

Abstract
Presently, there are few estimates of the number of molecules occupying membrane domains. Using a total internal reflection fluorescence microscopy (TIRFM) imaging approach, based on comparing the intensities of fluorescently labeled microdomains with those of single fluorophores, we measured the occupancy of DC-SIGN, a C-type lectin, in membrane microdomains. DC-SIGN or its mutants were labeled with primary monoclonal antibodies (mAbs) in either dendritic cells (DCs) or NIH3T3 cells, or expressed as GFP fusions in NIH3T3 cells. The number of DC-SIGN molecules per microdomain ranges from only a few to over 20, while microdomain dimensions range from the diffraction limit to > 1μm. The largest fraction of microdomains, appearing at the diffraction limit, in either immature DCs or 3T3 cells contains only 4-8 molecules of DC-SIGN, consistent with our preliminary super-resolution Blink microscopy estimates. We further show that these small assemblies are sufficient to bind and efficiently internalize a small (~50nm) pathogen, dengue virus, leading to infection of host cells.
Keywords: CD209, C-type lectins, membrane microdomains, microdomain occupancy, total internal reflection fluorescence microscopy, quantitative imaging, dengue
Introduction
Dendritic cells (DCs) are the most potent antigen-presenting cells (APCs) in the human immune system; they are professional APCs that can induce primary immune responses in naïve T lymphocytes (1-3). Pathogen recognition by DCs occurs through its membrane pattern recognition receptors (PRRs), which bind certain chemical structures on the surface of pathogens (4). One of the PRRs, DC-SIGN (dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin), binds to the mannose or fucose structures present on a variety of pathogens and thus stimulates diverse immune responses (5). Biochemical and biophysical assays have shown that DC-SIGN forms multimers (6-9), which greatly enhances its binding affinity to multivalent ligands (10). Furthermore, DC-SIGN forms clusters or microdomains on the surfaces of DCs, which function as entry portals for diverse pathogens including viruses, bacteria and yeasts (11-13). It is thus of potential clinical interest to investigate the organization and properties of such microdomains.
Wide-field microscope imaging reveals that DC-SIGN forms discrete microdomains on the surface of immature DCs as well as cell lines that ectopically express DC-SIGN (13). The dimensions of these microdomains range from the diffraction limit to over one micron (13). Moreover, application of super-resolution microscopy indicates that microdomains are composed of one or several smaller nanodomains or clusters (14), consistent with earlier studies by near-field scanning optical microscopy (NSOM) (12) and transmission electron microscopy (TEM) (11). Fluorescence microscopic techniques including fluorescence recovery after photobleaching (FRAP), line scan fluorescence correction spectroscopy (line scan-FCS) and quantum dot single particle tracking reveal that DC-SIGN in microdomains is largely immobile on ms to minute time scales and does not exchange with DC-SIGN in the surround on this time scale (15). Furthermore, the microdomain stability appears to originate in the extracellular rather than the intracellular region of DC-SIGN (16). In this study, we focus on quantifying the occupancy of DC-SIGN molecules in a single microdomain. For this purpose, we developed a quantitative imaging method based on total internal reflection fluorescence microscopy (TIRFM).
TIRFM was developed by Axelrod and coworkers in the early 1980s (17-20). TIR occurs when a light beam propagates through a medium of refractive index n1 and meets an interface with a second medium of refractive index n2 (n2 < n1), and the incident angle θ is larger than the critical angle θC (θC = arcsin (n2 / n1)). The incident light creates a thin evanescent wave, propagating parallel to the interface in the second medium, whose intensity decays exponentially with the distance from the interface. For fused silica or glass (n1 ≈ 1.5) and an aqueous solution (n2 ≈ 1.3), by adjusting the incident angle, the penetration depth of the evanescent wave can range from ~70 nm to ~300 nm. With such a thin layer of illumination, TIRFM enables exclusive detection of fluorescently labeled proteins or other molecules that are close to or on the plasma membranes of adherent cells, with minimal fluorescence background from more distal cytoplasmic molecules. The high signal-to-noise ratio of TIRFM enables the detection of single fluorophores, which would be much more difficult employing wide-field excitation. Consequently, TIRFM has been employed in diverse biological studies such as membrane protein/lipid dynamics (21-23), cell-substrate interactions (24-26), and exo- and endocytosis (27-29).
Our approach to estimate the number of DC-SIGN molecules in single microdomains on cell surfaces is based on the comparison of the brightnesses of single fluorophores and the brightnesses of DC-SIGN microdomains. DC-SIGN or its mutants were labeled with fluorescent primary monoclonal antibodies (mAbs) or expressed as GFP fusions in NIH3T3 cells. Our results indicate that the largest fraction of microdomains in either immature DCs or NIH3T3 cells, as imaged by TIRFM, contains an average of 4-8 molecules of DC-SIGN (one or two tetramers). In NIH3T3 cells, mutants lacking the cytoplasmic domain or the N-linked glycosylation site had somewhat larger domain occupancy numbers.
To examine the role of DC-SIGN microdomains in pathogen infection, we further studied the interactions between DC-SIGN microdomains and dengue viruses (DENVs). DENVs are mosquito-borne flaviviruses that cause up to 400 million infections each year and about 2.5 billion people are at risk of dengue infection globally (30, 31). Immature DCs in the epidermis, Langerhans cells, are targets of DENV infection (32). DC-SIGN is one of the conserved PRRs that DENV binds to at the cell membrane (33-36). Many reports have shown that ectopically expressed DC-SIGN enhances productive DENV infection in different human cell types; however, detailed molecular-level studies on interactions between DC-SIGN membrane assemblies on cells and DENV at the initial binding stage are lacking. In this study, we found DC-SIGN microdomains on NIH3T3 cells captured DENVs efficiently, and, in particular, nanodomains containing only a few DC-SIGN molecules were capable of binding and internalizing DENV.
Results
Overview
In this study we developed a quantitative imaging method centered on a prism-based TIRFM setup (Figure 1), for which the principles are described in detail in the Materials and Methods section. DC-SIGN microdomains on six cell sample types were quantitatively examined in this work: human immature DCs endogenously expressing wt DC-SIGN and NIH3T3 cells ectopically expressing five different DC-SIGN constructs including wt DC-SIGN, GFP-DC-SIGN, DC-SIGN-Δ37, DC-SIGN-N80Q and GFP-DC-SIGN-N80Q (Figure 2A). As imaged by objective-based TIRFM, all six cell types exhibited visible DC-SIGN microdomains; however, different cell types appeared qualitatively to possess microdomains with slightly different cell-surface distributions, densities and/or intensities (Figure 2 B-G). Similar images were obtained with through-prism TIRFM and these were used for subsequent analysis of domain occupancy (Figure 1 D, E). The spatially integrated intensities, corrected for the local evanescent excitation intensity, were measured for single, purified GFP molecules as well as for single AlexaFluor488 probes conjugated to monoclonal antibodies specific for DC-SIGN (Figure 3). Similar measurements were carried out for individual microdomains containing DC-SIGN on the six cell types and used with the single molecule data to produce histograms giving quantitative measures of the copy numbers of DC-SIGN molecules in the microdomains (see Eqs. 1-11 and Figure 4 and 5). Subtle differences in the results for different cell types are discussed below. However, the most striking conclusion is that for all cell types the copy numbers of DC-SIGN in the microdomains are usually very low (although a few microdomains contain more than ~ 20 DC-SIGN molecules).
Figure 1. Principle of the quantitative TIRFM setup.
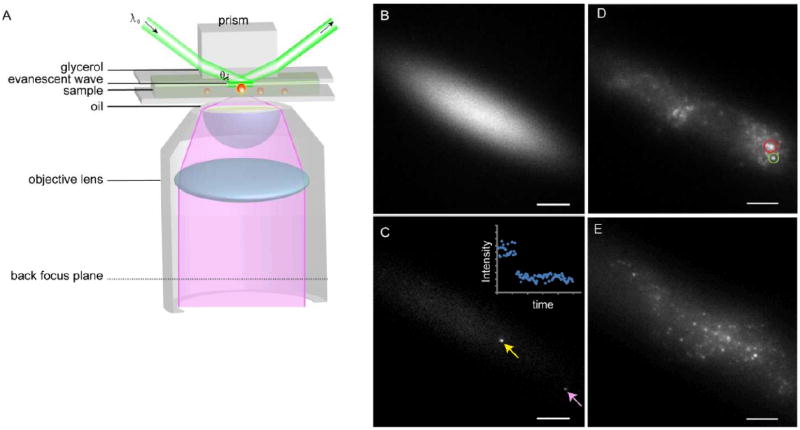
A) Schematic drawing of prism-based TIRFM. B) Prism-TIRFM creates an elliptical Gaussian illumination profile. The image was taken on a highly concentrated AlexaFluor488 solution. C) Fluorophores (yellow arrow) at the center of the Gaussian profile exhibit higher brightnesses compared to those at the periphery (pink arrow), due to the different local excitation intensity. The image was taken on a highly diluted, single-molecule level AlexaFluor488 solution. Inset, an example of single-step photobleaching of the AlexaFluor488. D) Prism-based TIRFM image of antibody-labeled DC-SIGN on A DC membrane. The green circle illustrates spots used for data analysis while red circle illustrates an ill-defined domain that was excluded from analysis. The latter domains were generally somewhat larger. E) Prism-based TIRF image of GFP-DC-SIGN expressed in NIH3T3 cells. Bars = 10 μm.
Figure 2. Constructs and sample cell images.
