
- 61.Hanley M.R., Jackson T.R., Vallejo M., Patterson S.I., Thastrup O., Lightman S., Rogers J., Henderson G., Pini A., Downes C.P. Neural function: metabolism and actions of inositol metabolites in mammalian brain [and discussion] Philos. Trans. R. Soc. London. B, Biol. Sci. 1988;320:381–398. doi: 10.1098/rstb.1988.0083. [DOI] [PubMed] [Google Scholar]
- 62.Brady S.T., Siegel G.J., Albers R.W., Price D.L. Elsevier Academic Press; 2012. Basic Neurochemistry: Principles of Molecular, Cellular and Medical Neurobiology. [Google Scholar]
- 63.Stokes C.E., Hawthorne J.N. Reduced phosphoinositide concentrations in anterior temporal cortex of Alzheimer-diseased brains. J. Neurochem. 1987;48:1018–1021. doi: 10.1111/j.1471-4159.1987.tb05619.x. [DOI] [PubMed] [Google Scholar]
- 64.Miller B.L., Moats R.A., Shonk T., Ernst T., Woolley S., Ross B.D. Alzheimer disease: depiction of increased cerebral myo-inositol with proton MR spectroscopy. Radiology. 1993;187:433–437. doi: 10.1148/radiology.187.2.8475286. [DOI] [PubMed] [Google Scholar]
- 65.Kalra S., Hanstock C.C., Martin W.R., Allen P.S., Johnston W.S. Detection of cerebral degeneration in amyotrophic lateral sclerosis using high-field magnetic resonance spectroscopy. Arch. Neurol. 2006;63:1144–1148. doi: 10.1001/archneur.63.8.1144. [DOI] [PubMed] [Google Scholar]
- 66.Teunissen C.E., Dijkstra C.D., Polman C.H., Hoogervorst E.L.J., von Bergmann K., Lütjohann D. Decreased levels of the brain specific 24S-hydroxycholesterol and cholesterol precursors in serum of multiple sclerosis patients. Neurosci. Lett. Aug. 2003;347(3):159–162. doi: 10.1016/s0304-3940(03)00667-0. [DOI] [PubMed] [Google Scholar]
- 67.van de Kraats C., Killestein J., Popescu V., Rijkers E., Vrenken H., Lutjohann D., Barkhof F., Polman C., Teunissen C. Oxysterols and cholesterol precursors correlate to magnetic resonance imaging measures of neurodegeneration in multiple sclerosis. Mult. Scler. J. 2014;20:412–417. doi: 10.1177/1352458513499421. [DOI] [PubMed] [Google Scholar]
- 68.Isotalo K., Kok E.H., Luoto T.M., Haikonen S., Haapasalo H., Lehtimäki T., Karhunen P.J. Upstream transcription factor 1 (USF1) polymorphisms associate with Alzheimer’s disease-related neuropathological lesions: Tampere Autopsy Study. Brain Pathol. 2012;22(6):765–775. doi: 10.1111/j.1750-3639.2012.00586.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pfrieger F.W. Outsourcing in the brain: do neurons depend on cholesterol delivery by astrocytes? Bioessays. 2003;25(1):72–78. doi: 10.1002/bies.10195. [DOI] [PubMed] [Google Scholar]
- 70.Sanchez H.B., Yieh L., Osborne T.F. Cooperation by sterol regulatory element-binding protein and Sp1 in sterol regulation of low density lipoprotein receptor gene. J. Biol. Chem. 1995;270:1161–1169. doi: 10.1074/jbc.270.3.1161. [DOI] [PubMed] [Google Scholar]
- 71.Dunah A.W., Jeong H., Griffin A., Kim Y.-M., Standaert D.G., Hersch S.M., Mouradian M.M., Young A.B., Tanese N., Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington’s disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- 72.Qiu Z., Norflus F., Singh B., Swindell M.K., Buzescu R., Bejarano M., Chopra R., Zucker B., Benn C.L., DiRocco D.P. Sp1 is up-regulated in cellular and transgenic models of Huntington disease, and its reduction is neuroprotective. J. Biol. Chem. 2006;281:16672–16680. doi: 10.1074/jbc.M511648200. [DOI] [PubMed] [Google Scholar]
- 73.Citron B.A., Dennis J.S., Zeitlin R.S., Echeverria V. Transcription factor Sp1 dysregulation in Alzheimer’s disease. J. Neurosci. Res. 2008;86(11):2499–2504. doi: 10.1002/jnr.21695. [DOI] [PubMed] [Google Scholar]
