

Abstract
The over-expression of NF-κB signalling in both muscle and immune cells contribute to the pathology in dystrophic muscle. The anti-inflammatory properties of glucocorticoids, mediated predominantly through monomeric glucocorticoid receptor inhibition of transcription factors such as NF-κB (transrepression), are postulated to be an important mechanism for their beneficial effects in Duchenne muscular dystrophy. Chronic glucocorticoid therapy is associated with adverse effects on metabolism, growth, bone mineral density and the maintenance of muscle mass. These detrimental effects result from direct glucocorticoid receptor homodimer interactions with glucocorticoid response elements of the relevant genes. Compound A, a non-steroidal selective glucocorticoid receptor modulator, is capable of transrepression without transactivation. We confirm the in vitro NF-κB inhibitory activity of compound A in H-2Kb-tsA58 mdx myoblasts and myotubes, and demonstrate improvements in disease phenotype of dystrophin deficient mdx mice. Compound A treatment in mdx mice from 18 days of post-natal age to 8 weeks of age increased the absolute and normalized forelimb and hindlimb grip strength, attenuated cathepsin-B enzyme activity (a surrogate marker for inflammation) in forelimb and hindlimb muscles, decreased serum creatine kinase levels and reduced IL-6, CCL2, IFNγ, TNF and IL-12p70 cytokine levels in gastrocnemius (GA) muscles. Compared with compound A, treatment with prednisolone, a classical glucocorticoid, in both wild-type and mdx mice was associated with reduced body weight, reduced GA, tibialis anterior and extensor digitorum longus muscle mass and shorter tibial lengths. Prednisolone increased osteopontin (Spp1) gene expression and osteopontin protein levels in the GA muscles of mdx mice and had less favourable effects on the expression of Foxo1, Foxo3, Fbxo32, Trim63, Mstn and Igf1 in GA muscles, as well as hepatic Igf1 in wild-type mice. In conclusion, selective glucocorticoid receptor modulation by compound A represents a potential therapeutic strategy to improve dystrophic pathology.
Keywords: Duchenne muscular dystrophy, glucocorticoids, glucocorticoid receptor, transrepression NF-κB, compound A
Introduction
Duchenne muscular dystrophy (DMD) results from mutations in the DMD gene, located at Xp21.2 [1], and subsequent absence of the subsarcolemmal protein dystrophin [2,3]. Dystrophin is a component of the dystrophin–glycoprotein complex (DGC) that provides a scaffold between the extracellular matrix (ECM) and the cytoskeleton. Impaired DGC function compromises the structural and mechanical integrity of skeletal muscle, alters signalling pathways within the muscle, leading to contraction-induced injury, reduces cell viability and ultimately causes myofibre necrosis and muscle inflammation [4]. The nature and sequence of events that occur during the initiation and perpetuation of this inflammatory process in dystrophic muscle remains controversial. Glucocor-ticoids (GCs) are the only therapy shown to alter the natural history of DMD [2,3,5] and modulation of the inflammatory response is thought to underlie their efficacy. The anti-inflammatory properties of GCs are due to GC-bound glucocorticoid receptor (GR) monomer interference with pro-inflammatory transcription factors such as NF-κB (transrepression), whereas adverse metabolic alterations and detrimental changes in muscle and bone homeostasis [6] are due to GC-bound GR dimer interactions with GC response elements (GRE) mediating the transcription of the relevant genes (transactivation) [7].
In DMD, NF-κB, a transcription factor for many pro-inflammatory genes, promotes muscle damage and fibrosis, impairs myofibre regeneration and accelerates satellite cell senescence [8–11]. Numerous studies support targeting of NF-κB as a therapeutic approach in DMD [12–21]. Compound A (CpdA), or 2-(4-acetoxyphenyl)-2-chloro-N -methyl-ethylammonium chloride [22]—a non-steroidal selective GR modulator (SGRM)—is the first natural compound with the ability to selectively induce GR-mediated transrepression. The molecular properties allowing CpdA to selectively signal through the GR, and its efficacy in animal models of inflammatory and immunological disorders, have been studied extensively [23–30]. CpdA has the theoretical potential to produce the beneficial effects of GCs in DMD, including NF-κB modulation [31], without the well-recognized adverse effects of GCs that often limit their therapeutic use. The aims of this study were to: test the beneficial potential of CpdA in the mdx mouse model of DMD; investigate the mechanisms by which these beneficial effects are achieved; and compare and contrast the effects of CpdA and prednisolone (PNSL), a classical GC, on mediators of muscle disease.
Materials and methods
Reagents/equipment were acquired through Life Technologies (Grand Island, NY, USA) unless stated otherwise. A complete description of the materials and methods is provided in the supplementary material.
Animals
Male wild-type (WT; C57BL/10ScSn/J ) mice and mdx (C57BL/10ScSn-mdx/J ) mice were obtained from the Jackson Laboratory (Bar Harbor, ME, USA) and housed/bred at the Veterans Affairs Animal Facility. All animal experiments were conducted in accordance with our IACUC guidelines under approved protocols.
Experimental design in animal studies and tissue preparation
All mouse studies followed a common schedule (Figure 1). Two studies were performed, with treatment groups outlined in Table S1 (see supplementary material). Intraperitoneal CpdA was favoured over oral CpdA because, at a dose of 7.5 mg/kg/day, intraperitoneal CpdA significantly reduced proinflammatory cytokine (IL-6, TNF, CCL2, IFNγ and IL-12p70) levels in gastrocnemius (GA) muscle. The 5.0 mg/kg/day dose of PNSL was chosen based on published FDA guidelines and our own experience with this dose in this model [32,33]. All animals were randomized to their treatment groups, based on body weight. Acquisition and analysis of all functional, imaging and histological data was performed in a blinded fashion.
Figure 1.
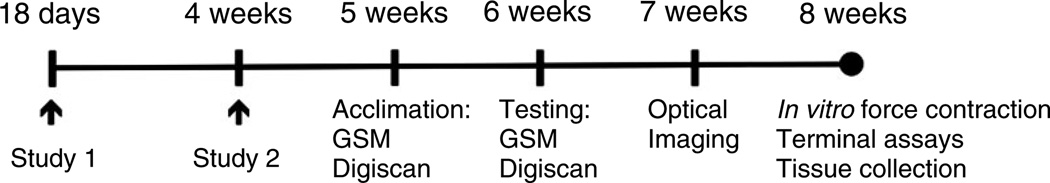
Animal studies experimental design. Study 1 commenced at 18 days and study 2 at 4 weeks. Treatments were given daily and weights measured at commencement of study and then weekly. Studies 1 and 2 differ with regard to assays performed and numbers per group. GSM, grip strength measurement; Digiscan, open field behavioural activity measurements. Details of treatments of groups are provided in Table S1 (see Supplementary material)
Grip strength measurement (GSM)
Forelimb and hindlimb GSMs were performed as previously described [34], but with an increase in the acclimation period from 3 to 5 consecutive days. Absolute and normalized [to body weight (BW)] GSMs were measured.
