

Significance
An HIV vaccine must induce antibodies [broadly neutralizing antibodies (bnAbs)] that neutralize many viral variants. Determining pathways of antibody affinity maturation that have led to specific bnAbs and tracking coevolution in an infected individual of virus and antibody will define characteristics of immunogens that might elicit broad responses. We have followed, in an infected individual, one round of coevolution of viral envelope with antibodies from a single lineage. Insertions into a loop (V5) in gp120 of autologous viruses allowed escape from neutralization by antibodies in this lineage; antibody affinity maturation shifted the relative orientation of the light- and heavy-chain variable domains, allowing binding to envelopes with augmented V5. The results illustrate a mechanism of affinity maturation through mutation outside the antigen combining site.
Keywords: protein evolution, HIV gp120, antibody structure, somatic hypermutation
Abstract
Rapidly evolving pathogens, such as human immunodeficiency and influenza viruses, escape immune defenses provided by most vaccine-induced antibodies. Proposed strategies to elicit broadly neutralizing antibodies require a deeper understanding of antibody affinity maturation and evolution of the immune response to vaccination or infection. In HIV-infected individuals, viruses and B cells evolve together, creating a virus−antibody “arms race.” Analysis of samples from an individual designated CH505 has illustrated the interplay between an antibody lineage, CH103, and autologous viruses at various time points. The CH103 antibodies, relatively broad in their neutralization spectrum, interact with the CD4 binding site of gp120, with a contact dominated by CDRH3. We show by analyzing structures of progenitor and intermediate antibodies and by correlating them with measurements of binding to various gp120s that there was a shift in the relative orientation of the light- and heavy-chain variable domains during evolution of the CH103 lineage. We further show that mutations leading to this conformational shift probably occurred in response to insertions in variable loop 5 (V5) of the HIV envelope. The shift displaced the tips of the light chain away from contact with V5, making room for the inserted residues, which had allowed escape from neutralization by the progenitor antibody. These results, which document the selective mechanism underlying this example of a virus−antibody arms race, illustrate the functional significance of affinity maturation by mutation outside the complementarity determining region surface of the antibody molecule.
Any candidate HIV vaccine will need to induce an immune response effective for a wide range of potential variants. Approximately 15–25% of chronically infected HIV patients develop such broadly neutralizing antibodies (bnAbs) (1–3). These target four distinct epitope regions on the envelope glycoprotein—the V1−V2 loop, the CD4 binding site, a set of residues near the base of the V3 loop on gp120, and the membrane proximal external segment of gp41 (4, 5).
A protective B-cell immune response requires antibody affinity maturation—selective proliferation of cells with antibody variable domain mutations that enhance antigen affinity (6–8). It is possible to study this process by isolating individual B cells from infected or vaccinated donors and cloning the recombined heavy- and light-chain variable regions of the antibodies (9–11). Understanding those patterns of antibody affinity maturation that give rise to the development of bnAbs could inform design of immunogens with increased likelihood of maturation along otherwise subdominant pathways (12).
Data now available from donors followed from the onset of HIV infection until the development of bnAbs clarify some of these issues. Recent findings from an African patient, CH505, who was followed from acute HIV-1 infection through bnAb development, illustrate the interplay, or arms race, between virus evolution and antibody maturation (13). The structure of a gp120 core, which excludes a number of variable loops and glycosylation sites, has been determined in complex with the antigen-binding fragment (Fab) of a mature, CD4-binding-site broadly neutralizing antibody, CH103, derived from the same individual (13). Fixation in this lineage of mutations in residues in both partners that lie outside their interface indicate that there may be factors driving affinity maturation that are not evident from the crystal structure.
Analysis of this CH103 antibody lineage (Fig. 1) indicates that the transmitter founder (T/F) virus binds with high affinity to the progenitor, or unmutated common ancestor (UCA), of the lineage but that virus envelopes (Envs) from later time points do not (13). Furthermore, the UCA has no detectable affinity for heterologous Envs, whereas the CH103 lineage antibodies show increasing affinity for heterologous Envs as maturation proceeds, and the more mature members of the lineage are more potent neutralizers of heterologous viruses (13). For example, Env binding and neutralization of a clade B virus (B.63521) both increased substantially as the antibody matured from UCA to a later intermediate, I2 (13). Thus, we have extensive sequence and biochemical information about the antibody−virus arms race in the CH505 individual.