- 74.Santpere G., Nieto M., Puig B., Ferrer I. Abnormal Sp1 transcription factor expression in Alzheimer disease and tauopathies. Neurosci. Lett. 2006;397(1–2):30–34. doi: 10.1016/j.neulet.2005.11.062. [DOI] [PubMed] [Google Scholar]
- 75.Kristjansdottir G., Sandling J.K., Bonetti A., Roos I.M., Milani L., Wang C., Gustafsdottir S.M., Sigurdsson S., Lundmark A., Tienari P.J. Interferon regulatory factor 5 (IRF5) gene variants are associated with multiple sclerosis in three distinct populations. J. Med. Genet. 2008;45:362–369. doi: 10.1136/jmg.2007.055012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gobin S.J.P., Montagne L., Zutphen M.V., Valk P.V.D., Elsen P.J.V.D., Groot C.J.A.D. Upregulation of transcription factors controlling MHC expression in multiple sclerosis lesions. Glia. 2001;36(1):68–77. doi: 10.1002/glia.1096. [DOI] [PubMed] [Google Scholar]
- 77.Sammon J.W., Jr. A nonlinear mapping for data structure analysis. Comput. IEEE Trans. 1969;100:401–409. [Google Scholar]
- 78.Wit E., McClure J. John Wiley & Sons; 2004. Statistics for Microarrays: Design, Analysis and Inference. [Google Scholar]
- 79.Durany N., Joseph J., Cruz-SÃ F.F., Carreras J. Phosphoglycerate mutase, 2, 3-bisphosphoglycerate phosphatase and creatine kinase activity and isoenzymes in human brain tumours. Br. J. Cancer. 1997;76:1139–1149. doi: 10.1038/bjc.1997.525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burbaeva G.S., Turishcheva M.S., Vorobyeva E.A., Savushkina O.K., Tereshkina E.B., Boksha I.S. Diversity of glutamate dehydrogenase in human brain. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2002;26(3):427–435. doi: 10.1016/s0278-5846(01)00273-1. [DOI] [PubMed] [Google Scholar]
- 81.Schmoll D., Führmann E., Gebhardt R., Hamprecht B. Significant amounts of glycogen are synthesized from 3-carbon compounds in astroglial primary cultures from mice with participation of the mitochondrial phosphoenolpyruvate carboxykinase isoenzyme. Eur. J. Biochem. 1995;227(1–2):308–315. doi: 10.1111/j.1432-1033.1995.tb20390.x. [DOI] [PubMed] [Google Scholar]
- 82.Cruz F., Scott S.R., Barroso I., Santisteban P., Cerdán S. Ontogeny and cellular localization of the pyruvate recycling system in rat brain. J. Neurochem. 1998;70(6):2613–2619. doi: 10.1046/j.1471-4159.1998.70062613.x. [DOI] [PubMed] [Google Scholar]
- 83.Beigneux A.P., Kosinski C., Gavino B., Horton J.D., Skarnes W.C., Young S.G. ATP-citrate lyase deficiency in the mouse. J. Biol. Chem. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Smith Q.R. Transport of glutamate and other amino acids at the blood-brain barrier. J. Nutr. 2000;130(4):1016S–1022S. doi: 10.1093/jn/130.4.1016S. [DOI] [PubMed] [Google Scholar]
- 86.Krusong K., Ercan-Sencicek A.G., Xu M., Ohtsu H., Anderson G.M., State M.W., Pittenger C. High levels of histidine decarboxylase in the striatum of mice and rats. Neurosci. Lett. 2011;495(2):110–114. doi: 10.1016/j.neulet.2011.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brown R.E., Stevens D.R., Haas H.L. The physiology of brain histamine. Prog. Neurobiol. 2001;63(6):637–672. doi: 10.1016/s0301-0082(00)00039-3. [DOI] [PubMed] [Google Scholar]
- 88.Nishibori M., Tahara A., Sawada K., Sakiyama J., Nakaya N., Saeki K. Neuronal and vascular localization of histamine N-methyltransferase in the bovine central nervous system. Eur. J. Neurosci. 2000;12(2):415–424. doi: 10.1046/j.1460-9568.2000.00914.x. [DOI] [PubMed] [Google Scholar]
