

Significance
One of the most widespread molecular switches in biochemical pathways is based on the isomerization of the amino acid proline, a process that normally is facilitated by enzymes known as “proline isomerases.” We show that cyclophilin A, one of the most common proline isomerases, acts by a simple mechanism, which we describe as an “electrostatic handle.” In this mechanism, the enzyme creates an electrostatic environment in its catalytic site that rotates a peptide bond in the substrate by pulling the electric dipole associated with the carbonyl group preceding the peptide bond itself. Our results thus identify a specific mechanism by which electrostatics is exploited in enzyme catalysis.
Keywords: enzyme catalysis, NMR spectroscopy
Abstract
Proline isomerization is a ubiquitous process that plays a key role in the folding of proteins and in the regulation of their functions. Different families of enzymes, known as “peptidyl-prolyl isomerases” (PPIases), catalyze this reaction, which involves the interconversion between the cis and trans isomers of the N-terminal amide bond of the amino acid proline. However, complete descriptions of the mechanisms by which these enzymes function have remained elusive. We show here that cyclophilin A, one of the most common PPIases, provides a catalytic environment that acts on the substrate through an electrostatic handle mechanism. In this mechanism, the electrostatic field in the catalytic site turns the electric dipole associated with the carbonyl group of the amino acid preceding the proline in the substrate, thus causing the rotation of the peptide bond between the two residues. We identified this mechanism using a combination of NMR measurements, molecular dynamics simulations, and density functional theory calculations to simultaneously determine the cis-bound and trans-bound conformations of cyclophilin A and its substrate as the enzymatic reaction takes place. We anticipate that this approach will be helpful in elucidating whether the electrostatic handle mechanism that we describe here is common to other PPIases and, more generally, in characterizing other enzymatic processes.
Different families of enzymes, often referred to as “peptidyl-prolyl isomerases” (PPIases), catalyze proline isomerization, a process that involves the interconversion between the cis and trans isomers of the N-terminal amide bond of the amino acid proline (1–3). This isomerization process is an intrinsically slow reaction, typically occurring on the time scale of several minutes under physiological conditions. Hence it often represents a rate-limiting step in biochemical reactions and indeed is used ubiquitously as a molecular switch in regulation (1–7).
The possible mechanisms by which PPIases speed up this reaction have been the subject of intense scrutiny (8–16), although consensus descriptions of such mechanisms have not yet emerged. A question of particular relevance is the specific manner in which the electrostatic field in the catalytic site may facilitate the isomerization reaction. To investigate this problem, we considered the case of cyclophilin A, a member of the cyclophilin family of PPIases (17–20). Previous studies have suggested that conformations resembling those typical of the cis-bound and the trans-bound states are populated through conformational fluctuations in the free state of the enzyme and therefore functional insights into its mechanism of action might be obtained from the study of the free state (21–23).
The approach that we followed in studying the mechanism of action of cyclophilin A is based on the simultaneous determination of the structures of the cis-bound and trans-bound states of the complex between the enzyme and its substrate as the catalytic process takes place. Our results reveal that the mechanism of the reaction involves the presence of an electrostatic field that acts on the N-terminal peptide bond of the proline residue in the substrate and induces the rotation of the electric dipole corresponding to the carbonyl group of the residue preceding the proline. In this sense, the carbonyl group represents a handle operated by an electrostatic field and helps overcome the isomerization barrier.
We investigated the conformational properties of cyclophilin A during the proline isomerization process by using NMR spectroscopy, which can provide atomic-resolution descriptions of the motions of macromolecules in solution (24–32). In our strategy, NMR data are used as replica-averaged structural restraints in molecular dynamics simulations. Such calculations, which in general can include NOE-derived distances (29), S2-order parameters (29), residual dipolar couplings (33–35), and chemical shifts (36–38), are particularly suitable when multiple conformations of a protein are present simultaneously in solution, because these conformations can be determined at the same time (29, 37).