Open-field behavioural activity
Voluntary locomotor activity was measured using an open-field behavioural activity assessment (Digiscan) apparatus (Omnitech Electronics, Columbus, OH, USA), as previously described [34]. Mice were acclimatized for 4 days before data collection every 10 min over a 1 h period each day for 4 consecutive days.
Measurement of histopathological parameters and serum creatine kinase (CK)
Assessment of pathology in WT and mdx mice following treatment was performed in paraffin-embedded right tibialis anterior (TA) cross-sections, using haema-toxylin and eosin (H&E) staining. Histological evaluations were performed in a blinded manner, using coded slides. Quantitative stereology was performed on a section of the entire TA (non-overlapping fields captured by a ×40 objective). Histological parameters were defined according to TREAT-NMD SOP criteria (Treat-NMD SOP No. DMD_M.1.2.007). CK determination was performed as previously described [34].
Quantitative real-time polymerase chain reaction (qPCR)
RNA was isolated from cells or frozen tissue using an integrated Trizol and RNeasy minikit (Qiagen, Valencia, CA, USA) protocol with DNase I treatment. cDNA was obtained using Reverse Transcription System (Promega, Madison, WI). All PCR reactions were carried out using Taqman Gene Expression Master Mix, except Spp1 and Hprt gene analysis (SYBR Green PCR Master Mix). Hprt was used as the housekeeping gene in all qPCR studies. Relative quantification was used in all except in atrophy studies, where relative expression software tool (REST) was used. Comparisons were made between vehicle-treated WT and mdx mice as well as between the vehicle and other treatment groups. For studies of the expression of genes determining muscle mass, GA muscle was used for all genes except Foxo3 , where the quadriceps was used due to inadequate mRNA from GA to complete all studies.
Taqman Gene Expression Assays, SYBR Green primer pairs, and thermal cycle conditions are presented in Table S2 (see supplementary material).
Flow cytometry
A fluorescence-activated cell sorting (FACS) system (BD Biosciences, San Jose, CA, USA) was used to quantify cytokines (IL-6, IL-10, CCL2, TNF, IFNγ, IL-12p70) in protein lysates from pulverized GA with the Cytometric Bead Array Mouse Inflammation Kit, measured according to the manufacturer’s instructions.
Cell culture
Conditionally immortalized H-2kb-tsA58 (H-2k) mdx myoblasts were cultured as previously described [35,36]. The growth medium contained Dulbecco’s modified Eagle’s medium (DMEM), 20% fetal bovine serum (FBS), 2% chick embryo extract (US Biologicals, Boston, MA, USA), 2% l-glutamine, 1% penicillin/streptomycin and 0.02 µg/ml IFNγ (Millipore, Billerica, MA, USA) at 33°C with 10% CO2. Differentiation into myotubes was achieved by removing the chick embryo extract, replacing the 20% FBS with 5% horse serum, and incubating at 37°C with 5% CO2 for at least 96 h. AtT-20/D16v–F2 murine pituitary corticotrophs (ATCC, Manassas, VA, USA) were cultured as previously described [37].
Western blotting
H-2K mdx myoblasts were pretreated with 1% DMSO, CpdA (4 µm) or PNSL (10 µm) for 4 h, followed by stimulation with 10 ng/ml TNF for 30 min (non-stimulated DMSO-treated groups were also included). Myotubes were treated for 24 h before stimulation for 24 h. Nuclear and cytoplasmic fractions were then obtained, using a p65 (RelA) Translocation Assay Kit (Five Photons Biochemicals, San Diego, CA, USA) according to the manufacturer’s instructions; 10 µl nuclear fraction and 15 µl cytoplasmic fraction from each treatment was used for the western blot, which was performed as previously described [38]. The primary and secondary antibodies used for immunoblotting are presented in Table S3 (see supplementary material). Densitometry analysis of p65 nuclear and cytoplasmic blots, normalized to both Actin and Lamin A, was used to assess p65 nuclear:cytoplasmic ratios.
Pituitary corticotroph studies
AtT-20 cells were treated with 1% DMSO, PNSL (1 or 10 µm) or CpdA (1 or 4 µm) for 24 h. The media were then collected for ACTH measurement with Mouse/Rat ACTH Ultra Sensitive lumELISA (Calbiotech, Spring Valley, CA, USA) according to the manufacturer’s instructions. Each well was then washed with phosphate-buffered saline (PBS) before the addition of Trizol for RNA extraction.
Live animal optical imaging
Quantification of inflammation in limb muscles of mice was achieved using live-animal optical imaging of cathepsin-B enzyme activity, using a caged near-infrared substrate as previously described [39], with n =7–10/group.
Osteopontin (OPN) ELISA assay
Quantification of OPN in protein lysates from GA muscles was performed using a commercial kit (Mouse Osteopontin Quantikine ELISA Kit, R&D Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions.
Statistical analysis
Two-tailed Student’s t-test was used for experiments involving two groups. One-way ANOVA with Dunnett’s post-test comparison against a control was used for experiments involving more than two groups. Relative Expression Software Tool or non-parametric one-way ANOVA with a Kruskal–Wallis comparison of relative quantification (RQ) values were used for qPCR. Two-way ANOVA with Bonferroni post-test comparison with vehicle (VEH)-treated controls was performed in weekly BW measurements and lengthening–contraction studies involving different time points. The relationships between OPN levels and percentage necrotic fibres in the GA muscles of mdx mice were analysed using Pearson correlation. Significance was defined as p < 0.05. Prism 5 statistical software was used for analyses.
Results
In vitro studies
CpdA reduces TNF-induced p65 nuclear translocation in H-2K mdx muscle cells
Western blot analysis was used to assess the effect of CpdA on p65 nuclear translocation in pretreated H-2K mdx muscle cells in response to 10 ng/ml TNF stimulation (Figure 2A, B). The nuclear:cytoplasmic ratios were reduced by −80% in control DMSO-treated/non-TNF-stimulated (TNF–) compared to DMSO-treated/TNF-stimulated (TNF+) myoblasts, and by −40% in both CpdA-treated/TNF-stimulated (CpdA) and PNSL-treated/TNF-stimulated (PNSL) myoblasts compared with TNF+ myoblasts. Lower ratios were also observed in myotubes: TNF– (−75%), CpdA (−48%) and PNSL (−50.0%). Reduced TNF-induced p65 nuclear translocation in H-2K muscle cells following CpdA treatment was also demonstrated using immunofluorescence (Figure S2).
Figure 2.