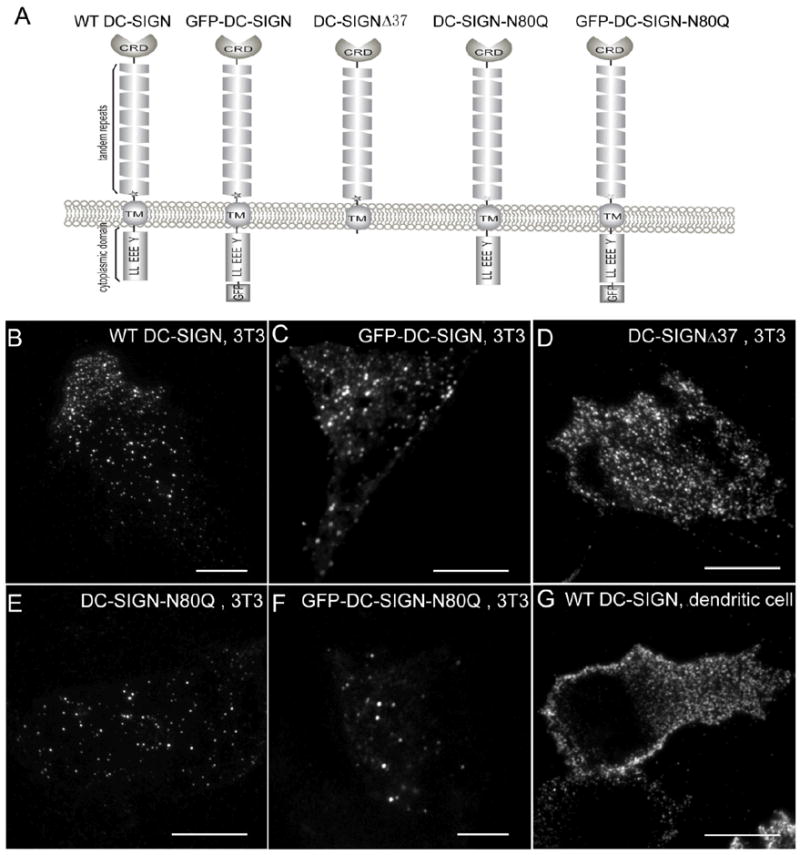
A) Schematic drawing of the different DC-SIGN constructs. B-F) Sample images of NIH3T3 cells expressing wt DC-SIGN (B), GFP-DC-SIGN (C), DC-SIGN-Δ37 (D), DC-SIGN-N80Q (E), and GFP-DC-SIGN-N80Q (F). G) A dendritic cell endogenously expressing DC-SIGN. Images were taken on a commercial Olympus objective-based TIRFM scope. Bars =10 μm.
Figure 3. Determination of single-fluorophore brightnesses.
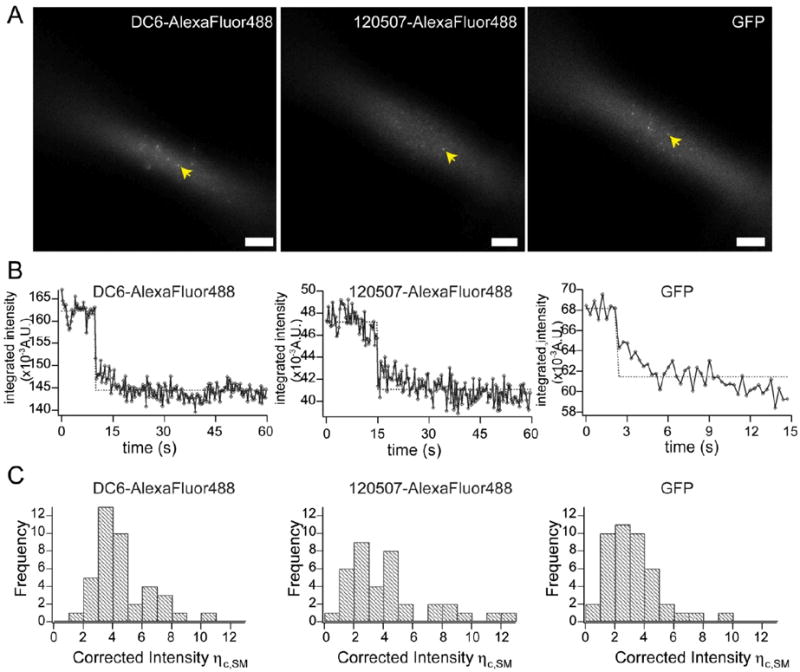
A) Sample images of single fluorophores (arrows) in prism-based TIRFM illumination. Left, single AlexaFluor488-conjugated DC6 mAb; middle, single AlexaFluor488-conjugated 120507 mAb; right, single GFP. B) Sample single-step photobleaching traces of single AlexaFluor488-conjugated DC6 mAb (left), single AlexaFluor488-conjugated 120507 mAb (middle), and single GFP (right). C) Histogram plots of the brightness distributions of single fluorophores. Left, single AlexaFluor488-conjugated DC6 mAb; middle, single AlexaFluor488-conjugated 120507 mAb; right, single GFP.
Figure 4. Histograms of the number of molecules per microdomain.
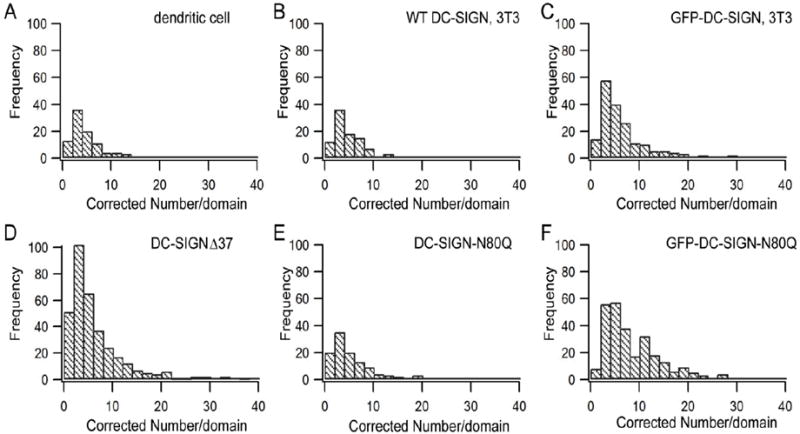
A) Dendritic cells. B-F) NIH3T3 cells expressing wt DC-SIGN (B), GFP-DC-SIGN (C), DC-SIGN-Δ37 (D), DC-SIGN-N80Q (E), and GFP-DC-SIGN-N80Q (F). Note that a few data points with numbers per domain > 40 are not shown in the plots.
Figure 5. Scatter plots of numbers per domain vs domain width.
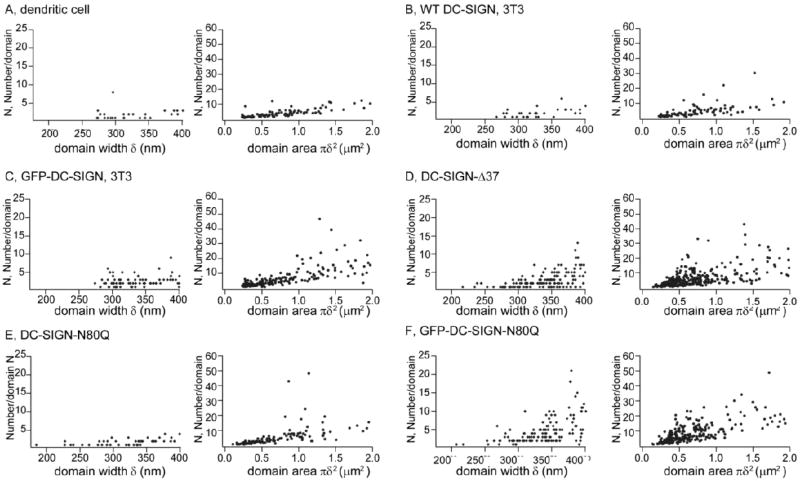
Left columns, scatter plots of numbers per domain with domain width δ < 400 nm. Right columns, scatter plots of numbers per domain vs domain area for the entire range of domain sizes. A) DC-SIGN in dendritic cells. B-F) NIH3T3 cells expressing wt DC-SIGN (B), GFP-DC-SIGN (C), DC-SIGN-Δ37 (D), DC-SIGN-N80Q (E), and GFP-DC-SIGN-N80Q (F).
Single fluorophore brightness
As described in Materials and Methods, spatially integrated intensities were measured from the images (Figure 3A) of single AlexaFluor488 probes conjugated to mAbs specific for DC-SIGN or of single, purified GFP molecules. The single molecule nature of such images was assessed from single step bleaching events (Figure 3B). The distributions of spot intensities for the three labels employed are given in Figure 3C.
wt DC-SIGN microdomains on native cell surfaces
In this study, we examined three sets of wt DC-SIGN microdomains on cell surfaces: 1) immature DCs endogenously expressing wt DC-SIGN; 2) NIH3T3 cells ectopically expressing wt DC-SIGN; 3) NIH3T3 cells ectopically expressing GFP-DC-SIGN. As summarized in Table 1, the first two sample types, DC-SIGN endogenously expressed in immature DCs and DC-SIGN expressed in NIH3T3 cells, gave similar averages for the numbers (N) of DC-SIGN molecules per microdomain (4 vs. 5). An unpaired t-test analysis gave a value of p = 0.2, indicating no evidence for a statistically significant difference in the distribution of N values for the two sample types (Figure 4 A and B). Thus, NIH3T3 cells expressing wt DC-SIGN appear to be a reasonable model for the purpose of quantifying the occupancy numbers of DC-SIGN in microdomains. Furthermore, because DCs were labeled with the mAb 120507 and NIH3T3 cells ectopically expressing wt DC-SIGN were labeled with the mAb DC6, these data provide evidence that the two different mAb had similar binding properties. The average N and its standard deviation for wt DC-SIGN on DCs (4 ± 3) were similar to the values previously measured by super-resolution Blink microscopy (7 ± 10) (14), which also employs antibody labeling, giving confidence in these estimates.
TABLE 1.