- 89.McBean G.J. The transsulfuration pathway: a source of cysteine for glutathione in astrocytes. Amino Acids. 2012;42:199–205. doi: 10.1007/s00726-011-0864-8. [DOI] [PubMed] [Google Scholar]
- 90.Vitvitsky V., Thomas M., Ghorpade A., Gendelman H.E., Banerjee R. A functional transsulfuration pathway in the brain links to glutathione homeostasis. J. Biol. Chem. 2006;281:35785–35793. doi: 10.1074/jbc.M602799200. [DOI] [PubMed] [Google Scholar]
- 91.Spector R., Coakley G., Blakely R. Methionine recycling in brain: a role for folates and vitamin B-12. J. Neurochem. 1980;34(1):132–137. doi: 10.1111/j.1471-4159.1980.tb04631.x. [DOI] [PubMed] [Google Scholar]
- 92.Lajtha A., Oja S.S., Schousboe A., Saransaari P. Springer; 2007. Handbook of Neurochemistry and Molecular Neurobiology: Amino Acids and Peptides in the Nervous System. [Google Scholar]
- 93.Wiesinger H. Neuroglia. Oxford University Press; New York: 1995. Glia-specific enzyme systems; pp. 488–499. [Google Scholar]
- 94.Regunathan S., Feinstein D.L., Raasch W., Reis D.J. Agmatine (decarboxylated arginine) is synthesized and stored in astrocytes. NeuroReport. 1995;6:1897–1900. doi: 10.1097/00001756-199510020-00018. [DOI] [PubMed] [Google Scholar]
- 95.Braissant O., Gotoh T., Loup M., Mori M., Bachmann C. L-arginine uptake, the citrulline–NO cycle and arginase II in the rat brain: an in situ hybridization study. Mol. Brain Res. Jul. 1999;70:231–241. doi: 10.1016/s0169-328x(99)00151-5. [DOI] [PubMed] [Google Scholar]
- 96.Iyo A.H., Zhu M.-Y., Ordway G.A., Regunathan S. Expression of arginine decarboxylase in brain regions and neuronal cells. J. Neurochem. 2006;96(4):1042–1050. doi: 10.1111/j.1471-4159.2005.03544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wiesinger H. Arginine metabolism and the synthesis of nitric oxide in the nervous system. Prog. Neurobiol. 2001;64(4):365–391. doi: 10.1016/s0301-0082(00)00056-3. [DOI] [PubMed] [Google Scholar]
- 98.Nakamura H., Itoh K., Kawabuchi M. NADPH-diaphorase and cytosolic urea cycle enzymes in the rat accessory olfactory bulb. J. Chem. Neuroanat. 1999;17:109–117. doi: 10.1016/s0891-0618(99)00031-9. [DOI] [PubMed] [Google Scholar]
- 99.Arnt-Ramos L.R., O’Brien W.E., Vincent S.R. Immunohistochemical localization of argininosuccinate synthetase in the rat brain in relation to nitric oxide synthase-containing neurons. Neuroscience. 1992;51:773–789. doi: 10.1016/0306-4522(92)90519-8. [DOI] [PubMed] [Google Scholar]
- 100.Shank R.P., Campbell G.LeM. Ornithine as a precursor of glutamate and GABA: uptake and metabolism by neuronal and glial enriched cellular material. J. Neurosci. Res. 1983;9:47–57. doi: 10.1002/jnr.490090107. [DOI] [PubMed] [Google Scholar]
- 101.Ma K., Deutsch J., Villacreses N.E., Rosenberger T.A., Rapoport S.I., Shetty H.U. Measuring brain uptake and incorporation into brain phosphatidylinositol of plasma myo-[2H6] inositol in unanesthetized rats: an approach to estimate in vivo brain phosphatidylinositol turnover. Neurochem. Res. 2006;31:759–765. doi: 10.1007/s11064-006-9080-4. [DOI] [PubMed] [Google Scholar]
- 102.Inhorn R.C., Bansal V.S., Majerus P.W. Pathway for inositol 1, 3, 4-trisphosphate and 1, 4-bisphosphate metabolism. Proc. Natl. Acad. Sci. U.S.A. 1987;84:2170–2174. doi: 10.1073/pnas.84.8.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fisher S.K., Novak J.E., Agranoff B.W. Inositol and higher inositol phosphates in neural tissues: homeostasis, metabolism and functional significance. J. Neurochem. 2002;82(4):736–754. doi: 10.1046/j.1471-4159.2002.01041.x. [DOI] [PubMed] [Google Scholar]