Results and Discussion
Simultaneous Determination of the cis-Bound and trans-Bound States.
To study the proline isomerization process catalyzed by cyclophilin A, we considered the model peptide substrate GSFGPDLRAGD (39, 40). We carried out chemical shift measurements in the bound state during the catalytic reaction (SI Text). In addition, we used NOESY measurements to obtain information about interproton distances (i.e., intermolecular NOE restraints) between the enzyme and the substrate; therefore NOEs were measured as averages over the cis-bound and the trans-bound conformations during the isomerization reaction (SI Text). We then performed molecular dynamics simulations with replica-averaged chemical shifts (37) and intermolecular NOE restraints (29), a technique that enables the information provided by NMR measurements to be incorporated in the structural determination procedure in a manner consistent with the maximum entropy principle (41–43). We used two replicas of the system; the initial structures were chosen with the proline in the model peptide in the cis conformation in the first replica and in the trans conformation in the second replica (SI Text). These calculations resulted in two (cis-bound and trans-bound) conformational ensembles (Fig. 1) with corresponding free-energy landscapes (Fig. 2). The agreement between experimental and calculated intermolecular NOEs and chemical shifts was excellent (Table S1 and Fig. 3). For comparison, we also carried out similar calculations for the free state of the enzyme (Fig. 2; see also Conformational Fluctuations in the Free State).
Fig. 1.
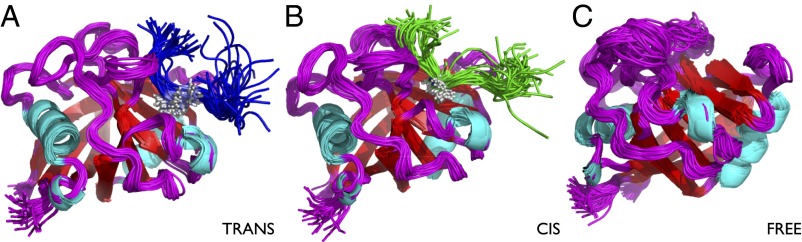
Ensembles of structures representing the conformational fluctuations of cyclophilin A in the trans-bound (A), the cis-bound (B), and the free (C) states. The ensembles have been determined using backbone chemical shifts as replica-averaged restraints (free state) and backbone chemical shifts and interchain NOEs replica-averaged restraints (bound state). The simulations were performed with a modified version of GROMACS, using the Amber99SB*-ILDN force-field and applying the CamShift and NOE restraints over two replicas (37). More details are provided in SI Text.
Fig. 2.

Cyclophilin A samples regions of its conformational space in the absence of the substrate similar to those sampled during the catalytic turnover. Free-energy surfaces for the bound state of cyclophilin A as a function of active site and protein core side chains are shown. (A) Free-energy landscape as a function of the χ3 dihedral angle of GLN63 and the χ2 dihedral angle of LEU98. (B) Free-energy landscape as a function of the χ1 dihedral angle of ARG55 and the χ2 dihedral angle of PHE112. The isolines are plotted at intervals of 2.0 kJ/mol. The contours represent the trans-bound– and cis-bound–specific basins; the red dots represent the free ensemble.
Fig. 3.

Comparison between the experimental and calculated chemical shifts for the bound-state ensemble of cyclophilin A. (A) Cα atoms, (B) Cβ atoms, (C) N atoms, and (D) HN atoms. r, correlation coefficient; sd, SE.
A Possible Electrostatic Handle Mechanism of Catalysis.
To formulate a hypothesis about the mechanism of catalysis, we analyzed the ensemble of conformations representing the bound state of cyclophilin A and its substrate. This ensemble can be divided into cis-bound and trans-bound subensembles. We then considered the overall electrostatic field in the active site of the enzyme (44), prompted by the observation that the presence of a conserved arginine residue at position 55 (R55) is known to play a key role in the function of cyclophilin A (13–15). More specifically, density functional theory (DFT) calculations (SI Text) of the electrostatic field acting on the glycine–proline peptide bond were carried out for the cis-bound and the trans-bound ensembles (Figs. S1–S3). Our results indicate that the z component, defined as the normal to the ring plane defined by the N, Cα, and Cγ atoms of the proline residue, is approximately the same for the cis-bound and the trans-bound states (Fig. 4).