Fig. 1.

CH103 B-cell clonal lineage tree. The UCA is shown on the left, with increasingly mature antibodies on the right. The top branch resulted in bnAbs (blue). Branches that did not lead to broadly neutralizing antibodies are excluded from the diagram.
To investigate the structural correlates of viral escape and of antibody affinity maturation in the CH103 lineage, we determined crystal structures of Fabs derived from the UCA and intermediate antibodies, I3 and I2. The structures show that the relative orientation of the heavy- and light-chain variable domains shifted during affinity maturation, changing the way the antibodies encountered gp120. The mutations that produced the shift occurred after the appearance of insertions into the gp120 V5 loop in Envs of the autologous virus. Binding measurements show that these insertions block binding by the UCA and that the shift in relative variable-domain orientation restores affinity. The shift depends largely on mutations in the light chain, and the timing of the relevant mutations appears to correlate with development of broad neutralization. Our analysis illustrates the importance of considering mutations outside the antibody−antigen interface when interpreting the contribution of mutations accumulated during antibody affinity maturation.
Results
Structure of CH103 UCA Implies Unfavorable Contacts with the gp120 V5 Loop.
The UCA crystallized in space group P21, with four molecules in the asymmetric unit (Table S1). The measured data extend only to a minimum Bragg spacing of 3.49 Å, but the fourfold noncrystallographic symmetry allowed us to improve the map by density modification. There was good density for the backbone of residues 2–214 in the heavy chain and residues 2–209 in the light chain, except for heavy-chain residues 97–100B [in heavy-chain complementarity determining region 3 (CDRH3)] and 129–133 in the constant domain.
The published structure of a CH103/gp120 complex is with a heterologous gp120 (ZM176.66). The UCA of the CH103 lineage also binds tightly to gp120 of the T/F virus (13). Because the heavy chain contacts the conserved CD4 binding loop, we have examined the UCA/gp120 interaction by superposing the UCA variable heavy chain (VH) on that of CH103. The superposition shows that during affinity maturation, a shift has occurred in the orientation of the light-chain variable domain (VL) with respect to VH (Fig. 2). The change appears to be an adaptation to insertions in the gp120 V5 loop during the first year of infection (Fig. 3 and Fig. S1), relieving unfavorable contacts with the UCA light chain. Structures and binding data described below support and amplify this general conclusion.
Fig. 2.

Superposition of the UCA Fab with the CH103/gp120 outer domain complex. Two orthogonal views of the variable region of the UCA Fab (heavy and light chains in brown and red, respectively) superposed onto the variable region of the CH103 Fab (heavy and light chains in blue and cyan, respectively) in complex with the gp120 outer domain (green) from ZM176.66 [Protein Data Bank (PDB) ID code 4JAN] (13). The V5 and D loops of gp120 and the framework 3 (FR3) region of the Fab light chain are marked. Image created in PyMol.
Fig. 3.

Relationship between timing of mutations in gp120 of CH505 autologous viruses and binding of intermediates in the CH103 lineage (13). Binding of UCA, I3.2, and I2 to gp120 with gp120 bearing D-loop and V5 mutations characteristic of the time points shown on the scale above the diagram. Bars just below the time scale represent times at which the indicated mutations became prevalent in the V5 and D loops, respectively. Plus (+) signs indicate strong binding; minus (−) signs, weak or unobservable binding to either V5-loop insertions alone (blue) or to a combination of V5 and D-loop mutations (dark red).
Structures of Intermediates I3.2, I3.1, and I2.
To confirm the significance and determine the approximate timing of the relative reorientation of the VH and VL domains apparent from comparing the UCA and CH103, we determined crystal structures of several intermediates. Intermediate 3 lies at a key node in the lineage, marking the point at which breadth of neutralization began to emerge. The light chain of I3 was not identified, however, so we studied two “synthetic” variants of I3, I3.2 and I3.1, in which we combined VHDJH of I3 with VLJL of the UCA and of I2, respectively (Fig. S2A) (14). We also determined the structure of I2 (Table S1).