Number of DC-SIGN molecules per domain as measured by quantitative TIRFM imaging
| Cell sample set | Sample size | |
|---|---|---|
| wt DC-SIGN, dendritic cell | 4 ± 3 | 91 |
| wt DC-SIGN, NIH3T3 cell | 5 ± 4 | 95 |
| GFP-DC-SIGN | 7 ± 9 | 189 |
| DC-SIGNΔ37 | 6 ± 6 | 340 |
| DC-SIGN-N80Q | 8 ± 13 | 118 |
| GFP-DC-SIGN-80Q | 9 ± 8 | 272 |
D/3D plugin in Image J.
The GFP-DC-SIGN data set gave an average value of N = 7, which is comparable to but somewhat larger than the values for the first two data sets and the differences are statistically significant. Two possible explanations for these differences are that the GFP tag promotes a small degree of DC-SIGN accumulation in microdomains or that the stoichiometry of mAb to DC-SIGN on cell surfaces is on average less than one, due to steric hindrance when multiple antibodies are bound to single DC-SIGN molecules within an oligomer.
Nonetheless, our results show that the number of DC-SIGN molecules in a given microdomain is almost always less than 20, indicating that, for the most part, cell surface microdomains contain a strikingly low number of DC-SIGN molecules. In vitro assays have shown that DC-SIGN is able to form oligomers, such as trimeric or tetrameric structures, and cross-linking measurements on cell surfaces have suggested that the fundamental unit of DC-SIGN is a tetramer (6-8, 37). With this assumption, our data indicate that DC-SIGN surface microdomains, as visualized in wide-field microscopy, contain in most cases only one or two DC-SIGN tetramers. Similarly, Figure 4C shows that GFP-DC-SIGN microdomains in NIH3T3 cells most frequently contain only one or a few DC-SIGN tetramers.
Single tetramers should give diffraction-limited spots when imaged with TIRFM. Therefore, we plotted the number of DC-SIGN molecules per microdomain vs. microdomain width. The domain widths, δ, were calculated by fitting the domain intensity distributions to two-dimensional Gaussian functions (Eq. 1). As shown in Figure 5 A-C (left panels), the number per microdomain is ≤10 for δ ≤ 400 nm; thus, if a tetramer is the basic unit, microdomains with diffraction-limited sizes usually consist of one or two of these units. The lower limits indicate a high frequency of even less than 4 DC-SIGN molecules in single microdomains; this observation may be a consequence of several factors that could result in underestimation of the copy numbers (see Discussion). By contrast, the presumed basic units of DC-SIGN tetramers in some cases did form larger sized domains. As shown in Figure 5 A-C (right panels), the microdomain sizes (proportional to the square root of domain area) range from the diffraction limit to ~1 μm and, for the larger sizes, some microdomains contain tens of DC-SIGN molecules, when it is ectopically expressed.
Effects of mutating DC-SIGN on microdomains
Previously we reported that deleting the tandem repeats region or truncation of the CRD of DC-SIGN results in loss of stable microdomains, while deleting the cytoplasmic domain (DC-SIGN-Δ37) or mutating the N-glycosylation site (DC-SIGN-N80Q) does not affect stable microdomain formation (16). In the studies described here, we further quantified the numbers of those domain-forming mutants of DC-SIGN molecules per microdomain, as compared to the values for wt DC-SIGN microdomains. Three sets of mutant DC-SIGN cell samples were examined: 1) DC-SIGN-Δ37; 2) DC-SIGN-N80Q; 3) GFP-DC-SIGN-N80Q. In general, these mutant forms exhibit slightly larger numbers of DC-SIGN molecules per microdomain compared to wt DC-SIGN.
As shown in Table 1, the average number per microdomain of DC-SIGN-Δ37, 6, and the average number per microdomain of DC-SIGN-N80Q, 8, are both higher than the average values of wt DC-SIGN (4 in DCs and 5 in NIH3T3 cells, respectively). For these comparisons, unpaired t-tests gave p values of 0.004 (DC-SIGN-Δ37 vs. wt DC-SIGN on DCs), 0.06 (DC-SIGN-Δ37 vs. wt DC-SIGN on NIH3T3 cells), 0.007 (DC-SIGN-N80Q vs. wt DC-SIGN on DCs), and 0.03 (DC-SIGN-N80Q vs. wt DC-SIGN on NIH3T3 cells). Histograms describing the distribution of molecular occupancy in microdomains are given in Figure 4 D-F. Similar to the wt proteins in DCs or expressed in NIH3T3 cells, DC-SIGN-N80Q copy numbers for microdomains with δ ≤ 400 nm were very low (Figure 5 E, left panel) but DC-SIGN-Δ37 and GFP-DC-SIGN-N80Q copy numbers for microdomains in this range, where δ is close to the pixel size (270 nm), were somewhat larger (Figure 5 D and F, left panels). The three mutant forms of DC-SIGN also tended to exhibit higher molecular copy numbers for larger microdomain sizes (Figure 5 D-F, right panels) compared to the wt DC-SIGN in DCs or NIH3T3 cells (Figure 5 A and B, right panels).
Role of DC-SIGN microdomains in DENV infection
DC-SIGN plays a crucial role in capturing pathogens ranging in size from viruses to yeasts. In a different study, we found that large size pathogens such as fungal particles require rearrangements and concentration of DC-SIGN nanodomains in pathogen-DC contact zones for stable binding and further processing (manuscript in preparation). In the studies described here, we examined the role of DC-SIGN micro/nanodomains in cell binding, internalization and infection by DENV, a very small pathogen. DENVs have an average size of ~ 50 nm in diameter, which is close to the size of DC-SIGN nanodomains (~70 nm) (14). Therefore, we hypothesized that DENV binding would not require rearrangements or concentration of DC-SIGN domains. Here we used NIH3T3 cells expressing DC-SIGN as a model system; similar results were seen with primary dendritic cells differentiated from human blood monocytes (data not shown). For all the experiments described below, DENVs were detected by immunofluorescence and the details of labeling are described in the Materials and Methods section.
As shown in Figure 6 A, individual DENV particles bound to cell surface DC-SIGN microdomains, and these domains did not change in appearance compared to controls. The majority of the DC-SIGN microdomains with DENV bound are still at the diffraction-limit size, and the brightnesses of those DENV-bound microdomains are comparable to the brightnesses of other surrounding microdomains without bound DENV (Supplemental Figure S2). In Figure 6 A the cells were pre-treated with endocytosis inhibitors (10 mM NaN3, 2 mM NaF, and 5 mM 2-deoxy-D-glucose) for 2 min before incubating with DENVs, to prevent fast internalization of DENVs after surface binding. After 10 minute incubation, bound DENVs showed high colocalization percentages with DC-SIGN microdomains (80 ± 14 %, averaged from hundreds of bound DENVs on 12 cell surfaces) (Figure 6 C), indicating that cell membrane DC-SIGN microdomains capture DENVs efficiently within minutes of exposure. The colocalization percentages did not reach 100%, which may be due to the fact that DENVs can bind other cell surface components such as heat-shock proteins or heparan sulfate (33). We did observe that a small fraction of DENVs, which did not colocalize with DC-SIGN microdomains, remained on cell surfaces even after 1-2 h incubation. We also observed that a small fraction of DENVs could bind to the surfaces of wt NIH3T3 cells not expressing DC-SIGN, but these viruses did not induce productive infection even after 72 hours of incubation (Supplemental Figure S3 A and B). While the endocytosis inhibition treatment might impair rearrangement of DC-SIGN microdomains, nonetheless, it is evident that cell surface DC-SIGN microdomains can capture DENVs within minutes of incubation.
Figure 6. Colocalization of DENVs with DC-SIGN micro/nano-domains.
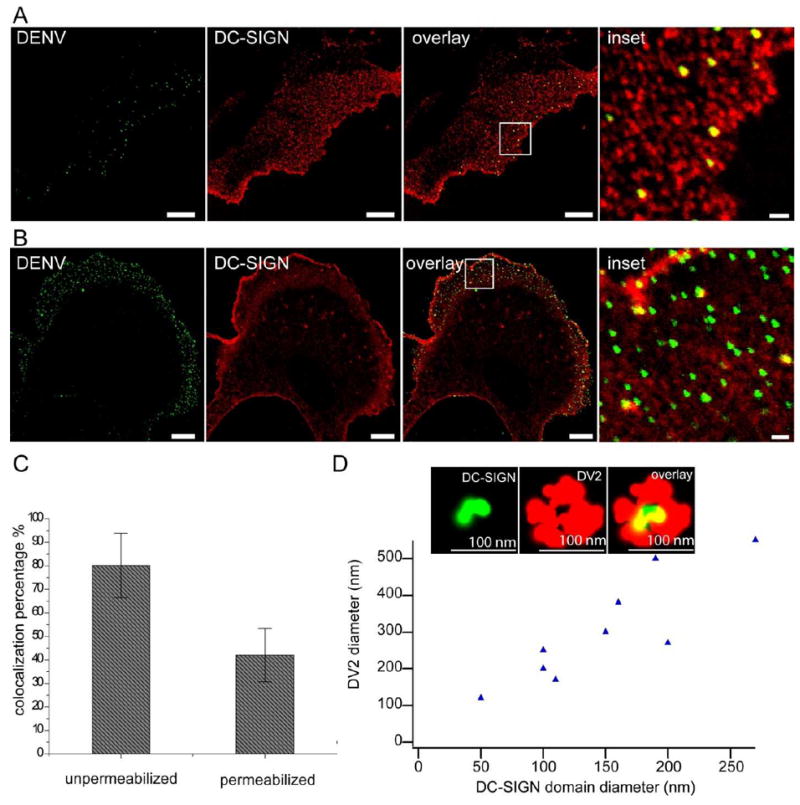
A) Representative confocal images of DENVs (left), DC-SIGN microdomains (middle left), overlay of the two (middle right), and an enlarged view of the inset in middle right (right), on cell surfaces. In this experiment, NIH3T3 cells expressing DC-SIGN were pretreated with endocytosis inhibitors (10 mM NaN3, 2 mM NaF, and 5 mM 2-deoxy-D-glucose) for 2 min, then incubated with DENV for 10 minutes prior to fixation. B) Representative confocal images of DENVs (left), DC-SIGN microdomains (middle left), and overlay of the two (middle right), and an enlarged view of the inset in middle right (right), after cell permeabilization. Bars in the first three panels, 10 μm. Bar in the fourth panel, 1 μm; in this experiment, DENV was incubated with 3T3 cells expressing DC-SIGN for 15 minutes prior to fixation and permeabilization. C) Colocalization percentages of DENVs with DC-SIGN microdomains before (left bar) and after (right bar) permeabilization. Error bar, standard deviation. D) Plot of DC-SIGN micro/nano-domain diameters with corresponding colocalized DENV (single or aggregates) diameters, from dSTORM images. Inset, a representative dSTORM image of a single DC-SIGN nanodomain (left), DENV particle (middle) and overlay of the two (right). Bars, 100 nm.