Fig. 4.

(A) Probability distributions of the electrostatic field (in megavolts per centimeter) components along the z axis. The cis-bound ensemble is shown in green and the trans-bound ensemble in blue. The electrostatic field was calculated from the electronic density derived by DFT (SI Text). (B) Illustration of the electrostatic handle mechanism (see also Movie S1). (C) Potential energy surfaces (in kilojoules per mole) of the Ace-Pro-Nme peptide in vacuo with and without an electric field of the magnitude found in the active site of cyclophilin A. The potential energy surfaces were calculated at the same level of accuracy (SI Text) as the electric field of A. The negative value of the z component of the field, which has same magnitude in both the cis-bound and the trans-bound states, has the effect of reducing the potential energy barrier between the cis (ω = 0) and trans (ω = 180°) in the positive direction (from 0° to 180°), whereas it increases the barrier in the negative direction (from 0° to −180°). (D) Schematic illustration of the electrostatic handle mechanism of proline isomerization. The electric field in the catalytic site acts on the electric dipole associated with the carbonyl group of the glycine preceding the proline in the substrate, thus causing a rotation in the ω angle of the peptide bond between the two residues.
Having determined the electrostatic field present in the active site of cyclophilin A during the catalytic process, we investigated its specific effect on the proline isomerization process. To obtain an initial insight into this effect, we performed DFT calculations (SI Text) on a model system, the N-acetyl-l-prolyl-N-methylamide (Ace-Pro-Nme) proline dipeptide, in vacuo and compared the potential energy surface of this system in the presence and absence of an electrostatic field corresponding to that found in the active site of cyclophilin A. The potential energy surfaces for the isomerization process as a function of the ω and ψ angles (Fig. 4) indicate that in the absence of electrostatic fields the trans isomer is about 20 kJ/mol more stable than the cis isomer, with the clockwise (ω = −90°) and counterclockwise (ω = 90°) energy barriers between the cis-bound and trans-bound states being of comparable height (“clockwise” and “counterclockwise” are defined for the trans-to-cis transition) (Fig. 4).
The potential energy surface of the proline dipeptide model in the presence of an electrostatic field of 50 MV/cm along the negative direction of the z axis is shown in Fig. 4. In this case the main effect of the electrostatic field is to reduce the energy barrier strongly, by about 30 kJ/mol, at ω = 90° while slightly increasing the energy barrier at ω = −90°. Furthermore this electrostatic field increases the stability of the cis-bound state by about 10 kJ/mol. Previous studies that used classic molecular dynamics simulations suggested that cyclophilin A catalyzes proline isomerization along a counterclockwise direction for the trans-to-cis transition (14, 15).
Here, our model calculations enable us to put forward the hypothesis that, perhaps not surprisingly, the source of this effect is the electrostatic field generated by the enzyme in its catalytic site and acting on the glycine–proline peptide bond. These results, more in detail, also suggest that the effect of cyclophilin A is to create an electrostatic handle that acts on the electric dipole of the glycine carbonyl group of the glycine–proline substrate (Movie S1), which is the only substantial electric dipole in proximity of the glycine–proline peptide bond, thus stabilizing the transition state in which the dipole is aligned with the field (ω = 90). The lowering of the barrier (i.e., the stabilization of the transition state) is compatible with the experimentally observed speed-up of four to five orders of magnitude (from minutes to milliseconds) of the isomerization process.
To investigate the presence of possible additional effects of the electrostatic field on the electron density in correspondence to the peptide bond, we performed a natural bond orbital analysis (SI Text) that clearly indicated that the electron density along the peptide bond is almost completely unaffected by the presence of the electrostatic field, showing that the nature of the chemical bond remains unchanged.