- 104.Braissant O., Henry H., Loup M., Eilers B., Bachmann C. Endogenous synthesis and transport of creatine in the rat brain: an in situ hybridization study. Mol. Brain Res. 2001;86(1–2):193–201. doi: 10.1016/s0169-328x(00)00269-2. [DOI] [PubMed] [Google Scholar]
- 105.Molloy G.R., Wilson C.D., Benfield P., de Vellis J., Kumar S. Rat brain creatine kinase messenger RNA levels are high in primary cultures of brain astrocytes and oligodendrocytes and low in neurons. J. Neurochem. 1992;59:1925–1932. doi: 10.1111/j.1471-4159.1992.tb11028.x. [DOI] [PubMed] [Google Scholar]
- 106.Molderings G.J., Haenisch B. Agmatine (decarboxylated l-arginine): physiological role and therapeutic potential. Pharmacol. Ther. 2012;133(3):351–365. doi: 10.1016/j.pharmthera.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 107.Bernstein H.-G., Stich C., Jäger K., Dobrowolny H., Wick M., Steiner J., Veh R., Bogerts B., Laube G. Agmatinase, an inactivator of the putative endogenous antidepressant agmatine, is strongly upregulated in hippocampal interneurons of subjects with mood disorders. Neuropharmacology. 2012;62:237–246. doi: 10.1016/j.neuropharm.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 108.Krauss M., Weiss T., Langnaese K., Richter K., Kowski A., Veh R.W., Laube G. Cellular and subcellular rat brain spermidine synthase expression patterns suggest region-specific roles for polyamines, including cerebellar pre-synaptic function. J. Neurochem. 2007;103(2):679–693. doi: 10.1111/j.1471-4159.2007.04770.x. [DOI] [PubMed] [Google Scholar]
- 109.Angulo M.C., Le Meur K., Kozlov A.S., Charpak S., Audinat E. GABA, a forgotten gliotransmitter. Prog. Neurobiol. 2008;86:297–303. doi: 10.1016/j.pneurobio.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 110.Laschet J., Grisar T., Bureau M., Guillaume D. Characteristics of putrescine uptake and subsequent GABA formation in primary cultured astrocytes from normal C57BL/6J and epileptic DBA/2J mouse brain cortices. Neuroscience. 1992;48(1):151–157. doi: 10.1016/0306-4522(92)90345-3. [DOI] [PubMed] [Google Scholar]
- 111.Bernstein H.-G., Müller M. The cellular localization of the l-ornithine decarboxylase/polyamine system in normal and diseased central nervous systems. Prog. Neurobiol. 1999;57:485–505. doi: 10.1016/s0301-0082(98)00065-3. [DOI] [PubMed] [Google Scholar]
- 112.Thompson S.G., Wong P.T.-H., Leong S.F., McGeer E.G. Regional distribution in rat brain of 1-pyrroline-5-carboxylate dehydrogenase and its localization to specific glial cells. J. Neurochem. 1985;45:1791–1796. doi: 10.1111/j.1471-4159.1985.tb10535.x. [DOI] [PubMed] [Google Scholar]
- 113.Vincent S.R., Das S., Maines M.D. Brain heme oxygenase isoenzymes and nitric oxide synthase are co-localized in select neurons. Neuroscience. 1994;63(1):223–231. doi: 10.1016/0306-4522(94)90018-3. [DOI] [PubMed] [Google Scholar]
- 114.Schipper H.M. Heme oxygenase-1: role in brain aging and neurodegeneration. Exp. Gerontol. 2000;35(6–7):821–830. doi: 10.1016/s0531-5565(00)00148-0. [DOI] [PubMed] [Google Scholar]
- 115.Ryter S.W., Tyrrell R.M. The heme synthesis and degradation pathways: role in oxidant sensitivity heme oxygenase has both pro- and antioxidant properties. Free. Radic. Biol. Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 116.Ipata P.L. Origin, utilization, and recycling of nucleosides in the central nervous system. Adv. Physiol. Educ. 2011;35:342–346. doi: 10.1152/advan.00068.2011. [DOI] [PubMed] [Google Scholar]