As expected from published binding and neutralization data and from the position of I2 in the lineage, the I2 structure is essentially identical to that of CH103. The latter has a three-residue deletion in CDRL1 (not present in CH104 and CH106—its closest siblings; see Fig. 1 and Fig. S2A). Superposition of I2 onto CH103 in the gp120 complex shows that the additional three residues can be accommodated without overlapping the V5 loop as configured in ZM176.66 (Fig. 4A).
Fig. 4.

Superposition of Fabs in the CH103 lineage. (A) Comparison of UCA, I3.1, I2, and CH103. Residues 62–75 of the FR3 of the light chain are in red, magenta, blue, and cyan for the UCA, I3.1, I2, and CH103, respectively. (B) Comparison of UCA and I3.2 (containing the UCA VLJL sequence). Residues 62–75 of the light-chain FR3 are in red and orange for the UCA and I3.2, respectively. The rest of the light chains are in gray; the heavy chains have been excluded. The gp120 outer domain (green) from ZM176.66 (PDB ID code 4JAN) (11) is also shown. (C) Superposition of the VH domains alone. UCA, I3.2, I3.1, I2, and CH103 are in red, orange, magenta, blue, and cyan, respectively. The complementarity determining region 3 (CDR3) is marked. All images created in PyMol.
Comparison of the I3.1 and I3.2 structures with each other and with those of I2 and the UCA of the lineage shows that the identity of the light chain is the principal determinant of the relative orientation of the two variable domains. When the VH structures are spatially aligned, VL of I3.1 superposes well on those of I2 and CH103 (Cα r.m.s.d. for framework regions of 0.54 Å and 0.73 Å, respectively), and VL of I3.2 superposes well on that of the UCA (framework Cα rmsd, 0.41 Å) (Fig. 4 A and B and Table S2); the Cα framework rmsd for the UCA or I3.2 with respect to I3.1, I2, or CH103 is generally greater than 0.9 Å, even though the individual VH and VL domains superpose with a significantly smaller rmsd (Tables S2 and S3). The UCA and I3.2 Fabs crystallized in different space groups, each with more than one molecule in the asymmetric unit, thus eliminating the possibility that crystal packing had influenced the relative VH−VL orientation in these antibodies and its divergence from the relative VH−VL orientation common to I3.1, I2, and CH103.
The conformations of CDRH3 in the free Fabs also correlate with the light-chain identity (Fig. 4C and Fig. S3). The CDRH3 conformation in the UCA is not compatible with gp120 binding (at least as in the CH103/gp120 complex). The high thermal parameters for residues in the CDRH3 loop (indeed, density for two of the four copies in the asymmetric unit of the UCA was too weak to fit) suggest, however, that the loop is adaptable and that other favorable interactions (e.g., those of the viral Envs that presumably drove affinity maturation) could compensate for the free energy cost of CDRH3 reorganization upon binding to CH505 gp120. We return to this point in Larger V5 Loops Interfere with Antibody Binding. The conformation of the CDRH3 loop in the more mature intermediates I3.1 and I2 and the mature antibody CH103 is clearly related to the light chain mutation, Tyr32LAsn, introduced between the UCA and intermediate I2. In the UCA, the tyrosine side chain contacts CDRH3 and displaces it from the configuration it assumes in I3.1, I2, and CH103 (Fig. S3). The side chain of the asparagine that substitutes for it appears to stabilize the mature CDRH3 conformation through a hydrogen bond network that includes the heavy-chain side chain of Asn100BH, conserved in all members of the lineage, the side chain of Glu50L in mature members of the CH103 lineage, and ultimately residues in the D and V5 loops of gp120 (Fig. S3). The Tyr32LAsn mutation thus cooperates with changes at the VH−VL interface in adapting the antibody to longer V5 loops in gp120.
Mutations at the VH−VL Interface.
Several changes in amino acid residues at the VH−VL interface correlate with the repositioning of the two domains with respect to each other. One relatively extended set of changes and rearrangements centers on light-chain residue 46, which has mutated from leucine in the UCA to valine in I2 and CH103. In the UCA structure, the leucine side chain bears on the ring of Tyr100D, which makes a hydrophobic bridge between Pro96H and Tyr49L at the domain interface. Mutation of residue 46L to valine releases the contact, leading to a concerted rearrangement that correlates not only with the overall reorientation of the light chain but also with a reconfiguration of CDRH3 (Fig. 5).
Fig. 5.