Western blot with densitometry analysis of p65 nuclear translocation in H-2K mdx (A) myoblasts and (B) myotubes. CpdA reduces TNF-induced p65 nuclear translocation in H-2K mdx myoblasts and myotubes. H-2K muscle cells were pretreated with 1% DMSO, 10 µm PNSL or 4 µm CpdA before stimulation with 10 ng/ml TNF, as described in Materials and methods. The study included a DMSO-treated, non-stimulated control. A minimum of three independent experiments were performed. Densitometry measurements are expressed as mean ± SEM. Statistical analysis of densitometry was performed using one-way ANOVA and Dunnett’s post-test comparison with the DMSO-treated, non-stimulated controls. TNF–, DMSO-treated, non-stimulated; TNF+, DMSO-treated, TNF-stimulated; PNSL, PNSL-treated, TNF-stimulated; CpdA, CpdA-treated, TNF-stimulated: *p < 0.05; **p < 0.01; ***p < 0.001
CpdA does not induce GR-related transactivation of genes
To confirm the absence of CpdA-induced GR–GRE interactions, we studied the effects of CpdA in vitro on the expression of Pomc and Sgk1 [37]—genes involved in the hypothalamic–pituitary–adrenal (HPA) axis feedback regulation—in AtT-20/D16v–F2 cells (Figure 3A, B). Pomc gene expression was not downregulated by CpdA (at 4 or 1 µm), whereas PNSL at 10 and 1 µm decreased expression of Pomc by −60% and −63%, respectively (Figure 3A). Similarly, Sgk1 gene expression was not different from control corticotrophs following CpdA treatment. PNSL at 10 and 1 µm up-regulated Sgk1 gene expression by 11- and 8-fold, respectively. ACTH secretion by AtT-20/D16v–F2 cells (Figure 3C) was not changed following either CpdA treatments, whereas both PNSL concentrations reduced ACTH levels by approximately −70%. No dose–response effect was observed for Pomc or Sgk1 gene expression or ACTH levels with PNSL.
Figure 3.

(A) Pomc gene expression, (B) Sgk1 gene expression and (C) media ACTH protein levels in experiments involving AtT-20/D16v–F2 pituitary corticotrophs. CpdA does not cause GR-related transactivation of genes (Pomc and Sgk1) involved in the feedback regulation of the hypothalamic–pituitary–adrenal (HPA) axis, and therefore preserves ACTH secretion in AtT-20/D16v–F2 murine corticotrophs. AtT-20/D16v–F2 cells were pretreated with 1% DMSO, 1 or 10 µm PNSL or 1 or 4 µm CpdA, as described in Materials and methods. Three independent experiments were performed for each study. Measurements are expressed as mean ± SEM; *p < 0.05; **p< 0.01; ***p < 0.001
In vivo studies with CpdA in mdx mice
The following results are described for study 1 and the experimental design is outlined in the supplementary material (Table S1, Figure S1).
CpdA does not alter tibial length or the normal pattern of BW gains in WT or mdx mice
Tibial length was used to assess the effects of drug treatment on long bone growth (Figure 4A, B). PNSL5.0 markedly reduced tibial length in both WT and mdx mice. This was not observed with CpdA7.5 treatment. The gain in BW of both WT and mdx mice (Figure 4C, D) treated with daily PNSL was reduced compared with the VEH-treated mice. In contrast, CpdA did not reduce BW gains except in mdx mice treated with CpdA7.5 at one single time point of 5 weeks.
Figure 4.

(A, B) Tibial length and (C, D) weekly BWs were compared between CpdA and PNSL in WT and mdx mice, in both of which CpdA preserves long bone growth and patterns of weight gain. All comparisons were made with VEH-treated WT or mdx controls. In all analyses, n = 15/group. Measurements are expressed as mean ± SEM; all drug doses in mg/kg/day: *p < 0.05; **p < 0.01; ***p< 0.001
CpdA increases GSMs in mdx mice
GSMs were performed at 6 weeks of age (Figure 5A – D). As expected, all GSM parameters were reduced in mdx compared to WT mice. In mdx mice, absolute and normalized forelimb GSMs were improved when compared to VEH treatment, following CpdA3.75 (+13% and +22%), CpdA7.5 (+21% and +30%) and PNSL5.0 (+15% and +50%) treatments. Improvement in absolute and normalized himdlimb GSM was also observed for mdx mice in the CpdA7.5 group (+19% and +28%), but only normalized hindlimb was improved following CpdA3.75 (+19%) and PNSL5.0 (+40%) treatments. In WT mice, CpdA7.5 did not affect GSM, whereas PNSL5.0 improved normalized forelimb (+12%) but reduced absolute hindlimb (−13%). Ex vivo measurements of maximal isometric force, specific force, and force following sequential lengthening-contractions in the EDL muscles of both mdx and WT mice were not changed following CpdA treatment (Figure S3).
Figure 5.

(A–D) GSM, (E–I) absolute voluntary locomotor activity assessed by open-field behavioural activity measurements, and (J–L) voluntary locomotor activity normalized to movement time in WT and mdx mice. CpdA has positive effects on grip strength measurement (GSM) and voluntary locomotor activity. Absolute and normalized (to BW at the start of the testing period) strength was improved by intrperitoneal CpdA 7.5 mg/kg/day in both forelimbs and hindlimbs, while PNSL oral 5.0 mg/kg/day reduced absolute hindlimb GSM in WT mice. In mdx mice, CpdA 7.5 mg/kg/day increased the horizontal activity normalized for movement time. All comparisons were made with VEH-treated controls. Differences in all GSM parameters existed between WT and mdx mice. All measurements are expressed as mean ± SEM; all groups, n = 15; all drug doses in mg/kg/day. See Materials and methods for details of drug treatments and GSM testing. GSMs were performed by the same individual, who was blinded to mouse strain and treatment group: *p < 0.05, **p < 0.01; ***p < 0.001
CpdA increases open-field locomotor activity in mdx mice
We used Digiscan to study the effects of CpdA on open-field behavioural activity. All measurement parameters were expressed as absolute values (Figure 5E – I). Quantification of horizontal activity, vertical activity and total distance over time for each mouse was obtained by normalizing to the movement time (Figure 5J – L). There was significant intragroup variation in all parameters. Differences were observed between WT and mdx mice, with WT mice having shorter rest times, greater horizontal activity and greater total distance. In WT mice, CpdA7.5 reduced horizontal activity and total distance. In mdx mice, the PNSL5.0-treated group had longer rest times. For the time that they were moving, WT mice exhibited greater horizontal and vertical activity than mdx mice. There was no difference in the distance/unit time between WT and mdx mice. CpdA and PNSL treatment in WT mice reduced total distance/s compared to the VEH-treated group. CpdA and PNSL treatment in mdx mice resulted in greater horizontal activity/s, but no difference in vertical activity/s or total distance/s.