If endocytosis was not initially inhibited, DENVs were quickly internalized within a few minutes. Figure 6B shows a representative image of NIH3T3 cells expressing DC-SIGN, incubated with DENVs for 15 min and stained for both surface bound and internalized DENVs after permeabilization. We examined 3 min, 5 min, 10 min and 15 min DENV incubation periods, and found DENV internalization even after 3 min (data not shown), consistent with the observation of van der Schaar et al. (38). Thus, cell surface DC-SIGN microdomains captured the majority of DENVs and those DENVs were efficiently internalized within minutes after binding. Furthermore, in Supplemental Figure S4, we show that the number of viral particles taken up is proportional to the relative expression levels of DC-SIGN in individual cells. This result further indicates that DC-SIGN domains play the major role in capturing DENV particles, leading to efficient internalization.
In the counting studies, most microdomains appearing at the diffraction limit contained only 4-8 DC-SIGN molecules and corresponded to single nanodomains. Furthermore, most DENV images were also diffraction-limited. Thus, it is likely that the dominant stoichiometry of DC-SIGN microdomains to bound DENVs is 1:1.
We further probed whether a single DC-SIGN nanodomain was sufficient to bind a single DENV by using dSTORM super-resolution imaging. Figure 6 D (inset), shows a single DC-SIGN nanodomain (~ 40 nm) that is colocalized at the center of a DENV particle, strongly suggesting that single DC-SIGN nanodomains with only a few DC-SIGN molecules are capable of capturing small sized viral pathogens. Note that a single DENV particle, about ~50 nm in diameter, with primary and secondary Abs bound would be imaged with a diameter of ~100 nm, consistent with what is observed. In addition, we also observed that larger apparent sizes of viral particles, probably due to DENV aggregation, correspondingly colocalized with larger sizes of DC-SIGN microdomains (Figure 6 D, plot).
Finally, productive DENV infection and viral replication in NIH3T3 cells expressing DC-SIGN was detected by both fluorescence activated cell sorting (FACS) and confocal imaging (Supplemental Figure S3 A, C and D). By contrast, wt NIH3T3 cells incubated with DENV for 24-72h showed little productive infection (Supplemental Figure S3 A and B). FACS results showed <1% wt NIH3T3 cells were infected (Supplemental Figure S3 A) when DC-SIGN was not expressed. Similarly, by confocal imaging, the chance to observe an infected cell was far below 1% (approximately less than 10 cells in a glass-bottom dish with ~50,000 cells). Supplemental Figure S3 B shows a representative image of wt NIH3T3 cells incubated with DENV at a multiplicity of infection (MOI) equal to 15, for 72h, and only a couple of viral particles were observed; i.e., no productive infection. We suspect that the very small percentage of infection as observed by FACS (0.72% in Supplemental Figure S3 A) mainly arose from dead cells with high autofluorescence, as observed by confocal imaging. By contrast, NIH3T3 cells expressing DC-SIGN showed massive viral replication observed by confocal imaging and over 90% infectivity by FACS (Figure S3 A, C and D). These results indicate that, in this system, DC-SIGN is capable of attaching DENV and this attachment leads to productive infection.
Discussion
In this study we developed a single molecule imaging approach, based on prism-TIRFM, to quantify the molecular density of cell surface DC-SIGN microdomains by comparing the brightnesses of single fluorophores with the brightnesses of single clusters. Our method is similar to that employed by Jaqaman et al. (39) to quantify antibody labeled CD36 receptor molecules on cell surfaces and to that used by Joglekar et al. (40) to count component proteins (as fluorescent protein fusions) in kinetochores. Fluorescence Correlation Spectroscopy (FCS) can also be employed, but this technique requires the diffusion of target molecules (41).
There are several physical and chemical effects that strongly suggest our values represent a lower limit to domain occupancy values. Physically, the exponential decay of the TIRF illumination field intensity as one moves away from the substrate-buffer interface will produce an uncertainty in the following way. The brightnesses of single fluorophores were measured on dyes that were immobilized on fibronectin-coated microscope slide surfaces, while the DC-SIGN domains that were characterized were on the ventral surface of cells, which could be some distance away from the interface but still in the TIRF illumination field. We make a rough estimate of this source of uncertainty as follows. The evanescent wave in our prism-based TIRF microscope undergoes an exponential decay characterized by the constant d = 85 nm (42). We assume that the single fluorophores immobilized on fibronectin were within 10 nm of the substrate resulting in a decrement of excitation intensity of ~11% due to the exponential decay of the evanescent wave. Interference reflection microscopy (IRM) images (Supplemental Figure S1) indicate that areas which were imaged grey, corresponding to so-called close contacts, are within ~ 30 nm of the substrate (43); at this position, the excitation intensity will have decreased by ~30 %. Thus, this effect would decrease the occupancy numbers by ~ 21%. In addition, there are several chemical effects that could result in an underestimate of the domain occupation number. Two labeling techniques were employed in this study: first, to label microdomains by using AlexaFluor488 conjugated mAb specific for DC-SIGN; and second, to genetically tag DC-SIGN with GFP at its cytoplasmic N-terminus. With respect to antibody labeling, even assuming saturation binding and an average dye to antibody ratio of ~1 (see Materials and Methods), the binding of mAb to DC-SIGN oligomers at saturation could be sterically limited. Therefore it is plausible that less than 4 mAbs can bind to one DC-SIGN tetramer. This fact may account for why domain occupancy numbers are about 2-fold higher for GFP-DC-SIGN fusions as compared to numbers obtained by mAb labeling. It is also possible that the two different mAbs employed have different monoclonal antibody: DC-SIGN stoichiometries at saturation.
With respect to fluorescent protein labeling, while using fluorescent protein fusions usually guarantees a 1:1 stoichiometry of label to the protein of interest, the possibility that either the fluorescent protein sterically interferes with the packing of DC-SIGN oligomers or that it enhances occupancy in the structure must be entertained. Such effects could produce underestimates or overestimates, respectively. It should also be noted that we excluded from analysis a portion of domains that were irregularly shaped and usually larger; such domains generally have larger occupancy numbers. We assume that other factors such as homo-FRET between labels are insignificant. Overall, though, the agreement between antibody and GFP labeling supports the conclusion that the number of DC-SIGN molecules per microdomain, although possibly underestimated by a factor of no more than ~4, is surprisingly small.
One technical issue of the approach described here is that the point spread function (PSF) was undersampled in our system setup. The Nyquist criterion requires pixel sizes ≤ (1/2) the diffraction limited spot size. In our TIRFM setup, the pixel size is limited by the relatively large pixel size of EMCCD chip. However, the objects imaged in this study, i.e., single molecules on glass substrates or DC-SIGN domains on cell surfaces, were widely separated in space so resolution is not important for the intensity measurements. The relatively large pixel size does affect the accuracy of the size of the domains to a small extent. We simulated DC-SIGN domains under full sampling and undersampling situations, and our simulation results show that undersampling of PSF leads to an increase of 3-7% of the domain width, as compared to the domain width obtained by full sampling. Thus, undersampling does not appear to be a major source of error.
Our results indicate that the smallest microdomains imaged by wide-field methods are occupied by only 4-8 copies of DC-SIGN on average, which, based on the literature (6-9), corresponds to one or two tetramers. These basic units may be collected to form larger microdomains; however, given that super-resolution studies indicate nanodomains are randomly disposed, as judged by the Ripley’s K or Hopkins statistic (14), on the membrane, it may be that a stochastic clustering of nanodomains when convoluted with the point spread function gives rise to the appearance of larger microdomains (unpublished calculations). Both cytoplasmic truncation and a glycosylation site mutation result in somewhat higher molecular occupancy numbers within the apparent microdomains. This result can be compared with the report by Serrano-Gómez et al., which shows that the N80Q mutation resulted in higher order oligomer formation of DC-SIGN (8). Since DC-SIGN captures pathogens ranging in sizes from viruses to yeasts, our results suggest that higher order oligomers are not essential to DC-SIGN’s pattern recognition function.
Importantly, the values obtained from this study are consistent with previous Blink microscopy results (14), giving confidence in our result and approach. Estimation of the numbers of DC-SIGN molecules in a microdomain by Blink was based on the assumption that a fluorophore would give the same number of Blink localizations, regardless of whether the fluorophore is a single dye (conjugated to Fab) on a glass surface or the fluorophore conjugated to Fab is on a single DC-SIGN molecule in cell surface microdomains. Clearly, this approach needs corroboration, because blinking of a single dye is a stochastic process and thus one dye may give more or fewer blink localizations during one Blink image data acquisition. In contrast, the quantitative TIRFM imaging approach described in this paper directly compares the brightnesses of single fluorophores with the brightnesses of single DC-SIGN domains, which does not depend on the blink events of single fluorophores. Therefore, the quantitative TIRFM imaging approach is a more straightforward method, which can be carried out on common TIRF microscopes and this approach does not require the setup of a superresolution imaging system. On the other hand, our result is consistent with the quantitation result by Blink, indicating that Blink microscopy may also be employed for approximate counting measurements, in addition to providing super-resolution imaging.