The heavy−light chain variable domain interface. VH−VL interface of the superposed CH103 and I3.2 Fabs. The interface regions are colored according to the Fab (CH103 heavy chain is in blue; CH103 light chain, in cyan; I3 heavy chain, in orange, UCA light chain, in light orange). The light-chain cartoons are surrounded by a gray surface. On the right are zoomed images of two regions at the interface, illustrating changes in interdomain contacts due to mutations that occurred between the early intermediates and the bnAbs. Images created in PyMol.
Distal to the gp120 interface, residues 38L and 39H face each other across the interdomain contact. In the UCA, both are glutamine; they engage in reciprocal side-chain hydrogen bonding between NE of one and OE of the other. In I2 (and CH103), residues 39H and 38L are leucine and valine, respectively. The heavy-chain mutation, Gln39HLeu, occurred between I3 and I2; the timing of the light-chain mutation is uncertain, but either Val or Gln could fit. The transition from a hydrogen-bond network to a more closely packed hydrophobic contact appears to contribute to the relative orientational adjustment of the two apposed domains (Fig. 5).
Larger V5 Loops Interfere with Antibody Binding.
To test whether V5 loop insertions in the autologous viruses correlate with the change in relative orientation of the light and heavy chains in the CH103 antibodies, we mutated the V5 loop of a heterologous gp120 core protein (from HIV-1 clade A strain 92ug037.8) to that of the CH505 T/F virus and measured binding of Fabs from the CH103 lineage by biolayer interferometry (Fig. S2B). This substitution resulted in a V5 loop two residues shorter than its counterparts in wild-type 92ug037.8 and ZM176.66, the gp120 used for crystallization with CH103 (Fig. S2B).
The gp120 core with a shorter V5 loop bound with higher affinity to the UCA and I3.2 than did the wild-type core (for which those Fabs showed essentially no detectable binding), but the loop substitution did not affect binding to either I3.1 or I2 (Table 1 and Fig. S4). This result is consistent with our crystal structures and with our inference that the antibodies have evolved in response to insertions in the V5 loops of autologous viruses, allowing the more mature antibodies to tolerate larger V5 loops without steric interference.
Table 1.
92ug037.8 gp120 core binding to CH103 Fabs
| HIV 92ug037.8 | Fab | Kd, ×10−6 M |
| gp120 core WT | UCA | NB |
| gp120 core V5 mutant | UCA | 191 ± 40 |
| gp120 core WT | I3.2 | >200 |
| gp120 core V5 mutant | I3.2 | 6.34 ± 0.15 |
| gp120 core WT | I3.1 | 7.73 ± 0.14 |
| gp120 core V5 mutant | I3.1 | 10.5 ± 0.5 |
| gp120 core WT | I2 | 4.62 ± 0.09 |
| gp120 core V5 mutant | I2 | 5.25 ± 0.14 |
Biolayer interferometry data, analyzed as described in SI Methods. V5 loop residues “GNINES” of wild-type 92ug037.8 gp120 core were replaced with “KNNT” of CH505 T/F virus Env. Averages and SEs are for three runs with different gp120 concentrations. Kd values derived by global fit of all data for each experiment. NB, no detectable binding under experimental conditions.
When we deleted two residues (Asn-Ile; see green highlighted segment in Fig. S2B) from the V5 loop of 92ug037.8 gp120, to give a V5 loop of the same length and charge as in the CH505 T/F virus, we did not detect enhanced affinity for the UCA or I3.2. Thus, even some shorter V5 loops fail to allow tight binding, presumably because of their conformational properties or their interaction with the D loop.
I3.2, which combines the heavy chain of I3 with the UCA light chain, bound 92ug037.8 gp120 bearing the T/F loop substantially more strongly than did the UCA, with Kd equivalent to I3.1 and I2. The CDRH3 conformation in I3.2, unlike the conformation in the UCA, is essentially the same as in the more mature forms, but displaced about 2 Å away from the light chain. A comparison of structures suggests that the heavy-chain mutations in CDRH1 might account for stabilizing this binding-competent conformation of CDRH3.
Reconstructing a Step in the HIV−Antibody Arms Race.