CpdA reduces muscle inflammation in mdx mice
Muscle inflammation in vivo was quantified using live-animal optical imaging to measure photon intensity (an indicator of cathepsin-B enzyme activity) [39] in the limb muscles of mdx mice, using ProSense-680 (Figure 6). VEH-treated mdx mice had higher photon intensity measurements in both the forelimbs and hindlimbs when compared to VEH-treated WT mice. Treatment of mdx mice with CpdA7.5 and PNSL5.0 attenuated photon intensity in both the forelimb and hindlimb muscles when compared to VEH-treated mdx mice.
Figure 6.

Quantification of inflammation in (A) forelimb and (B) hindlimb muscles by measurement of cathepsin-B activity via live-animal optical imaging of WT and mdx mice. CpdA reduces photon intensity, a surrogate marker for inflammation, in both the forelimb and hindlimb muscles of mdx mice. Measurements were performed at 7 weeks of age and expressed as photon intensity (photon count/mm2). Comparisons were made between VEH-treated WT and VEH-treated mdx groups. Within mdx groups, comparisons were made with the VEH-treated group. All measurements are expressed as mean ± SEM; WT VEH, n = 7; mdx VEH, n = 8; mdx CpdA 7.5, n = 10; mdx PNSL 5.0, n = 9; all drug doses in mg/kg/day: *p < 0.05; **p < 0.01; ***p < 0.001
CpdA and PNSL effects on histopathological parameters in the TA muscle of mdx mice
Histological assessment of H&E sections were performed on the TA muscles harvested from 8 week-old mdx mice. Figure 7 shows representative H&E sections from the TA muscle and the percentage change (from the mean) in each parameter between VEH-treated mdx mice and the other mdx groups. Treatment with CpdA7.5 reduced degenerative myofibres (−52%), regeneration (−43%) and inflammatory infiltration (−55%). PNSL5.0 increased the percentage of degenerative fibres (+54%), which was associated with reduced regenerative fibres (−59%). Despite the increased necrosis, there was reduced inflammatory infiltration (−27%).
Figure 7.

Percentage of (A) degenerative fibres, (B) regenerative fibres and (C) inflammatory foci in the TA muscle of mdx mice, with representative H&E-stained sections from VEH-, CpdA- and PNSL-treated mice. CpdA did not improve histopathological parameters in the tibialis anterior (TA) muscles of mdx mice when treatment was commenced before the peak necrosis period (18 days). Comparisons were made with the VEH-treated group and percentage changes in parameters were calculated from the means. Histopathological parameters were scored on one cross-section of an entire TA at ×40 magnification in a blinded fashion, using an accepted standard operating protocol from Treat-NMD, as described in Materials and methods. Measurements are expressed as mean ± SEM; all groups, n = 12; all drug doses in mg/kg/day: *p < 0.05; **p < 0.01; ***p < 0.001
CpdA reduces inflammatory cytokine protein levels in GA muscles
The effects of CpdA and PNSL on inflammation in the muscles of mdx mice were quantified by analysing gene expression in GA muscles for the cytokines Il6 , Tnf and Ccl2 (Figure 8A). Gene expression was measured using qPCR and expressed as RQ. There were no differences in RQ between mdx mice groups for gene expression any of the cytokines. All genes discriminated between VEH-treated WT and mdx mice. Protein levels of six inflammatory cytokines were measured in protein lysates from the same GA muscles via FACS (Figure 8B). IL-6, CCL2, TNF, IFNγ and IL-12p70 levels were all reduced in mdx mice treated with CpdA3.75, CpdA7.5 or PNSL5.0. IL-10 levels were not reduced in any of the treatment groups. All cytokines discriminated between VEH-treated WT and mdx mice, except for IL-10.
Figure 8.

(A) GA cytokine gene expression, (B) GA cytokine protein levels assessed via FACS, (C) serum CK levels, (D) GA Spp1 gene expression, (E) GA OPN protein levels and (F) correlation between percentage degenerative fibres in the tibialis anterior and OPN protein levels in the GA were measured and calculated as described in Materials and methods. Inflammatory cytokine protein levels and osteopontin (OPN) gene (Spp1) expression in GA muscles, as well as serum creatine kinase (CK) levels, are reduced by CpdA in mdx mice. Comparisons were made between VEH-treated WT and VEH-treated mdx mice. Within the mdx groups, comparisons are made with the VEH-treated mdx group. Measurements are expressed as mean ± SEM; for CK levels, n = 15/group; cytokine gene expression, n = 5/group; cytokine protein levels, n = 12/group; OPN qPCR, WT, n = 5 and mdx groups, n = 8; OPN protein levels, WT, n = 6 and mdx groups, n = 10. All measurements are expressed as mean ± SEM; all drug doses in mg/kg/day: *p < 0.05; **p < 0.01; ***p < 0.001
CpdA reduces serum CK levels in mdx mice
Serum CK levels were markedly elevated in VEH-treated mdx mice compared to VEH-treated WT mice (Figure 8C). Within the mdx mice groups, all treatments reduced serum CK levels when compared to VEH-treated mdx mice (CpdA3.75, −67%; CpdA7.5, −48%; PNSL5.0, −82%).
CpdA and PNSL have opposite effects on OPN protein levels in the GA muscles of mdx mice
To study the relationship between the effects of CpdA on OPN (Spp1 ) gene expression (Figure 8D) and protein levels (Figure 8E) in the GA muscles of mdx mice, we used qPCR and an OPN ELISA. OPN gene expression was down-regulated in mdx mice treated with CpdA7.5, but not with PNSL5.0. OPN protein levels were higher in the PNSL group compared to the CpdA group. Gene expression and protein levels of OPN were lower in VEH-treated WT mice compared to the VEH-treated mdx mice. There was a significant correlation between OPN protein levels in the GA muscles of and the percentage of necrotic myofibres in the TA muscles of mdx mice (r2 = 0.4147, p < 0.001; Figure 8F).
CpdA does not affect the weights of the GA, TA and EDL muscles in mdx mice
PNSL5.0 reduced the weights of the GA, TA and EDL muscles in both WT (Figure 9A) and mdx mice (Figure 9B). CpdA7.5 did not cause any decrease in TA or EDL weights in either WT or mdx mice. The average weights of the GA (−23% and −29%) and TA (−21% and −22%) muscles were reduced in PNSL5.0-treated WT and mdx mice. The GA muscles of WT mice were also reduced (−8%) in the CpdA7.5 group, although this was not observed for CpdA in the mdx mice.
Figure 9.