In this study, we also found that DC-SIGN domains on NIH3T3 cells captured DENV efficiently, and, apparently one nanodomain was sufficient for the capture. Bound DENV was internalized within minutes after attachment and viral replication was observed within 24 h following viral uptake. In contrast, incubation of wt NIH3T3 cells, not expressing DC-SIGN, with DENV led to only small amounts of attachment and no internalization of bound virus. These results strongly suggest that small numbers of DC-SIGN can mediate both binding and subsequent interiorization and viral replication. This conclusion follows from the observations that microdomains imaged at the diffraction limit are colocalized with DENV and these microdomains most likely contain only 4-8 DC-SIGN molecules. Indeed, using super-resolution microscopy, instances can be imaged where one DENV can be found associated with one DC-SIGN nanodomain.
Our results indicate that, in dendritic cells, DC-SIGN is arranged in nanodomains containing only one or two tetramers on average. This raises the semantic and conceptual issue of whether this degree of occupancy represents a nanodomain or simply a molecular cluster. It is conceivable, given the stability of the cluster array (15), that proteomic/lipidomic studies of isolated domains may reveal a distinct molecular composition associated with DC-SIGN which would qualify these clusters as true nanodomains.
This study and previous ones (11, 12, 14) beg the question of why this small functional unit has evolved. Functionally speaking, DC-SIGN is a DC receptor for diverse pathogens ranging in size from small viruses to larger yeasts. It is possible that a cluster consisting of one or two (that are closely spaced) tetramers would provide a nanoscale platform having the requisite avidity to successfully bind (or rebind in the case of dissociation) small viruses such as the ~50 nm diameter dengue virus (44, 45). While the dengue virus envelope is highly populated with the E glycoprotein, a single cluster of one or two DC-SIGN tetramers could also bind HIV since its envelope glycoproteins, which are present in low numbers, are capable of lateral rearrangement into a single focus on the envelope during maturation and/or binding to target lymphocytes (46). However, following binding of viruses like dengue, which must be endocytosed, presumably some process must occur to release the virus-nanocluster complex for lateral transport to the internalization portal, such as a coated pit (38). This release might occur because the pathogen displaces DC-SIGN ectodomain interactions that laterally stabilize the cluster analogous to the SIGLEC hypothesis (47, 48). Indeed, when larger pathogens, such as yeasts, bind DC, there is evidence that the native nanodomain arrangement is altered suggesting that some lateral mobility has occurred to accommodate the yeast (manuscript in preparation).
Materials and Methods
Reagents
Two labeling techniques were employed in this study: first, to label microdomains by using AlexaFluor488 conjugated mAb specific for DC-SIGN; and second, to genetically tag DC-SIGN with GFP at its cytoplasmic N-terminus. DC6 mAb was prepared in-house as described elsewhere (15). 120507 mAb was obtained commercially (R&D Systems, Minneapolis, MN). AlexaFluor488 (Invitrogen) was conjugated to the two types of mAb as described (16); the final molar ratio of dye to antibody was ≈ 1, as assessed by absorption spectroscopy. A fusion protein consisting of GST and GFP was expressed in E. Coli and purified as described elsewhere (49). Dynamic light scattering data were consistent with the preparation being mostly single molecules of GST-GFP. NaN3 was purchased from FisherBiotech (Fair Lawn, NJ). Anti-mouse IgG-Atto488, NaF and 2-deoxy-D-glucose were purchased from Sigma-Aldrich (St. Louis, MO). For labeling of DENVs, 2H2 mAb was purchased from EMD Millipore (Billerica, MA) and conjugated to AlexaFluor488 as described above. H-200 IgG was purchased from Santa Cruz (Dallas, Texas), and anti-rabbit (Fab’)2 AlexaFluor647 was purchased from Cell Signaling (Danvers, MA).
Fibronectin-coated microscope slides
Glass microscope slides (1” × 1.5” × 1 mm) were cleaned by boiling in ICN detergent (MP Biomedicals, Solon, OH), bath sonicating, extensive washing with deionized H2O, and drying at 120 °C. The slides were then sterilized with 70% ethanol, dried, treated with 10 μg/ml fibronectin (Invitrogen) in phosphate-buffered saline (PBS) at 4 °C overnight under sterile conditions. Fibronectin-coated slides were then washed with PBS before being used as cell substrates. For identifying single molecule brightness in our study, very small amounts (≪ 1 μg/ml) of GFP or fluorescently labeled antibodies were added to the fibronectin solutions before substrate treatment and imaged later under the same conditions as cell measurements; these samples were not sterile.
Dendritic cells
Human monocyte-derived immature dendritic cells were prepared as described elsewhere (13, 15). The cells were seeded on fibronectin-coated microscope slides and cultured in RPMI-1640 media containing 10% fetal bovine serum (FBS), 500 U/ml of human IL-4 and 800 U/ml of human GM-CSF (Peprotech, Rocky Hill, NJ) for about one week in a 37 °C incubator with 5% CO2. After the monocytes developed into immature dendritic cells, slides containing adherent cells were removed from the growth media, washed several times with PBS, treated with 4% paraformaldehyde (PFA) in PBS for 20 min at room temperature for cell fixation and then washed with PBS again. The cells were then blocked with 0.1% bovine serum albumin (BSA) for 15 min, stained with 8 μg/ml AlexaFluor488-conjugated 120507 monoclonal antibodies (see below) in 0.1 % BSA/PBS for 20 min, and washed thoroughly with PBS.
NIH3T3 cells
Plasmid construction has been described previously (15, 16). NIH3T3 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% FBS, 50 U/ml penicillin G and 50 μg/ml streptomycin at 37°C in 5% (v/v) CO2. One day before transfection, fibronectin-coated slides were put into 60 mm cell culture dishes with cell growth media as mentioned above. Cells were then seeded on the surface of the slides at 3 × 105 cells/dish. On the day of transfection, plasmids were introduced into the cells by following the standard protocol employing Lipofectamine (Invitrogen) at 2 μg cDNA/dish. After transfection, cells were maintained in normal growth medium for at least two days in a 37 °C incubator. On the day of image acquisition, cells were fixed (see above). After fixation, cells were either directly used for imaging (for GFP constructs) or blocked with 0.1% BSA and then stained with 8-16 μg/ml AlexaFluor488-conjugated DC6 mAb (wt DC-SIGN) or 120507 mAb (DC-SIGN-Δ37 and DC-SIGN-N80Q) in 0.1% BSA/PBS for 20 min, and washed thoroughly with PBS. Previous work has shown that 10 μg/ml DC6 mAb is sufficient to saturate cell-surface DC-SIGN (14). Similar measurements with 120507 mAb indicated that 8-16 ug/ml of this mAb is also sufficient for cell-surface saturation (data not shown).
Sample preparation for TIRFM imaging
For objective-based TIRFM, cells were prepared as described above except that they were grown on MatTek dishes (MatTek Corp., Ashland, MA). For prism-based TIRFM, microscope coverslips (#0 thickness, 24 mm × 60 mm) were cleaned as described above for microscope slides. Two small strips of Scotch transparent tape, used as the ‘spacer’, were adhered to the long edges of fibronectin-coated slides containing bound cells or single fluorescent molecules. A thin layer of UV-curing optical glue (NOA81, Norland Products Inc.) was applied to the tape tops prior to adding the coverslips to make the sandwiches. An aluminum foil mask was used to cover the center of the sandwich sample to avoid any photodamage from the UV light. Then the whole sample was illuminated with UV light for approximately one minute to harden the glue. The inner volumes of the samples were filled with PBS, and the two open edges were sealed with thin layers of vacuum grease before mounting the samples, with the coverslip adjacent to the objective, on the stage of the inverted microscope.
Objective-based TIRFM
For comparative purposes, initial imaging of different cell types was carried by using a conventional objective-based TIRFM instrument. The system is based on an Olympus IX81 microscope, equipped with two Melles Griot diode lasers (491nm and 561nm, respectively). Images were taken by using a 100×, 1.49 NA objective and a conventional CCD camera.
Prism-based TIRFM
The principle and basic setup of a prism-based TIRFM system (Fig. 1 A) has been described in detail elsewhere (18, 50). For the measurements described here, fluorescence was excited by the evanescent wave generated by internally reflecting the 488 nm line of an argon ion laser (Coherent Innova 90-3). Unless otherwise specified, the power of the incident beam was 300 μW. The beam was passed through a fused silica prism optically coupled to the microscope slides with glycerol, so that the incidence angle at the interface of the microscope slide and inner volume was greater than the critical angle. The incident beam was always s-polarized, the evanescent wave depth was ≈ 85 nm, and the 1/e2-radii of the elliptically shaped Gaussian illumination area were approximately 65 μm × 20 μm. Evanescently excited fluorescence was collected through a 60×, 1.4 NA oil immersion objective on an inverted microscope (Zeiss Axiovert 35) coupled below the sandwich coverslip, passed through an appropriate dichroic mirror and barrier filter, and imaged by an EMCCD camera (Andor iXon DU-871E) driven by Andor iQ software. The pixel size of this camera was 16 μm which, when a 60× 1.4 NA objective is employed, means that the point spread function is under sampled. However, because we are measuring the emitted intensity of single molecules or domains that are widely separated as opposed to creating detailed images of those objects, this was not considered to be a limitation. Unless otherwise specified, the EMCCD was cooled to -70 °C, the gain was set at 260, and images were collected at 512 × 512 pixels per frame with a 300 ms exposure time. Depending on the photostability of the fluorophores, 100-500 frames per video were recorded to observe single-step photobleaching and calculate the brightness of single fluorophores. For imaging DC-SIGN microdomains on cell surfaces, a single frame was collected.