- 117.Melø T.M., Nehlig A., Sonnewald U. Neuronal–glial interactions in rats fed a ketogenic diet. Neurochem. Int. 2006;48(6–7):498–507. doi: 10.1016/j.neuint.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 118.Hasselbalch S.G., Knudsen G.M., Jakobsen J., Hageman L.P., Holm S., Paulson O.B. Brain metabolism during short-term starvation in humans. J. Cereb. Blood Flow Metab. 1994;14:125–131. doi: 10.1038/jcbfm.1994.17. [DOI] [PubMed] [Google Scholar]
- 119.Chechik T., Roeder L.M., Tildon J.T., Poduslo S.E. Ketone body enzyme activities in purified neurons, astrocytes and oligodendroglia. Neurochem. Int. 1987;10:95–99. doi: 10.1016/0197-0186(87)90179-3. [DOI] [PubMed] [Google Scholar]
- 120.Ellis C.E., Murphy E.J., Mitchell D.C., Golovko M.Y., Scaglia F., Barceló-Coblijn G.C., Nussbaum R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking α-synuclein. Mol. Cell. Biol. 2005;25:10190–10201. doi: 10.1128/MCB.25.22.10190-10201.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pope S., Land J.M., Heales S.J.R. Oxidative stress and mitochondrial dysfunction in neurodegeneration; cardiolipin a critical target? Biochim. Biophys. Acta. 2008;1777(7–8):794–799. doi: 10.1016/j.bbabio.2008.03.011. [DOI] [PubMed] [Google Scholar]
- 122.Ma K., Langenbach R., Rapoport S.I., Basselin M. Altered brain lipid composition in cyclooxygenase-2 knockout mouse. J. Lipid Res. 2007;48:848–854. doi: 10.1194/jlr.M600400-JLR200. [DOI] [PubMed] [Google Scholar]
- 123.Pahan K., Sheikh F.G., Khan M., Namboodiri A.M., Singh I. Sphingomyelinase and ceramide stimulate the expression of inducible nitric-oxide synthase in rat primary astrocytes. J. Biol. Chem. 1998;273:2591–2600. doi: 10.1074/jbc.273.5.2591. [DOI] [PubMed] [Google Scholar]
- 124.Mitoma J., Kasama T., Furuya S., Hirabayashi Y. Occurrence of an unusual phospholipid, phosphatidyl-l-threonine, in cultured hippocampal neurons exogenous 1-serine is required for the synthesis of neuronal phosphatidyl-1-serine and sphingolipids. J. Biol. Chem. 1998;273:19363–19366. doi: 10.1074/jbc.273.31.19363. [DOI] [PubMed] [Google Scholar]
- 125.Hirabayashi Y., Furuya S. Roles of l-serine and sphingolipid synthesis in brain development and neuronal survival. Prog. Lipid Res. 2008;47(3):188–203. doi: 10.1016/j.plipres.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 126.Yamanaka T., Hanada E., Suzuki K. Acid sphingomyelinase of human brain. Improved purification procedures and characterization. J. Biol. Chem. 1981;256:3884–3889. [PubMed] [Google Scholar]
- 127.Lying-Tunell U., Lindblad B.S., Malmlund H.O., Persson B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol. Scand. 1981;63(6):337–350. doi: 10.1111/j.1600-0404.1981.tb00788.x. [DOI] [PubMed] [Google Scholar]
- 128.Patil K.R., Nielsen J. Uncovering transcriptional regulation of metabolism by using metabolic network topology. Proc. Natl. Acad. Sci. U.S.A. 2005;102:2685–2689. doi: 10.1073/pnas.0406811102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cvijovic M., Olivares-Hernández R., Agren R., Dahr N., Vongsangnak W., Nookaew I., Patil K.R., Nielsen J. BioMet Toolbox: genome-wide analysis of metabolism. Nucleic Acids Res. 2010;38(suppl. 2):W144–W149. doi: 10.1093/nar/gkq404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B. JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 2004;32(Suppl. 1):D91–D94. doi: 10.1093/nar/gkh012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zambelli F., Pesole G., Pavesi G. Pscan: finding over-represented transcription factor binding site motifs in sequences from co-regulated or co-expressed genes. Nucleic Acids Res. 2009;37:W247–W252. doi: 10.1093/nar/gkp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lubow J.M., Piñón I.G., Avogaro A., Cobelli C., Treeson D.M., Mandeville K.A., Toffolo G., Boyle P.J. Brain oxygen utilization is unchanged by hypoglycemia in normal humans: lactate, alanine, and leucine uptake are not sufficient to offset energy deficit. Am. J. Physiol. Endocrinol. Metab. 2006;290:E149–E153. doi: 10.1152/ajpendo.00049.2005. [DOI] [PubMed] [Google Scholar]