(A, B) Muscle weights and (C, D) atrophy-related gene expression in gastrocnemius (GA) muscles (quadriceps muscles for Foxo3 analysis) were compared in WT and mdx mice, in both of which CpdA has less effect on muscle mass and causes fewer adverse changes in the expression of genes related to the maintenance of muscle mass than PNSL. All comparisons are made with VEH-treated WT or mdx controls. In all analyses, n = 15/group, except in gene expression studies in GA muscles, where WT, n = 5 and mdx,n = 8–10. For Foxo3 gene expression studies in quadriceps muscles, WT and mdx,n = 8 or 9. Measurements are expressed as mean ± SEM. GA and tibialis anterior (TA) weights were an average of both left and right muscles, while extensor digitorum longus (EDL) weights were only taken from the left muscle, due to use of the right EDL for in vitro force contraction studies. All drug doses in mg/kg/day: *p < 0.05; **p < 0.01; ***p < 0.001
CpdA and PNSL have differential effects on the expression of atrophy-related genes in GA muscles of WT and mdx mice
To compare the effects of CpdA and PNSL in normal and dystrophic muscles, the expression of a number of atrophy-related genes was analysed by qPCR in GA muscles and liver of WT (Figure 9C) and mdx mice (Figure 9D). In WT mice, PNSL up-regulated Foxo1 , Foxo3, Fbxo32, Trim63 and Mstn mRNA, while down-regulating Igf1 in both muscle and hepatic tissue. CpdA did not cause any changes to the expression of these genes in WT mice. In mdx mice, the effect of PNSL on the gene expression was less marked, with only up-regulation in Foxo1 and Foxo3 in muscle and down-regulation of hepatic Igf1 . For CpdA, Trim63 was down-regulated in mdx mice.
Discussion
This is the first study demonstrating the efficacy of CpdA in any muscle disease, including DMD. Previous studies have focused on its ability to inhibit NF-κB in immune cells. NF-κB activation occurs via significantly different mechanisms in muscle and immune cells. Contraction-induced injury in muscle is one such example. CpdA possesses anti-inflammatory properties via selective transrepression through the GR. We have shown that CpdA is an inhibitor of NF-κB in both muscle in vitro (C2C12 and H-2K mdx cells) and immune cells in vivo. Our results support conclusions from previous studies [15] that confirm the contribution of NF-κB signalling to the pathogenesis of DMD.
In study 1, CpdA improved absolute and normalized GSM in the forelimbs and hindlimbs of mdx mice.
The improvements in absolute GSM were comparable to and, in the case of the hindlimb, better than that observed in the PNSL-treated group. PNSL5.0 increased necrosis in the TA and did not improve absolute GSM in the hindlimb. Weight-loss in PNSL-treated mice contributed to the perceived improvement in normalized GSM. There were no differences in ex vivo force contraction measurements using the EDL muscle. The difference in GSM and ex vivo force contraction results may reflect the relative contribution of different muscle groups in the performance of the GSM assay, and differences in the sensitivities of the assays. Moreover, GSM is dependent on behavioural aspects of the mice and CpdA may influence behaviour to improve performance in the GSM assay.
There were no statistical differences in histopathology between control and treatment mdx mice groups. The variation in the severity of pathology between individual mice and between muscles of the same mdx mouse [40], as well as the low-level chronic myonecrosis in untreated sedentary mdx mice [41], makes statistical comparison very difficult [34,42]. When comparing the mean values, differences were observed between findings in studies 1 and 2. PNSL increased necrosis and decreased regeneration in study 1, while reducing necrosis and increasing the number of regenerative fibres in study 2 (data not shown). CpdA reduced the amount of necrosis in both studies, but improved regeneration only in study 2. The recognized role that early inflammatory infiltration plays in the removal of necrotic myofibres and signalling for initiating regeneration may explain these differences [43]. In study 1, early PNSL and CpdA treatment may produce negative alterations in the signalling networks between muscle and immune cells to diminish regenerative capacity.
PNSL increased muscle necrosis in study 1, while reducing it in study 2. Some possible explanations include, for example, that mast cells rapidly accumulate and degranulate (by 8 h) in response to damage [40]. They play a pro-inflammatory role in the muscle of adult mdx mice [40,44,45] but their role in very young mdx muscles is not known. Nonetheless, the differential impact of PNSL and CpdA treatments on mast cell degranulation should be considered.
Neutrophil accumulation in damaged muscle occurs early to achieve debris removal and degradation at the site of injury. GCs, unlike CpdA, enhance neutrophil survival by modifying pro-apoptotic signals [46] and this may represent another potential advantage of CpdA and other SGRMs over GCs.
In damaged muscle, the macrophage population response transitions from Th-1-responsive promoting satellite cell activation and proliferation, while inhibiting differentiation, to Th-2-responsive for differentiation, growth and repair [47–49]. Both CpdA and GCs inhibit the master Th-1 transcription factor, T-bet, by a transrepression mechanism [50]. However, GCs inhibit, while CpdA induces, GATA-3 (the master Th-2 transcription) activity, both via opposite effects on p38 MAPK-induced GATA-3 phosphorylation and its nuclear translocation [51]. While both GCs and CpdA ultimately favour a Th-2 over a Th-1 response, GCs achieve this via preferential inhibition of the Th-1 response while CpdA actively promotes the Th-2 response.
Muscle proteolysis through the ubiquitin– proteasome system is a dominant feature of GC-induced muscle atrophy preferentially affecting fast-twitch (type-II) muscle (GA, TA and EDL) fibres [52]. Fbxo32 and Trim63 are two ubiquitin ligases that target protein for degradation in the proteasome machinery and are up-regulated by GCs through Foxo1 , a transcription factor serving as a major switch for enhanced atrophy-related gene expression. As expected, PNSL up-regulated Foxo1 , Foxo3 , Fbxo32 and Trim63 gene expression in WT GA muscles. CpdA did not alter the expression of these genes in WT or mdx mice. Interestingly, only Foxo1 and Foxo3 gene expression was increased in PNSL-treated mdx mice. Enhanced NF-κB activity independently causes muscle atrophy possibly through increased expression of Trim63 [53,54]. NF-κB inhibition by PNSL in mdx GA muscles may account for the more favourable Fbxo32 and Trim63 gene expression profile observed in our study. The combined neutral effect of CpdA in normal muscle and inhibition of NF-κB in dystrophic muscle may explain the net down-regulation in Trim63 gene expression in mdx mice. Muscle catabolism through the autophagy–lysosome pathway is mediated by Foxo3 through Bnip3 [55]. The up-regulation of Foxo3 by PNSL, and the associated increased autophagy, in both WT and mdx may represent a mechanism for the observed relative reduction in myofibre damage and inflammation.