Molecular Counting Data analysis
To determine the numbers of DC-SIGN molecules in microdomains, the brightnesses of microdomains and single fluorescent molecules were compared. Ideally, this approach requires a 1:1 labeling ratio of single fluorophores to single DC-SIGN molecules, as well as identical optical parameters for both single molecule and microdomain imaging. The extent to which deviations from these ideal conditions might have skewed the reported results is discussed above.
Prism-based TIRFM employing an incident argon ion laser beam creates a surface-associated evanescent wave with an intensity profile in the sample plane that has an elliptically Gaussian shape (20) as shown in Figure 1B. The evanescent excitation intensity is higher at the middle than at positions away from the center; thus, fluorophores towards the middle of the illuminated area will be brighter than those in peripheral regions. To account for this effect, the local background luminescence arising from substrate impurities and close to single fluorescent molecules or microdomains was employed as an indicator of the local excitation light intensity. This procedure can in general be applied to any illumination scheme, although it is particularly important for prism-based TIRFM.
For single fluorescent molecules, first, a time sequence of 100-500 images was acquired. ImageJ software was then used to roughly localize the single molecules and generate plots of the time-dependent, total intensities within selected regions of interest surrounding the molecules (Figure 1C). These plots were examined to determine the time of the single-step bleach (for GFP) or the last single-step bleach (for mAb). A frame preceding the single or last bleach and having an integrated spot intensity approximately equal to the time-average immediately before this bleach was then selected for further analysis, as described below.
Single fluorescent molecules or single DC-SIGN microdomains were localized in individual frames by a particle finding algorithm in the ‘localizer’ plug-in of Igor Pro software (kindly provided by Dr. Peter Dedecker at the University of Leuven, Belgium; the plugin can be downloaded at: www.igorexchange.com/project/Localizer). The criteria by which spots were identified are described elsewhere (16, 51). The counts per pixel surrounding the kth localized centroid for single molecules, Zsm(i,j,k), or the mth localized centroid for single microdomains, Zdomain(I,j,m), where the integers i and j denote the pixel position, were then fit to two-dimensional Gaussian functions; i.e.,
| (1) |
The free parameters were A(k,m) (the amplitude of the emitted fluorescence), i0(k,m) and j0(k,m) (a more accurate center), μ(k,m) (the spatial width), and B(k,m) (the local emitted background luminescence). In the following discussion, we use the parameter s to denote the length of one pixel with s equal to one; hence, s2 also equals one. Parameters i, j, i0(k,m), j0(k,m), and μ(k,m) have the units of s. Zsm(i,j,k), Zdomain(i,j,m), A(k,m), B(k,m), and C have the units of (detected counts)(s)-2(τ)-1, where τ was the exposure time (300 ms). More explicitly, the A(k,m) have the units of (detected fluorescence counts)(s)-2(τ)-1 and the B(k,m) have the units of (detected background luminescence counts)(s)-2(τ)-1. C, representing the sum of the background luminescence which is independent of the excitation intensity and the dark count of a single pixel on the EMCCD camera, was measured far from the evanescently illuminated area to have an average of 165 (detected counts)(s)-2(τ)-1 and fixed at this number during curve-fitting. This analysis gave three lists (for the different single molecule types) containing the best-fit values of A(k), B(k), i0(k), j0(k) and μ(k), where the index k denotes the kth single molecule of a given type. The analysis also gave six lists (for the different microdomain types) containing the best-fit values of A(m), B(m), i0(m), j0(m) and μ(m), where the index m denotes the mth microdomain of a given type.
For a given single fluorescent molecule or DC-SIGN microdomain, the local evanescent excitation intensity was accounted for in the following manner. The intensity-dependent background luminescence’s can be expressed as
| (2) |
where I(k) and I(m) are the local excitation intensities for the kth single molecule or the mth microdomain, respectively, in units of (excitation photons)(s)-2(τ)-1, and β is a proportionality constant independent of k and m with the units of (detected background luminescence counts)(excitation photons)-1. Single molecule and microdomain powers (spatially integrated fluorescence emission intensities) were calculated for each single molecule or domain, from the best-fit values of A(k,m) and μ(k,m), as
| (3) |
in units of (detected fluorescence counts)/τ. These parameters are also given by
| (4) |
where Q is a proportionality constant independent of k and m, with the units of (s2)(detected fluorescence counts)(excitation photons)-1 and N(m) is the number of DC-SIGN molecules in the mth microdomain. Corrected powers, which account for the local excitation intensity, were calculated for each single molecule or microdomain as
| (5) |
Thus, referring to Eqs. 2 and 4, one finds that
| (6) |
The corrected powers do not depend on the local excitation intensity and have the units of (s2) (detected fluorescence counts)(detected background luminescence counts)-1.
In the idealized case in which other sources of noise are not present, for GFP, ηsm,c(k) should not depend on the particular kth single molecule (Eq. 6). Thus, an average, ηGFP,c, was calculated as
| (7) |
where ηsm,c(k) denotes the measured corrected power for the kth single GFP molecule and TGFP denotes the number of single GFP molecule images analyzed. Then, for each microdomain in which the fluorescence was reported via GFP, the number of DC-SIGN molecules in this microdomain was computed as (see Eqs. 6)
| (8) |
For the two different mAbs, the situation is a bit more complex. Computing experimentally obtained, average corrected powers for the two types of mAbs gives
| (9) |
where ηsm,c(k) denotes the measured corrected power for the kth single mAb of a given type and TmAb denotes the number of single mAb molecule images analyzed. Because the experimental values of ηsm,c(k) were measured for the last single-step bleach, ηmAB,c = [ηmAB,c]expt is the measured, average corrected power for a single AlexaFluor488 conjugated to a mAb. In general, for an average labeling ratio of γ ≈ 1 of AlexaFluor488 probes per mAb, and assuming a Poisson distribution for the number of fluorophores per mAb, the average corrected power is, theoretically,
| (10) |
Thus, because γ ≈ 1 and because only frames immediately prior to the last single step bleach for the mAbs were used, the fact that some mAb have 0, 1, 2 or more conjugated fluorophores can be accounted for. Worth noting is that a similar procedure can be used when γ ≠ 1, by multiplying [ηmAb,c]expt by γ. For each microdomain in which the fluorescence was reported via a mAb, the number of DC-SIGN molecules in this microdomain was computed as (see Eqs. 6)
| (11) |
The spot widths (for single molecules) or microdomain widths are denoted by δsm(k) and δdomain(m), respectively, and were calculated in nm as δsm(k) = μ(k)σ or δdomain(m) = μ(m)σ where σ = (16 μm)/(60) = 270 nm was the pixel size (16 μm is the pixel dimension of the camera and the objective was 60X). Apparent microdomain areas were determined as Adomain(m) = πδdomain2(m). As noted in Figure 1D, large ill-defined microdomains were excluded from analysis as it was impossible to ascertain whether such domains were a collection of smaller microdomains.
DENV
In this study we used DENV serotype 2 strain S-16803 (denoted as DENV in this paper), which was produced in C636 insect cells as previously described (52). The titer of the infectious virus stock is 1.57 × 107 FFU/ml.
Confocal imaging and colocalization analysis
For DENV and DC-SIGN microdomain colocalization analysis, NIH3T3 cells expressing DC-SIGN plated on 35 mm MatTek dishes were first incubated with endocytosis inhibitors (10 mM NaN3, 2 mM NaF, and 5 mM 2-deoxy-D-glucose) for 2 min, then incubated with DENVs at 15.7 MOI for 10 min, thoroughly washed several times with Dulbecco’s phosphate-buffered saline (DPBS) and fixed with 2% paraformaldehyde (PFA) for 20 min. After fixation, the cell dishes were separated into two groups: nonpermeabilized and permeabilized. Nonpermeabilized cells were used to image only cell-surface DENV and DC-SIGN microdomains for surface colocalization analysis. For this group, the cells were washed three times with DPBS, and submerged in 1% normal mouse serum (NMS) in DPBS for 30 min for blocking. Permeabilized cells were used to image both surface and internalized DENVs and DC-SIGN. For this group, the cells were washed three times with DPBS, submerged in Perm Buffer (2% BSA, 0.1% saponin, 0.02% NaN3 in sterile DPBS) and washed twice with Perm Buffer. After permeabilization the cells were incubated with blocking buffer (1% NMS in Perm Buffer) for 30 min. After blocking, antibodies for staining DENVs or DC-SIGN were diluted either in 1% NMS in DPBS for nonpermeabilized cells, or in 1% NMS in Perm Buffer for permeabilized cells. The cells were stained with anti-DENV 2H2-AlexaFluor488 at saturation concentration for 1 h at 37°C, washed thoroughly several times with DPBS, incubated with primary anti-DC-SIGN H-200 IgG at 6 μg/ml for 20 min, washed thoroughly several times with DPBS, treated with anti-rabbit (Fab’)2 AlexaFluor647 for 20 min, and finally washed thoroughly several times with DPBS.
Confocal imaging of DENV and DC-SIGN on NIH3T3 cells was carried out on a Fluoview FV1200 laser scanning microscope (Olympus) with an oil-immersion 60x NA 1.35 objective. An excitation wavelength of 488 nm at 2% power was used to image DENVs stained by 2H2-AlexaFluor488 mAb and an excitation wavelength of 635 nm at 2% power was used to image DC-SIGN stained with secondary anti-rabbit AlexaFluor 647 (Fab’)2. The emission detection wavelength ranges were 500-550 nm and 650-710 nm, respectively. Image frames contained 1024 × 1024 pixels, with a pixel size of 69 nm.