Validation of the Electrostatic Handle Mechanism with a Thioamide-Substituted Peptide.
To test the electrostatic handle mechanism suggested by the model calculations described above, we selectively altered the electrostatic properties of the handle by replacing the CO group with a CS group in the glycine residue preceding the proline in the substrate. The replacement of an oxygen atom by a sulfur atom modifies the substrate primarily by reducing the electrostatic dipole of the handle (i.e., the CO group in the wild-type peptide and the CS group in the modified peptide) and thus is expected to reduce the catalytic activity of cyclophilin A. Indeed, a calculation of the electrostatic potential charge on the Ace-Pro-Nme proline dipeptide in vacuo indicates that replacing a CO group by a CS group reduces the value of the dipole from 0.65 eÅ for the CO bond to 0.35 eÅ for the CS bond. Assuming that the average electrostatic field in the active site of cyclophilin A is 40 MV/cm, one can estimate the increase in the isomerization barrier associated with the CS replacement to be ∼10 kJ/mol, corresponding to a slowing down of the isomerization process by approximately two orders of magnitude.
We verified that the thioamide modification alters the binding affinity only marginally (Fig. S4), but, consistently with the above prediction, the absence of exchange peaks in the homonuclear NOESY spectrum (Fig. 5) and the absence of cross-peaks in the ZZ-exchange spectrum (Fig. S5) correspond to a lack of proline isomerization in the thioamide-substituted peptide.
Fig. 5.

Homonuclear NOESY spectra of the model peptide (A) and thioamide-substituted peptide (B) in the presence of 20 μM cyclophilin A after a 200-ms mixing time. Exchange peaks, which indicate isomerization, are visible for the model peptide but not for the thioamide-substituted peptide. The sulfur atom is shown in yellow in B.
Further Support for the Electrostatic Handle Mechanism from the R55A Mutant.
To characterize better the specific contribution to the total electrostatic field of the arginine residue at position 55 (R55), which has been proposed to be key in the catalytic process (22, 23), the DFT calculations were repeated over the same ensemble of bound structures but with an R55A mutation. The analysis of the electric field distributions in this case is consistent with the observation of an almost complete loss of enzymatic activity of this mutation (22). Indeed, the z component of the electrostatic field is strongly reduced (Fig. 6). In the R55A variant, the electrostatic field lowers the isomerization barrier by less than 15 kJ/mol, to about half the value in the wild-type R55. These calculations confirmed that R55 plays a key role in the catalysis by generating the electrostatic field that turns the carbonyl group of glycine and by keeping the proline in place by a hydrogen bond with its side chain. Overall, the global effect of the electrostatic field is to reduce the counterclockwise barrier, thus making the isomerization process much more accessible, as well as stabilizing the alternative isomerization state.
Fig. 6.

Comparison between the probability distributions of the electrostatic field component (in megavolts per centimeter) along the z axis for the R55A mutant in the cis-bound and trans-bound states. The corresponding distributions for wild-type R55 (Fig. 4A) are shifted to the left by about 30 MV/cm.
Other PPIases.
To investigate whether the electrostatic handle mechanism is specific for cyclophilin A or is used more generally by other PPIases, we calculated the electrostatic field acting on the carbonyl group in three other structures representing the three major families of PPIases: immunophilins (including cyclophilins), FK506-binding proteins (FKBPs), and parvulins (3). Our results were consistent with those found for cyclophilin A: −19 MV/cm for cyclophilin B (PDB ID code 1VAI), −33 MV/cm for an FKBP (PDB ID code 4ITZ), and −10 MV/cm for Pin1 (PDB ID code 1PIN), a parvulin (Fig. 4A). These values of the electrostatic field indicate, but do not prove, that the electrostatic handle mechanism may be common among PPIases, although other effects also may contribute to the isomerization process in different cases (1–3).