Our results support previous studies confirming the pathological role of GC-induced augmentation of myostatin production in GC myopathy [56]. Myostatin negatively regulates muscle mass by inhibiting satellite cell proliferation and differentiation, reducing protein synthesis and altering muscle metabolism [57]. The up-regulation of Mstn gene expression by PNSL in WT mice was not observed with CpdA treatment. In sarcopenia, myostatin promotes the generation of ROS where NF-κB activity is elevated, and this is reversed in old Mstn null mice [58]. No differences were found in Mstn gene expression in the 8 week-old PNSL-treated and VEH-treated mdx mice in our study. NF-κB inhibition by PNSL may contribute to its neutral effect on Mstn gene expression in this group of mice.
IGF-1, produced predominantly in the liver but also in local tissues (muscle and bone), is a growth factor that stimulates myoblast proliferation and differentiation during myogenesis and muscle regeneration and mediates hypertrophy of growing myofibres [59,60]. GCs suppresses the GH–IGF-1 axis (resulting in reduced hepatic IGF-1 production) and also has a negative effect on muscle IGF-1 production [61]. IGF-1, in turn, plays a critical role in antagonizing the catabolic action of GCs [62]. In our study, PNSL reduced IGF-1 gene expression in WT (liver and GA) and mdx mice (liver), while CpdA had no effect on IGF-1 gene expression.
Increased OPN, predominantly from immune cells and myoblasts, is a striking feature of dystrophic muscles [63]. The presence of OPN appears to: correlate with necrosis [64]; be integral in the facilitation and modulation of the immune response; be essential for regeneration [65]; and mediate fibrosis [63]. In our study, OPN gene (Spp1 ) expression and protein levels in GA muscles were higher in mdx mice compared to WT mice. Interestingly, CpdA treatment in mdx mice, associated with reduced necrosis, reduced Spp1 expression relative to VEH-treated mdx mice. In contrast, PNSL treatment was associated with more necrosis, unaltered Spp1 and higher OPN protein level. Our results support a correlation (r2 = 0.4147) between higher OPN levels and myonecrosis in dystrophic muscle.
We have demonstrated that selective signalling through the GR using CpdA modulates detrimental pathways such as NF-κB, while exerting positive influences on modifier genes (Foxo1 , Foxo3 , Fbxo32 , Trim63 , Mstn, Igf1 and Spp1 ) and improving a range of outcome measures in mdx mice. Further studies investigating the relative contributions of GR signalling in dystrophic muscle would enhance our understanding of the disease and promote more targeted approaches to developing therapies.
Supplementary Material
Acknowledgements
The authors would like to thank Tanya Cohen, Will Coley, Arpana Sali and Aditi Phadke for technical assistance; and Jyoti Jaiswal and Will Coley for helpful discussions. KN is supported by the National Institutes of Health (2R24HD050846-06; R01-AR050478, 5U54HD053177 and K26OD011171), the Muscular Dystrophy Association (translational grant), the US Department of Defense (W81XWH-05-1-0616) and a pilot grant from Parent Project Muscular Dystrophy (PPMD). Tony Huynh would like to thank and acknowledge PPMD for providing the Peter B Weis-man Fellowship for the purposes of completing this work as part of his thesis.
Footnotes
Author contributions
TH, KU, CRH, MH, CJN, GH, MDG, and KN were involved in the conceptualization, design and planning of experiments; TH, KU, JLQ, KST, JHV, and QY were involved in performing the experiments; TH was the principle author of the manuscript; while KU, KST, CRH, MH, GH, MDG and KN provided editing support.
SUPPLEMENTARY MATERIAL ON THE INTERNET
The following supplementary material may be found in the online version of this article:
Supplementary Materials and Methods
Supplementary Results
Figure S1. Dose–response and cell viability curves in myoblasts and myotubes for CpdA and PNSL
Figure S2. Qualitative analysis using immunofluorescence of p65 nuclear translocation in H-2K mdx myoblasts and myotubes
Figure S3Ex vivo measurements of maximal isometric force, specific force and force following sequential lengthening–contractions in EDL muscles of WT and mdx mice
Table S1. Treatment groups for compound A efficacy trials
Table S2. qPCR reagents and thermal cycle conditions
Table S3. Primary and secondary antibodies used in western blot analysis
References
- 1.Hoffman EP, Brown RH, Jr, et al. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. doi: 10.1016/0092-8674(87)90579-4. [DOI] [PubMed] [Google Scholar]
- 2.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol. 2010;9:77–93. doi: 10.1016/S1474-4422(09)70271-6. [DOI] [PubMed] [Google Scholar]
- 3.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol. 2010;9:177–189. doi: 10.1016/S1474-4422(09)70272-8. [DOI] [PubMed] [Google Scholar]
- 4.Gumerson JD, Michele DE. The dystrophin-glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;2011:210797. doi: 10.1155/2011/210797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Manzur AY, Kuntzer T, Pike M, et al. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev. 2008:CD003725. doi: 10.1002/14651858.CD003725.pub3. [DOI] [PubMed] [Google Scholar]
- 6.Pivonello R, De Martino MC, De Leo M, et al. Cushing’s syndrome. Endocrinol Metab Clin North Am. 2008;37:135–149. doi: 10.1016/j.ecl.2007.10.010. ix. [DOI] [PubMed] [Google Scholar]
- 7.De Bosscher K, Haegeman G. Minireview: latest perspectives on antiinflammatory actions of glucocorticoids. Mol Endocrinol. 2009;23:281–291. doi: 10.1210/me.2008-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans NP, Misyak SA, Robertson JL, et al. Immune-mediated mechanisms potentially regulate the disease time-course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. PMR. 2009;1:755–768. doi: 10.1016/j.pmrj.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Monici MC, Aguennouz M, Mazzeo A, et al. Activation of nuclear factor-κB in inflammatory myopathies and Duchenne muscular dystrophy. Neurology. 2003;60:993–997. doi: 10.1212/01.wnl.0000049913.27181.51. [DOI] [PubMed] [Google Scholar]
- 10.Kumar A, Boriek AM. Mechanical stress activates the nuclear factor-KB pathway in skeletal muscle fibers: a possible role in Duchenne muscular dystrophy. FASEB J. 2003;17:386–396. doi: 10.1096/fj.02-0542com. [DOI] [PubMed] [Google Scholar]