Colocalization analysis was carried out with a plugin named JACop on ImageJ (53). After pre-processing and setting the thresholds in both green and red channels, Manders’ coefficients were calculated and read out as indications for colocalization percentages. Manders’ overlap coefficients are based on Pearson’s correlation coefficients (54), and range from 0 (no colocalization) to 1 (100% colocalization). Since, in our images the numbers of DENVs were much less than the numbers of DC-SIGN microdomains on cell surfaces, we used M1, the ratio of green DENV signals that overlap with red DC-SIGN signals to the total green signals. Calculation of the number of DENV particles was done by using the Mosaic Particle Tracker 2D/3D plug-in in Image J (55).
dSTORM
Cell sample preparation for dSTORM was similar to the sample preparation for colocalization imaging on nonpermeabilized cells. The main difference arose from the fact that Atto488 is better suited for dSTORM than AlexaFluor488. Therefore, we used a primary mouse mAb 12C1 against DENVs, and a secondary anti-mouse Atto488 IgG to stain DENVs. Staining of DC-SIGN was performed by using primary H-200 IgG and secondary anti-rabbit AlexaFluor647 (Fab’)2. As dSTORM is more sensitive compared to conventional confocal imaging, the blocking step was prolonged to over 1.5 h and washing steps were more extensive. At the time of observation, the cells were merged in fresh-prepared STORM imaging buffer with cysteamine (MEA), kindly provided by Nikon, and dSTORM imaging was performed on a Nikon N-STORM super-resolution microscope (Nikon). Data analysis was carried out in situ on the N-STORM scope computer after imaging.
Infectivity assay by FACS
Two different concentrations of DENVs were used (15.7 and 1.57 MOI). NIH3T3 cells stably expressing DC-SIGN were plated on 35 mm cell culture dishes, and incubated with the two concentrations of DENVs for 24 h, 48 h, and 72 h, respectively. At the time of observation, cells were washed several times with DPBS, trypsinized, centrifuged at 1500 rpm for 5 min, resuspended in DPBS, centrifuged at 1500 rpm for 5 min, and resuspended in 2% PFA for 20 min. After fixation, the cells were washed with DPBS, and permeabilized with Perm buffer. Staining of DENVs using 2H2-AlexaFluor488 and staining of DC-SIGN using H-200/anti-rabbit AlexaFluor647 was performed as described above in the confocal imaging section. The cells were finally transferred into a 96-well plate and infectivity assays were carried out on a Guava easyCyte 8HT flow cytometer (Guava). The data analysis was performed using Guava Express software (Guava).
Infectivity assay by confocal imaging
NIH3T3 cells expressing DC-SIGN plated on 35 mm MatTek glass-bottom dishes were incubated with the two concentrations of DENVs introduced above for 24 h, 48 h, and 72 h. Fixation and staining were carried out as described above, except without trypsinization. Confocal imaging of infected cells was carried out on a Zeiss780 laser scanning microscope (Carl Zeiss).
Supplementary Material
Acknowledgments
We thank Dr. Peter Dedecker at the University of Leuven (Belgium) for providing the Igor plug-in for microdomain size and brightness analysis and helpful discussion. We also thank Drs. Yang Liu and Leaf Huang at UNC Eshelman School of Pharmacy for their assistance with the light scattering measurements. We also thank Dr Rukie de Alwis, Dr Wahala M P B Wahala, Anne Broadwater, Yang Zhou and Kizzmekia S Corbett for their great support in DENV preparation and infection assay. Confocal and TIRF imaging was carried out in the UNC-Olympus Imaging Research Center. P. Liu is grateful to Dr Marc R Ridilla for his help with a Mathematica simulation and helpful discussions on manuscript preparation. This work was supported primarily by NIH grant GM-041402 (K.J. and N.L.T.) and also by the NIH Cell Migration Consortium NIH GM 64346 (K.J.), NSF grant MCB-0641087 (N.L.T.), and NIAID RO1-AI107731 (A. M. de S.).
References
- 1.Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392(6673):245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- 2.Lipscomb MF, Masten BJ. Dendritic Cells: Immune Regulators in Health and Disease. Physiological Reviews. 2002;82(1):97–130. doi: 10.1152/physrev.00023.2001. [DOI] [PubMed] [Google Scholar]
- 3.Adema GJ, Hartgers F, Verstraten R, de Vries E, Marland G, Menon S, Foster J, Xu Y, Nooyen P, McClanahan T, Bacon KB, Figdor CG. A dendritic-cell-derived C-C chemokine that preferentially attracts naive T cells. Nature. 1997;387(6634):713–717. doi: 10.1038/42716. [DOI] [PubMed] [Google Scholar]
- 4.Siamon G. Pattern Recognition Receptors: Doubling Up for the Innate Immune Response. Cell. 2002;111(7):927–930. doi: 10.1016/s0092-8674(02)01201-1. [DOI] [PubMed] [Google Scholar]
- 5.Guo Y, Feinberg H, Conroy E, Mitchell DA, Alvarez R, Blixt O, Taylor ME, Weis WI, Drickamer K. Structural basis for distinct ligand-binding and targeting properties of the receptors DC-SIGN and DC-SIGNR. Nat Struct Mol Biol. 2004;11(7):591–598. doi: 10.1038/nsmb784. [DOI] [PubMed] [Google Scholar]
- 6.Su SV, Hong P, Baik S, Negrete OA, Gurney KB, Lee B. DC-SIGN binds to HIV-1 glycoprotein 120 in a distinct but overlapping fashion compared with ICAM-2 and ICAM-3. J Biol Chem. 2004;279(18):19122–19132. doi: 10.1074/jbc.M400184200. [DOI] [PubMed] [Google Scholar]
- 7.Feinberg H, Guo Y, Mitchell DA, Drickamer K, Weis WI. Extended neck regions stabilize tetramers of the receptors DC-SIGN and DC-SIGNR. J Biol Chem. 2005;280(2):1327–1335. doi: 10.1074/jbc.M409925200. [DOI] [PubMed] [Google Scholar]
- 8.Serrano-Gomez D, Sierra-Filardi E, Martinez-Nunez RT, Caparros E, Delgado R, Munoz-Fernandez MA, Abad MA, Jimenez-Barbero J, Leal M, Corbi AL. Structural requirements for multimerization of the pathogen receptor dendritic cell-specific ICAM3-grabbing non-integrin (CD209) on the cell surface. J Biol Chem. 2008;283(7):3889–3903. doi: 10.1074/jbc.M706004200. [DOI] [PubMed] [Google Scholar]
- 9.Yu QD, Oldring AP, Powlesland AS, Tso CK, Yang C, Drickamer K, Taylor ME. Autonomous tetramerization domains in the glycan-binding receptors DC-SIGN and DC-SIGNR. J Mol Biol. 2009;387(5):1075–1080. doi: 10.1016/j.jmb.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Snyder GA, Ford J, Torabi-Parizi P, Arthos JA, Schuck P, Colonna M, Sun PD. Characterization of DC-SIGN/R interaction with human immunodeficiency virus type 1 gp120 and ICAM molecules favors the receptor’s role as an antigen-capturing rather than an adhesion receptor. J Virol. 2005;79(8):4589–4598. doi: 10.1128/JVI.79.8.4589-4598.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cambi A, de Lange F, van Maarseveen NM, Nijhuis M, Joosten B, van Dijk EM, de Bakker BI, Fransen JA, Bovee-Geurts PH, van Leeuwen FN, Van Hulst NF, Figdor CG. Microdomains of the C-type lectin DC-SIGN are portals for virus entry into dendritic cells. J Cell Biol. 2004;164(1):145–155. doi: 10.1083/jcb.200306112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koopman M, Cambi A, de Bakker BI, Joosten B, Figdor CG, van Hulst NF, Garcia-Parajo MF. Near-field scanning optical microscopy in liquid for high resolution single molecule detection on dendritic cells. FEBS Lett. 2004;573(1-3):6–10. doi: 10.1016/j.febslet.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 13.Neumann AK, Thompson NL, Jacobson K. Distribution and lateral mobility of DC-SIGN on immature dendritic cells--implications for pathogen uptake. J Cell Sci. 2008;121(Pt 5):634–643. doi: 10.1242/jcs.022418. [DOI] [PubMed] [Google Scholar]
- 14.Itano MS, Steinhauer C, Schmied JJ, Forthmann C, Liu P, Neumann AK, Thompson NL, Tinnefeld P, Jacobson K. Super-Resolution Imaging of C-Type Lectin and Influenza Hemagglutinin Nanodomains on Plasma Membranes Using Blink Microscopy. Biophysical Journal. 2012;102(7):1534–1542. doi: 10.1016/j.bpj.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Itano MS, Neumann AK, Liu P, Zhang F, Gratton E, Parak WJ, Thompson NL, Jacobson K. DC-SIGN and influenza hemagglutinin dynamics in plasma membrane microdomains are markedly different. Biophys J. 2011;100(11):2662–2670. doi: 10.1016/j.bpj.2011.04.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu P, Wang X, Itano MS, Neumann AK, Jacobson K, Thompson NL. The Formation and Stability of DC-SIGN Microdomains Require its Extracellular Moiety. Traffic. 2012;13(5):715–726. doi: 10.1111/j.1600-0854.2012.01337.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Axelrod D. Cell-substrate contacts illuminated by total internal reflection fluorescence. J Cell Biol. 1981;89(1):141–145. doi: 10.1083/jcb.89.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Axelrod D, Burghardt TP, Thompson NL. Total internal reflection fluorescence. Annu Rev Biophys Bioeng. 1984;13:247–268. doi: 10.1146/annurev.bb.13.060184.001335. [DOI] [PubMed] [Google Scholar]