The values of the electrostatic field shown in Fig. 4A for individual structures also illustrate the importance of determining an ensemble of conformations representing the dynamics of the enzyme because individual structures may exhibit low values of the electrostatic field just by chance, thus making it difficult to identify the importance of the electrostatic field in the catalytic mechanism.
Conformational Fluctuations in the Free State.
We then applied the approach we used for the bound states, i.e., using molecular dynamics simulations with chemical shift restraints (but this time without NOE restraints), to characterize the free conformations of cyclophilin A (Fig. 1C). In this case also, the agreement between experimental and calculated chemical shifts was excellent (Fig. S6). Moreover, in the absence of the substrate, residual dipolar couplings (RDCs) were readily obtained for cyclophilin A (SI Text). The free-state ensemble thus was validated by using RDC data, which were not used in the structure calculations (Fig. S7). We found that the Q factor for the X-ray structure of PDB ID code 1OCA (45) is 0.45, whereas that of the ensemble is 0.31.
From relaxation-dispersion measurements of the free (21, 22, 39) and bound (46) states of cyclophilin A, it has been suggested that the conformational fluctuations of these states are similar (21–23). Recently presented crystal structures of the free state (23) showed that two populations could be characterized in terms of different rotameric states of a specific set of amino acids. An analysis of the ensembles determined in this work demonstrates that in the free state of cyclophilin A the cis-bound–like and the trans-bound–like conformations are in conformational exchange (i.e., these functionally relevant conformations already are being sampled in the absence of the substrate). These results are illustrated by plotting the free-energy surface for the free state of cyclophilin A as a function of the rotameric state of four amino acids belonging either to the active site or to the core of the protein. The cis-bound and trans-bound ensembles are clearly included in the free-energy surface of the free enzyme (Fig. 2). Further analysis of the conformational fluctuations of the side chains shows that, in particular for S99 and F113, the free ensemble that we determined is fully consistent with previous results (23) (Fig. S8). The coexistence of cis-bound–like and trans-bound–like conformations in the free state of cyclophilin A is a defining trait of the high conformational mobility of this enzyme in the absence of a substrate.
Concluding Remarks.
To characterize the mechanism by which cyclophilin A catalyzes proline isomerization, we simultaneously determined the cis-bound and trans-bound states of the enzyme as the catalytic reaction takes place (Fig. 1). This result was obtained by using NMR spectroscopy in combination with molecular dynamics simulations. In our approach, the NMR measurements are used as replica-averaged structural restraints in molecular dynamics simulations (29, 37, 41–43). Because the experimental information is used to restrain the average values corresponding to the measured quantities over multiple copies of the protein molecules, it is possible to take into account the conformational flexibility of the molecules themselves (Figs. 1 and 2).
By analyzing the electrostatic field in the catalytic site in the ensembles of conformations that we determined, which represent the cis-bound and trans-bound states of the peptide substrate in complex with cyclophilin A, we identified an electrostatic handle mechanism underlying the catalytic process (Fig. 4 and Movie S1). We then validated this mechanism by studying the proline isomerization process of a modified version of the substrate, in which we performed a targeted change in the electric dipole representing the handle. We obtained this result by replacing the oxygen atom of the carbonyl group of the amino acid preceding the proline with a sulfur atom, a specific substitution that concerns a single atom in the substrate and conserves the group in the periodic table. As expected, this rationally designed substitution, which significantly reduces the electric dipole of the handle but leaves the other properties of the substrate essentially unchanged, suppressed significantly the catalytic activity of cyclophilin A (Fig. 5).
These results also provide further insights into the possible roles of dynamics in catalysis (21–23, 27, 47–50) when no chemical bond is formed or broken, because the conformational fluctuations in the bound state, which resemble those previously described in the free state (21–23), enable the population of structures that are particularly effective in reducing the isomerization barrier by providing the appropriate electrostatic fields (Fig. 4A).
More generally, our findings illustrate that the combination of NMR spectroscopy with molecular dynamics simulations and quantum mechanical calculations has the potential of identifying the specific mechanisms by which enzymes use electrostatic fields for catalysis.