- 133.Madsen P.L., Cruz N.F., Sokoloff L., Dienel G.A. Cerebral oxygen/glucose ratio is low during sensory stimulation and rises above normal during recovery; excess glucose consumption during stimulation is not accounted for by lactate efflux from or accumulation in brain tissue. J. Cereb. Blood Flow Metab. 1999;19:393–400. doi: 10.1097/00004647-199904000-00005. [DOI] [PubMed] [Google Scholar]
- 134.Nybo L., Nielsen B., Blomstrand E., Møller K., Secher N. Neurohumoral responses during prolonged exercise in humans. J. Appl. Physiol. 2003;95:1125–1131. doi: 10.1152/japplphysiol.00241.2003. [DOI] [PubMed] [Google Scholar]
- 135.Wahren J., Ekberg K., Fernqvist-Forbes E., Nair S. Brain substrate utilisation during acute hypoglycemia. Diabetologia. 1999;42:812–818. doi: 10.1007/s001250051231. [DOI] [PubMed] [Google Scholar]
- 136.Gruetter R., Seaquist E.R., Ugurbil K. A mathematical model of compartmentalized neurotransmitter metabolism in the human brain. Am. J. Physiol. Endocrinol. Metab. 2001;281:E100–E112. doi: 10.1152/ajpendo.2001.281.1.E100. [DOI] [PubMed] [Google Scholar]
- 137.Lebon V., Petersen K.F., Cline G.W., Shen J., Mason G.F., Dufour S., Behar K.L., Shulman G.I., Rothman D.L. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J. Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Öz G., Berkich D.A., Henry P.-G., Xu Y., LaNoue K., Hutson S.M., Gruetter R. Neuroglial metabolism in the awake rat brain: CO2 fixation increases with brain activity. J. Neurosci. 2004;24:11273–11279. doi: 10.1523/JNEUROSCI.3564-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Hertz L., Peng L., Dienel G.A. Energy metabolism in astrocytes: high rate of oxidative metabolism and spatiotemporal dependence on glycolysis/glycogenolysis. J. Cereb. Blood Flow Metab. 2006;27:219–249. doi: 10.1038/sj.jcbfm.9600343. [DOI] [PubMed] [Google Scholar]
- 140.Siegel G.J., Agranoff B.W., Albers R.W., Molinoff P.B. Raven Press; 1994. Basic Neurochemistry: Molecular, Cellular, and Medical Aspects. [Google Scholar]
- 141.Wiesinger H., Hamprecht B., Dringen R. Metabolic pathways for glucose in astrocytes. Glia. 1997;21:22–34. doi: 10.1002/(sici)1098-1136(199709)21:1<22::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 142.Ben-Yoseph O., Camp D.M., Robinson T.E., Ross B.D. Dynamic measurements of cerebral pentose phosphate pathway activity in vivo using [1, 6–13C2, 6, 6–2H2] glucose and microdialysis. J. Neurochem. 1995;64:1336–1342. doi: 10.1046/j.1471-4159.1995.64031336.x. [DOI] [PubMed] [Google Scholar]
- 143.Aureli T., Di Cocco M.E., Calvani M., Conti F. The entry of [1-13C] glucose into biochemical pathways reveals a complex compartmentation and metabolite trafficking between glia and neurons: a study by 13C NMR spectroscopy. Brain Res. 1997;765:218–227. doi: 10.1016/s0006-8993(97)00514-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The reconstructed brain metabolic network (iMS570) in Excel format.
The reconstructed brain metabolic network (iMS570) in SBML format.
Detailed results of computational binding site analysis.