- 19.Burghardt TP, Axelrod D. Total internal reflection/fluorescence photobleaching recovery study of serum albumin adsorption dynamics. Biophys J. 1981;33(3):455–467. doi: 10.1016/S0006-3495(81)84906-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burghardt TP, Thompson NL. Evanescent Intensity of a Focused Gaussian Light-Beam Undergoing Total Internal-Reflection in a Prism. Opt Eng. 1984;23(1):62–67. [Google Scholar]
- 21.Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, Armitage JP. Stoichiometry and turnover in single, functioning membrane protein complexes. Nature. 2006;443(7109):355–358. doi: 10.1038/nature05135. [DOI] [PubMed] [Google Scholar]
- 22.Douglass AD, Vale RD. Single-molecule microscopy reveals plasma membrane microdomains created by protein-protein networks that exclude or trap signaling molecules in T cells. Cell. 2005;121(6):937–950. doi: 10.1016/j.cell.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pinton P, Tsuboi T, Ainscow EK, Pozzan T, Rizzuto R, Rutter GA. Dynamics of glucose-induced membrane recruitment of protein kinase C beta II in living pancreatic islet beta-cells. J Biol Chem. 2002;277(40):37702–37710. doi: 10.1074/jbc.M204478200. [DOI] [PubMed] [Google Scholar]
- 24.Burmeister JS, Olivier LA, Reichert WM, Truskey GA. Application of total internal reflection fluorescence microscopy to study cell adhesion to biomaterials. Biomaterials. 1998;19(4-5):307–325. doi: 10.1016/s0142-9612(97)00109-9. [DOI] [PubMed] [Google Scholar]
- 25.Axelrod D. Total internal reflection fluorescence microscopy in cell biology. Traffic. 2001;2(11):764–774. doi: 10.1034/j.1600-0854.2001.21104.x. [DOI] [PubMed] [Google Scholar]
- 26.Small JV, Kaverina I. Microtubules meet substrate adhesions to arrange cell polarity. Curr Opin Cell Biol. 2003;15(1):40–47. doi: 10.1016/s0955-0674(02)00008-x. [DOI] [PubMed] [Google Scholar]
- 27.Bowser DN, Khakh BS. Two forms of single-vesicle astrocyte exocytosis imaged with total internal reflection fluorescence microscopy. Proc Natl Acad Sci U S A. 2007;104(10):4212–4217. doi: 10.1073/pnas.0607625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bezzi P, Gundersen V, Galbete JL, Seifert G, Steinhauser C, Pilati E, Volterra A. Astrocytes contain a vesicular compartment that is competent for regulated exocytosis of glutamate. Nat Neurosci. 2004;7(6):613–620. doi: 10.1038/nn1246. [DOI] [PubMed] [Google Scholar]
- 29.Marchaland J, Cali C, Voglmaier SM, Li H, Regazzi R, Edwards RH, Bezzi P. Fast subplasma membrane Ca2+ transients control exo-endocytosis of synaptic-like microvesicles in astrocytes. J Neurosci. 2008;28(37):9122–9132. doi: 10.1523/JNEUROSCI.0040-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whitehead SS, Blaney JE, Durbin AP, Murphy BR. Prospects for a dengue virus vaccine. Nat Rev Microbiol. 2007;5(7):518–528. doi: 10.1038/nrmicro1690. [DOI] [PubMed] [Google Scholar]
- 31.Bhatt S, Gething PW, Brady OJ, Messina JP, Farlow AW, Moyes CL, Drake JM, Brownstein JS, Hoen AG, Sankoh O, Myers MF, George DB, Jaenisch T, Wint GR, Simmons CP, et al. The global distribution and burden of dengue. Nature. 2013;496(7446):504–507. doi: 10.1038/nature12060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu SJ, Grouard-Vogel G, Sun W, Mascola JR, Brachtel E, Putvatana R, Louder MK, Filgueira L, Marovich MA, Wong HK, Blauvelt A, Murphy GS, Robb ML, Innes BL, Birx DL, et al. Human skin Langerhans cells are targets of dengue virus infection. Nat Med. 2000;6(7):816–820. doi: 10.1038/77553. [DOI] [PubMed] [Google Scholar]
- 33.Flipse J, Wilschut J, Smit JM. Molecular mechanisms involved in antibody-dependent enhancement of dengue virus infection in humans. Traffic. 2013;14(1):25–35. doi: 10.1111/tra.12012. [DOI] [PubMed] [Google Scholar]
- 34.Pokidysheva E, Zhang Y, Battisti AJ, Bator-Kelly CM, Chipman PR, Xiao C, Gregorio GG, Hendrickson WA, Kuhn RJ, Rossmann MG. Cryo-EM reconstruction of dengue virus in complex with the carbohydrate recognition domain of DC-SIGN. Cell. 2006;124(3):485–493. doi: 10.1016/j.cell.2005.11.042. [DOI] [PubMed] [Google Scholar]
- 35.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfherer C, Finke J, Sun W, Eller MA, Pattanapanyasat K, Sarasombath S, Birx DL, Steinman RM, Schlesinger S, Marovich MA. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. 2003;197(7):823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL, Arenzana-Seisdedos F, Despres P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. Embo Rep. 2003;4(7):723–728. doi: 10.1038/sj.embor.embor866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mitchell DA, Fadden AJ, Drickamer K. A novel mechanism of carbohydrate recognition by the C-type lectins DC-SIGN and DC-SIGNR - Subunit organization and binding to multivalent ligands. Journal of Biological Chemistry. 2001;276(31):28939–28945. doi: 10.1074/jbc.M104565200. [DOI] [PubMed] [Google Scholar]
- 38.van der Schaar HM, Rust MJ, Chen C, van der Ende-Metselaar H, Wilschut J, Zhuang X, Smit JM. Dissecting the cell entry pathway of dengue virus by single-particle tracking in living cells. PLoS Pathog. 2008;4(12):e1000244. doi: 10.1371/journal.ppat.1000244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jaqaman K, Kuwata H, Touret N, Collins R, Trimble WS, Danuser G, Grinstein S. Cytoskeletal control of CD36 diffusion promotes its receptor and signaling function. Cell. 2011;146(4):593–606. doi: 10.1016/j.cell.2011.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Joglekar AP, Salmon ED, Bloom KS. Counting kinetochore protein numbers in budding yeast using genetically encoded fluorescent proteins. Method Cell Biol. 2008;85:127. doi: 10.1016/S0091-679X(08)85007-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwille P. Fluorescence correlation spectroscopy and its potential for intracellular applications. Cell Biochem Biophys. 2001;34(3):383–408. doi: 10.1385/CBB:34:3:383. [DOI] [PubMed] [Google Scholar]
- 42.Pero JK, Haas EM, Thompson NL. Size dependence of protein diffusion very close to membrane surfaces: Measurement by total internal reflection with fluorescence correlation spectroscopy. J Phys Chem B. 2006;110(22):10910–10918. doi: 10.1021/jp056990y. [DOI] [PubMed] [Google Scholar]
- 43.Izzard CS, Lochner LR. Cell-to-substrate contacts in living fibroblasts: an interference reflexion study with an evaluation of the technique. J Cell Sci. 1976;21(1):129–159. doi: 10.1242/jcs.21.1.129. [DOI] [PubMed] [Google Scholar]
- 44.Mosquera JA, Hernandez JP, Valero N, Espina LM, Anez GJ. Ultrastructural studies on dengue virus type 2 infection of cultured human monocytes. Virol J. 2005;2:26. doi: 10.1186/1743-422X-2-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kuhn RJ, Zhang W, Rossmann MG, Pletnev SV, Corver J, Lenches E, Jones CT, Mukhopadhyay S, Chipman PR, Strauss EG, Baker TS, Strauss JH. Structure of dengue virus: implications for flavivirus organization, maturation, and fusion. Cell. 2002;108(5):717–725. doi: 10.1016/s0092-8674(02)00660-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chojnacki J, Staudt T, Glass B, Bingen P, Engelhardt J, Anders M, Schneider J, Muller B, Hell SW, Krausslich HG. Maturation-dependent HIV-1 surface protein redistribution revealed by fluorescence nanoscopy. Science. 2012;338(6106):524–528. doi: 10.1126/science.1226359. [DOI] [PubMed] [Google Scholar]
- 47.O’Reilly MK, Paulson JC. Siglecs as targets for therapy in immune-cell-mediated disease. Trends Pharmacol Sci. 2009;30(5):240–248. doi: 10.1016/j.tips.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7(4):255–266. doi: 10.1038/nri2056. [DOI] [PubMed] [Google Scholar]
- 49.McLean MA, Rajfur Z, Chen Z, Humphrey D, Yang B, Sligar SG, Jacobson K. Mechanism of chromophore assisted laser inactivation employing fluorescent proteins. Anal Chem. 2009;81(5):1755–1761. doi: 10.1021/ac801663y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thompson NL, Pearce KH, Hsieh HV. Total internal reflection fluorescence microscopy: application to substrate-supported planar membranes. Eur Biophys J. 1993;22(5):367–378. doi: 10.1007/BF00213560. [DOI] [PubMed] [Google Scholar]
- 51.Serge A, Bertaux N, Rigneault H, Marguet D. Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat Meth. 2008;5(8):687–694. doi: 10.1038/nmeth.1233. [DOI] [PubMed] [Google Scholar]
- 52.Wahala WM, Kraus AA, Haymore LB, Accavitti-Loper MA, de Silva AM. Dengue virus neutralization by human immune sera: role of envelope protein domain III-reactive antibody. Virology. 2009;392(1):103–113. doi: 10.1016/j.virol.2009.06.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bolte S, Cordelieres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc-Oxford. 2006;224:213–232. doi: 10.1111/j.1365-2818.2006.01706.x. [DOI] [PubMed] [Google Scholar]
- 54.Manders EM, Stap J, Brakenhoff GJ, van Driel R, Aten JA. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J Cell Sci. 1992;103(Pt 3):857–862. doi: 10.1242/jcs.103.3.857. [DOI] [PubMed] [Google Scholar]
- 55.Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151(2):182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.