Methods
Expression and purification of recombinant 13C,15N-labeled cyclophilin A were carried out as described in SI Text. The measurements of chemical shifts and residual dipolar couplings in the free and peptide-bound states (including the bound state with the thioamide-substituted peptide) were carried out as described in SI Text. Molecular dynamics simulations with replica-averaged NMR restraints and quantum mechanical calculations were carried out as described in SI Text.
Supplementary Material
Acknowledgments
We are grateful to Dr. Francesco Aprile for useful discussions. Part of this research was performed using the facilities of the Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the Office of Biological and Environmental Research of the Department of Energy located at Pacific Northwest National Laboratory, Richland, WA. C.C. was supported by a Marie Curie Intra European Fellowship.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404220111/-/DCSupplemental.
References
- 1.Fischer G, Bang H, Mech C. Detection of enzyme catalysis for cis-trans-isomerization of peptide-bonds using proline-containing peptides as substrates. Biomed Biochim Acta. 1984;43:1101–1111. [PubMed] [Google Scholar]
- 2.Göthel SF, Marahiel MA. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell Mol Life Sci. 1999;55(3):423–436. doi: 10.1007/s000180050299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lu KP, Finn G, Lee TH, Nicholson LK. Prolyl cis-trans isomerization as a molecular timer. Nat Chem Biol. 2007;3(10):619–629. doi: 10.1038/nchembio.2007.35. [DOI] [PubMed] [Google Scholar]
- 4.Andreotti AH. Native state proline isomerization: An intrinsic molecular switch. Biochemistry. 2003;42(32):9515–9524. doi: 10.1021/bi0350710. [DOI] [PubMed] [Google Scholar]
- 5.Eakin CM, Berman AJ, Miranker AD. A native to amyloidogenic transition regulated by a backbone trigger. Nat Struct Mol Biol. 2006;13(3):202–208. doi: 10.1038/nsmb1068. [DOI] [PubMed] [Google Scholar]
- 6.Eckert B, Martin A, Balbach J, Schmid FX. Prolyl isomerization as a molecular timer in phage infection. Nat Struct Mol Biol. 2005;12(7):619–623. doi: 10.1038/nsmb946. [DOI] [PubMed] [Google Scholar]
- 7.Sarkar P, Saleh T, Tzeng SR, Birge RB, Kalodimos CG. Structural basis for regulation of the Crk signaling protein by a proline switch. Nat Chem Biol. 2011;7(1):51–57. doi: 10.1038/nchembio.494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fanghänel J, Fischer G. Insights into the catalytic mechanism of peptidyl prolyl cis/trans isomerases. Front Biosci. 2004;9:3453–3478. doi: 10.2741/1494. [DOI] [PubMed] [Google Scholar]
- 9.Fischer S, Michnick S, Karplus M. A mechanism for rotamase catalysis by the FK506 binding protein (FKBP) Biochemistry. 1993;32(50):13830–13837. doi: 10.1021/bi00213a011. [DOI] [PubMed] [Google Scholar]
- 10.Jakob RP, Schmid FX. Molecular determinants of a native-state prolyl isomerization. J Mol Biol. 2009;387(4):1017–1031. doi: 10.1016/j.jmb.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 11.Sarkar P, Reichman C, Saleh T, Birge RB, Kalodimos CG. Proline cis-trans isomerization controls autoinhibition of a signaling protein. Mol Cell. 2007;25(3):413–426. doi: 10.1016/j.molcel.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Trzesniak D, van Gunsteren WF. Catalytic mechanism of cyclophilin as observed in molecular dynamics simulations: Pathway prediction and reconciliation of X-ray crystallographic and NMR solution data. Protein Sci. 2006;15(11):2544–2551. doi: 10.1110/ps.062356406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zydowsky LD, et al. Active site mutants of human cyclophilin A separate peptidyl-prolyl isomerase activity from cyclosporin A binding and calcineurin inhibition. Protein Sci. 1992;1(9):1092–1099. doi: 10.1002/pro.5560010903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamelberg D, McCammon JA. Mechanistic insight into the role of transition-state stabilization in cyclophilin A. J Am Chem Soc. 2009;131(1):147–152. doi: 10.1021/ja806146g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leone V, Lattanzi G, Molteni C, Carloni P. Mechanism of action of cyclophilin A explored by metadynamics simulations. PLoS Comput Biol. 2009;5(3):e1000309. doi: 10.1371/journal.pcbi.1000309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li G, Cui Q. What is so special about Arg 55 in the catalysis of cyclophilin A? insights from hybrid QM/MM simulations. J Am Chem Soc. 2003;125(49):15028–15038. doi: 10.1021/ja0367851. [DOI] [PubMed] [Google Scholar]
- 17.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. Cyclophilin: A specific cytosolic binding protein for cyclosporin A. Science. 1984;226(4674):544–547. doi: 10.1126/science.6238408. [DOI] [PubMed] [Google Scholar]
- 18.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature. 1989;337(6206):476–478. doi: 10.1038/337476a0. [DOI] [PubMed] [Google Scholar]
- 19.Wang P, Heitman J. The cyclophilins. Genome Biol. 2005;6(7):226. doi: 10.1186/gb-2005-6-7-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davis TL, et al. Structural and biochemical characterization of the human cyclophilin family of peptidyl-prolyl isomerases. PLoS Biol. 2010;8(7):e1000439. doi: 10.1371/journal.pbio.1000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eisenmesser EZ, Bosco DA, Akke M, Kern D. Enzyme dynamics during catalysis. Science. 2002;295(5559):1520–1523. doi: 10.1126/science.1066176. [DOI] [PubMed] [Google Scholar]
- 22.Eisenmesser EZ, et al. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438(7064):117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 23.Fraser JS, et al. Hidden alternative structures of proline isomerase essential for catalysis. Nature. 2009;462(7273):669–673. doi: 10.1038/nature08615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wüthrich K. Protein structure determination in solution by nuclear magnetic resonance spectroscopy. Science. 1989;243(4887):45–50. doi: 10.1126/science.2911719. [DOI] [PubMed] [Google Scholar]
- 25.Bax A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003;12(1):1–16. doi: 10.1110/ps.0233303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mittermaier A, Kay LE. New tools provide new insights in NMR studies of protein dynamics. Science. 2006;312(5771):224–228. doi: 10.1126/science.1124964. [DOI] [PubMed] [Google Scholar]
- 27.Boehr DD, Dyson HJ, Wright PE. An NMR perspective on enzyme dynamics. Chem Rev. 2006;106(8):3055–3079. doi: 10.1021/cr050312q. [DOI] [PubMed] [Google Scholar]
- 28.Kalodimos CG. NMR reveals novel mechanisms of protein activity regulation. Protein Sci. 2011;20(5):773–782. doi: 10.1002/pro.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lindorff-Larsen K, Best RB, Depristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433(7022):128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 30.Markwick PRL, et al. Enhanced conformational space sampling improves the prediction of chemical shifts in proteins. J Am Chem Soc. 2010;132(4):1220–1221. doi: 10.1021/ja9093692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Robustelli P, Stafford KA, Palmer AG., 3rd Interpreting protein structural dynamics from NMR chemical shifts. J Am Chem Soc. 2012;134(14):6365–6374. doi: 10.1021/ja300265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shaw DE, et al. Atomic-level characterization of the structural dynamics of proteins. Science. 2010;330(6002):341–346. doi: 10.1126/science.1187409. [DOI] [PubMed] [Google Scholar]
- 33.Clore GM, Schwieters CD. How much backbone motion in ubiquitin is required to account for dipolar coupling data measured in multiple alignment media as assessed by independent cross-validation? J Am Chem Soc. 2004;126(9):2923–2938. doi: 10.1021/ja0386804. [DOI] [PubMed] [Google Scholar]