- 11.Messina S, Vita GL, Aguennouz M, et al. Activation of NF-κB pathway in Duchenne muscular dystrophy: relation to age. Acta Myol. 2011;30:16–23. [PMC free article] [PubMed] [Google Scholar]
- 12.Reay DP, Yang M, Watchko JF, et al. Systemic delivery of NEMO binding domain/IKKy inhibitory peptide to young mdx mice improves dystrophic skeletal muscle histopathology. Neurobiol Dis. 2011;43:598–608. doi: 10.1016/j.nbd.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peterson JM, Kline W, Canan BD, et al. Peptide-based inhibition of NF-κB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophy. Mol Med. 2011;17:508–515. doi: 10.2119/molmed.2010.00263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang Y, Reay DP, Salay MN, et al. Inhibition of the IKK/NF-kB pathway by AAV gene transfer improves muscle regeneration in older mdx mice. Gene Ther. 2010;17:1476–1483. doi: 10.1038/gt.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Acharyya S, Villalta SA, Bakkar N, et al. Interplay of IKK/NF-κB signaling in macrophages and myofibers promotes muscle degeneration in Duchenne muscular dystrophy. J Clin Invest. 2007;117:889–901. doi: 10.1172/JCI30556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Messina S, Bitto A, Aguennouz M, et al. The soy isoflavone genistein blunts nuclear factor-κB, MAPKs and TNFα activation and ameliorates muscle function and morphology in mdx mice. Neuromusc Disord. 2011;21:579–589. doi: 10.1016/j.nmd.2011.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Evans NP, Call JA, Bassaganya-Riera J, et al. Green tea extract decreases muscle pathology and NF-κB immunostaining in regenerating muscle fibers of mdx mice. Clin Nutr. 2010;29:391–398. doi: 10.1016/j.clnu.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siegel AL, Bledsoe C, Lavin J, et al. Treatment with inhibitors of the NF-κB pathway improves whole body tension development in the mdx mouse. Neuromusc Disord. 2009;19:131–139. doi: 10.1016/j.nmd.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 19.Pan Y, Chen C, Shen Y, et al. Curcumin alleviates dystrophic muscle pathology in mdx mice. Mol Cells. 2008;25:531–537. [PubMed] [Google Scholar]
- 20.Messina S, Bitto A, Aguennouz M, et al. Nuclear factor-KB blockade reduces skeletal muscle degeneration and enhances muscle function in mdx mice. Exp Neurol. 2006;198:234–241. doi: 10.1016/j.expneurol.2005.11.021. [DOI] [PubMed] [Google Scholar]
- 21.Messina S, Altavilla D, Aguennouz M, et al. Lipid peroxidation inhibition blunts nuclear factor-κB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am J Pathol. 2006;168:918–926. doi: 10.2353/ajpath.2006.050673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Bosscher K, Van den Berghe W, Beck IM, et al. A fully dissociated compound of plant origin for inflammatory gene repression. Proc Natl Acad Sci USA. 2005;102:15827–15832. doi: 10.1073/pnas.0505554102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dewint P, Gossye V, De Bosscher K, et al. A plant-derived ligand favoring monomeric glucocorticoid receptor conformation with impaired transactivation potential attenuates collagen-induced arthritis. J Immunol. 2008;180:2608–2615. doi: 10.4049/jimmunol.180.4.2608. [DOI] [PubMed] [Google Scholar]
- 24.Gossye V, Elewaut D, Van Beneden K, et al. A plant-derived glucocorticoid receptor modulator attenuates inflammation without provoking ligand-induced resistance. Ann Rheum Dis. 2009;69:291–296. doi: 10.1136/ard.2008.102871. [DOI] [PubMed] [Google Scholar]
- 25.Gossye V, Elewaut D, Bougarne N, et al. Differential mechanism of NF-κB inhibition by two glucocorticoid receptor modulators in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum. 2009;60:3241–3250. doi: 10.1002/art.24963. [DOI] [PubMed] [Google Scholar]
- 26.Robertson S, Allie-Reid F, Vanden Berghe W, et al. Abrogation of glucocorticoid receptor dimerization correlates with dissociated glucocorticoid behavior of compound a. J Biol Chem. 2010;285:8061–8075. doi: 10.1074/jbc.M109.087866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Loo G, Sze M, Bougarne N, et al. Antiinflammatory properties of a plant-derived, non-steroidal dissociated glucocorticoid receptor modulator in experimental autoimmune encephalomyelitis. Mol Endocrinol. 2010;24:310–322. doi: 10.1210/me.2009-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reber LL, Daubeuf F, Plantinga M, et al. A dissociated glucocor-ticoid receptor modulator reduces airway hyperresponsiveness and inflammation in a mouse model of asthma. J Immunol. 2012;188:3478–3487. doi: 10.4049/jimmunol.1004227. [DOI] [PubMed] [Google Scholar]
- 29.Rauner M, Goettsch C, Stein N, et al. Dissociation of osteogenic and immunological effects by the selective glucocorticoid receptor agonist, compound A, in human bone marrow stromal cells. Endocrinology. 2011;152:103–112. doi: 10.1210/en.2010-0456. [DOI] [PubMed] [Google Scholar]
- 30.Rauch A, Gossye V, Bracke D, et al. An anti-inflammatory selective glucocorticoid receptor modulator preserves osteoblast differentiation. F A S E B J. 2011;25:1323–1332. doi: 10.1096/fj.10-173393. [DOI] [PubMed] [Google Scholar]
- 31.De Bosscher K, Vanden Berghe W, Haegeman G. Crosstalk between nuclear receptors and nuclear factor-κB. Oncogene. 2006;25:6868–6886. doi: 10.1038/sj.onc.1209935. [DOI] [PubMed] [Google Scholar]
- 32.Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. F A S E B J. 2008;22:659–661. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- 33.Sali A, Guerron AD, Gordish-Dressman H, et al. Glucocorticoid-treated mice are an inappropriate positive control for long-term preclinical studies in the mdx mouse. PLoS One. 2012;7:e34204. doi: 10.1371/journal.pone.0034204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spurney CF, Gordish-Dressman H, Guerron AD, et al. Preclinical drug trials in the mdx mouse: assessment of reliable and sensitive outcome measures. Muscle Nerve. 2009;39:591–602. doi: 10.1002/mus.21211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morgan JE, Beauchamp JR, Pagel CN, et al. Myogenic cell lines derived from transgenic mice carrying a thermolabile T antigen: a model system for the derivation of tissue-specific and mutation-specific cell lines. Dev Biol. 1994;162:486–498. doi: 10.1006/dbio.1994.1103. [DOI] [PubMed] [Google Scholar]