- 34.Lange OF, et al. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320(5882):1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- 35.Montalvao RW, De Simone A, Vendruscolo M. Determination of structural fluctuations of proteins from structure-based calculations of residual dipolar couplings. J Biomol NMR. 2012;53(4):281–292. doi: 10.1007/s10858-012-9644-3. [DOI] [PubMed] [Google Scholar]
- 36.Robustelli P, Kohlhoff K, Cavalli A, Vendruscolo M. Using NMR chemical shifts as structural restraints in molecular dynamics simulations of proteins. Structure. 2010;18(8):923–933. doi: 10.1016/j.str.2010.04.016. [DOI] [PubMed] [Google Scholar]
- 37.Camilloni C, Robustelli P, De Simone A, Cavalli A, Vendruscolo M. Characterization of the conformational equilibrium between the two major substates of RNase A using NMR chemical shifts. J Am Chem Soc. 2012;134(9):3968–3971. doi: 10.1021/ja210951z. [DOI] [PubMed] [Google Scholar]
- 38.Neudecker P, et al. Structure of an intermediate state in protein folding and aggregation. Science. 2012;336(6079):362–366. doi: 10.1126/science.1214203. [DOI] [PubMed] [Google Scholar]
- 39.Schlegel J, et al. Solution characterization of the extracellular region of CD147 and its interaction with its enzyme ligand cyclophilin A. J Mol Biol. 2009;391(3):518–535. doi: 10.1016/j.jmb.2009.05.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bahmed K, et al. Extracellular cyclophilin-A stimulates ERK1/2 phosphorylation in a cell-dependent manner but broadly stimulates nuclear factor kappa B. Cancer Cell Int. 2012;12(1):19. doi: 10.1186/1475-2867-12-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cavalli A, Camilloni C, Vendruscolo M. Molecular dynamics simulations with replica-averaged structural restraints generate structural ensembles according to the maximum entropy principle. J Chem Phys. 2013;138(9):094112. doi: 10.1063/1.4793625. [DOI] [PubMed] [Google Scholar]
- 42.Pitera JW, Chodera JD. On the use of experimental observations to bias simulated ensembles. J Chem Theory Comput. 2012;8:3445–3451. doi: 10.1021/ct300112v. [DOI] [PubMed] [Google Scholar]
- 43.Roux B, Weare J. On the statistical equivalence of restrained-ensemble simulations with the maximum entropy method. J Chem Phys. 2013;138(8):084107. doi: 10.1063/1.4792208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warshel A, et al. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106(8):3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 45.Ottiger M, Zerbe O, Güntert P, Wüthrich K. The NMR solution conformation of unligated human cyclophilin A. J Mol Biol. 1997;272(1):64–81. doi: 10.1006/jmbi.1997.1220. [DOI] [PubMed] [Google Scholar]
- 46.Bosco DA, et al. Dissecting the microscopic steps of the cyclophilin A enzymatic cycle on the biological HIV-1 capsid substrate by NMR. J Mol Biol. 2010;403(5):723–738. doi: 10.1016/j.jmb.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 47.Kamerlin SCL, Warshel A. At the dawn of the 21st century: Is dynamics the missing link for understanding enzyme catalysis? Proteins. 2010;78(6):1339–1375. doi: 10.1002/prot.22654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glowacki DR, Harvey JN, Mulholland AJ. Taking Ockham’s razor to enzyme dynamics and catalysis. Nat Chem. 2012;4(3):169–176. doi: 10.1038/nchem.1244. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz SD, Schramm VL. Enzymatic transition states and dynamic motion in barrier crossing. Nat Chem Biol. 2009;5(8):551–558. doi: 10.1038/nchembio.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nashine VC, Hammes-Schiffer S, Benkovic SJ. Coupled motions in enzyme catalysis. Curr Opin Chem Biol. 2010;14(5):644–651. doi: 10.1016/j.cbpa.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.