- 36.Jat PS, Noble MD, Ataliotis P, et al. Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc Natl Acad Sci USA. 1991;88:5096–5100. doi: 10.1073/pnas.88.12.5096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reiter MH, Vila G, Knosp E, et al. Opposite effects of serum- and glucocorticoid-regulated kinase-1 and glucocorticoids on POMC transcription and ACTH release. Am J Physiol Endocrinol Metab. 2011;301:E336–E341. doi: 10.1152/ajpendo.00155.2011. [DOI] [PubMed] [Google Scholar]
- 38.Rayavarapu S, Coley W, Cakir E, et al. Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin-deficient mdx mouse. Mol Cell Proteom. 2013;12:1061–1073. doi: 10.1074/mcp.M112.023127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baudy AR, Sali A, Jordan S, et al. Non-invasive optical imaging of muscle pathology in mdx mice using cathepsin caged near-infrared imaging. Mol Imaging Biol. 2011;13:462–470. doi: 10.1007/s11307-010-0376-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radley HG, Grounds MD. Cromolyn administration (to block mast cell degranulation) reduces necrosis of dystrophic muscle in mdx mice. Neurobiol Dis. 2006;23:387–397. doi: 10.1016/j.nbd.2006.03.016. [DOI] [PubMed] [Google Scholar]
- 41.Grounds MD, Radley HG, Lynch GS, et al. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol Dis. 2008;31:1–19. doi: 10.1016/j.nbd.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Willmann R, De Luca A, Benatar M, et al. Enhancing translation: guidelines for standard pre-clinical experiments in mdx mice. Neuromuscul Disord. 2012;22:43–49. doi: 10.1016/j.nmd.2011.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tidball JG. Inflammation in skeletal muscle regeneration. In: Schiaffino S, editor. In Skeletal Muscle Repair and Regeneration. Springer Science and Business Media; Dordrecht: 2008. pp. 243–268. [Google Scholar]
- 44.Marques MJ, Ventura Machado R, Minatel E, et al. Disodium cromoglycate protects dystrophin-deficient muscle fibers from leak-iness. Muscle Nerve. 2008;37:61–67. doi: 10.1002/mus.20892. [DOI] [PubMed] [Google Scholar]
- 45.Granchelli JA, Avosso DL, Hudecki MS, et al. Cromolyn increases strength in exercised mdx mice. Res Commun Mol Pathol Pharmacol. 1996;91:287–296. [PubMed] [Google Scholar]
- 46.Saffar AS, Ashdown H, Gounni AS. The molecular mechanisms of glucocorticoids-mediated neutrophil survival. Curr Drug Targets. 2011;12:556–562. doi: 10.2174/138945011794751555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Serrano AL, Mann CJ, Vidal B, et al. Cellular and molecular mechanisms regulating fibrosis in skeletal muscle repair and disease. Curr To p D e v B i o l. 2011;96:167–201. doi: 10.1016/B978-0-12-385940-2.00007-3. [DOI] [PubMed] [Google Scholar]
- 48.Mann CJ, Perdiguero E, Kharraz Y, et al. Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle. 2011;1:21. doi: 10.1186/2044-5040-1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McLennan IS. Degenerating and regenerating skeletal muscles contain several subpopulations of macrophages with distinct spatial and temporal distributions. J Anat. 1996;188(1):17–28. [PMC free article] [PubMed] [Google Scholar]
- 50.Liberman AC, Druker J, Refojo D, et al. Glucocorticoids inhibit GATA-3 phosphorylation and activity in T cells. FA S E B J. 2009;23:1558–1571. doi: 10.1096/fj.08-121236. [DOI] [PubMed] [Google Scholar]
- 51.Liberman AC, Antunica-Noguerol M, Ferraz-de-Paula V, et al. Compound A, a dissociated glucocorticoid receptor modulator, inhibits T-bet (Th1) and induces GATA-3 (Th2) activity in immune cells. PLoS One. 2012;7:e35155. doi: 10.1371/journal.pone.0035155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ruff RL, Weissmann J. Endocrine myopathies. Neurol Clin. 1988;6:575–592. [PubMed] [Google Scholar]
- 53.Cai D, Frantz JD, Tawa NE, Jr, et al. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 54.Mourkioti F, Kratsios P, Luedde T, et al. Targeted ablation of IKK2 improves skeletal muscle strength, maintains mass, and promotes regeneration. J Clin Invest. 2006;116:2945–2954. doi: 10.1172/JCI28721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy. 2008;4:524–526. doi: 10.4161/auto.5905. [DOI] [PubMed] [Google Scholar]
- 56.Ma K, Mallidis C, Bhasin S, et al. Glucocorticoid-induced skeletal muscle atrophy is associated with upregulation of myostatin gene expression. Am J Physiol Endocrinol Metab. 2003;285:E363–E371. doi: 10.1152/ajpendo.00487.2002. [DOI] [PubMed] [Google Scholar]
- 57.Amthor H, Hoogaars WM. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr Gene Ther. 2012;12:245–259. doi: 10.2174/156652312800840577. [DOI] [PubMed] [Google Scholar]
- 58.Sriram S, Subramanian S, Sathiakumar D, et al. Modulation of reactive oxygen species in skeletal muscle by myostatin is mediated through NF-κB. Aging Cell. 2011;10:931–948. doi: 10.1111/j.1474-9726.2011.00734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clemmons DR. Role of IGF-I in skeletal muscle mass maintenance. Trends Endocrinol Metab. 2009;20:349–356. doi: 10.1016/j.tem.2009.04.002. [DOI] [PubMed] [Google Scholar]
- 60.Shavlakadze T, Chai J, Maley K, et al. A growth stimulus is needed for IGF-1 to induce skeletal muscle hypertrophy in vivo . J Cell Sci. 2010;123:960–971. doi: 10.1242/jcs.061119. [DOI] [PubMed] [Google Scholar]
- 61.Gayan-Ramirez G, Vanderhoydonc F, Verhoeven G, et al. Acute treatment with corticosteroids decreases IGF-1 and IGF-2 expression in the rat diaphragm and gastrocnemius. Am J Respir Crit Care Med. 1999;159:283–289. doi: 10.1164/ajrccm.159.1.9803021. [DOI] [PubMed] [Google Scholar]
- 62.Schakman O, Gilson H, Kalista S, et al. Mechanisms of muscle atrophy induced by glucocorticoids. Horm Res. 2009;72(suppl 1):36–41. doi: 10.1159/000229762. [DOI] [PubMed] [Google Scholar]
- 63.Vetrone SA, Montecino-Rodriguez E, Kudryashova E, et al. Osteo-pontin promotes fibrosis in dystrophic mouse muscle by modulating immune cell subsets and intramuscular TGFβ. J Clin Invest. 2009;119:1583–1594. doi: 10.1172/JCI37662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zanotti S, Gibertini S, Di Blasi C, et al. Osteopontin is highly expressed in severely dystrophic muscle and seems to play a role in muscle regeneration and fibrosis. Histopathology. 2011;59:1215–1228. doi: 10.1111/j.1365-2559.2011.04051.x. [DOI] [PubMed] [Google Scholar]
- 65.Uaesoontrachoon K, Wasgewatte Wijesinghe DK, Mackie EJ, et al. Osteopontin deficiency delays inflammatory infiltration and the onset of muscle regeneration in a mouse model of muscle injury. Dis Model Mech. 2012;6:197–205. doi: 10.1242/dmm.009993. e-pub. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.