The T/F V5 swapped into 92ug037.8 gp120 imparts reasonable affinity for the UCA, but not at a level equivalent to the affinity of either wild-type or loop-swapped 92ug037.8 gp120 for more mature antibodies in the CH103 lineage. To study binding in the context of the autologous gp120, we expressed the T/F CH505 gp120 core and introduced observed mutations into the V5 and D loops. These loops form a continuous patch on the surface of gp120; CH103 contacts both, and some interactions bridge the two. The mutations we made in both D and V5 loops mimic those that occurred in the autologous virus in the interval between the T/F and week 78 (Fig. 3 and Fig. S1). For the V5 loop, we focused on insertions of DT, ETF, and EGKNN, respectively, because they spanned time points before and after expanding heterologous neutralization (13).
The binding data, determined by biolayer interferometry, are summarized in Table 2 and shown schematically in Fig. 3. The V5 insertions alone decreased affinity for each of the Fabs tested; the decrease was greatest for the UCA, which was affected even when just two residues, DT, were added to V5. The three- and five-amino acid insertions in V5 had stronger effects: we detected no binding for the UCA and marginal binding (Kd > 100 μM) for I3.2. The two- and three-residue insertions, DT and ETF, had little influence on the binding of I2 (and presumably CH103), but the five-residue EGKNN insertion did reduce I2 binding substantially. D-loop mutations that occurred between weeks 30 and 53, driven by escape from other antibodies (15), increased affinity for the CH103 intermediates and UCA. None of the mutations we examined affected binding of the broadly neutralizing antibody VRC01, which approaches the CD4 binding site of gp120 from a different direction than the CH103 antibodies (16) (Table 2).
Table 2.
CH505 gp120 core binding to CH103 Fabs
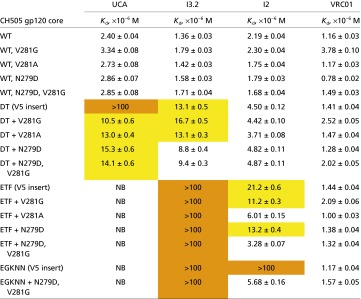 |
Biolayer interferometry data, analyzed as described in SI Methods. Averages and SEs are for three runs with different gp120 concentrations. Kd values derived by a global fit of all data for each experiment. For full kinetic data, see Table S4. NB, no binding detected. Orange highlight, Kd > 100 μM; yellow, 100 μM > Kd > 10 μM.
Sampling from subject CH505 was more frequent for viruses than for B-cells. Although the timing of antibody mutations is therefore uncertain, we can still compare the timing of changes in the V5 and D loops (Fig. 3 and Fig. S1) with the binding data in Table 2. Viral Envs were sampled at 30 wk, 53 wk, 78 wk, and 100 wk (also earlier and later time points not relevant here). The DT insertion in V5 was prevalent by week 30; the ETF insertion “took over” by week 53; longer insertions were prevalent around week 78. The D-loop mutations appeared to be relatively well fixed by week 53, without much further change until after week 100. Thus, around week 30, the prevalent gp120 sequences had a two-residue insertion in V5, and a single mutation in loop D. By week 53, double mutations had appeared in loop D, together with a three-residue insertion in V5. Of the three antibodies we examined (Table 2), only I2 would have retained binding after week 53. We cannot date I2 precisely, as the antibody mutation rate is unlikely to be linear with time, but a time between weeks 78 and 100 is likely. These temporal correlations are consistent with the qualitative conclusions described above. Insertions in the V5 loop led to escape from neutralization by the UCA of this lineage; exposure to the modified gp120 then drove affinity maturation that included adjusting the VH−VL interface to avoid overlap of light-chain CDRs and framework 3 region (FR3) with V5.
Superposition of the various structures, using the VH domains as reference, provides plausible molecular explanations for data in Table 2. The overlays suggest unfavorable contacts of the DE loop (residues 65L−67L) of the UCA and the gp120 V5 loop (Fig. S5), assuming the latter to have a conformation similar to that of ZM176.66 (two residues longer than that of the T/F and hence of the same length as V5 with the DT insertion). Residues at the tips of CDRL1 and CDRL2, Tyr32L and Asp51L, would also have unfavorable contacts that a shorter V5 might relax (Fig. S3).
The D-loop mutations partially compensate, in some of the combinations in Table 2, for reduced affinity caused by V5 insertions. Each of the D-loop mutants we tested enhanced affinity of the UCA for CH505 gp120 with a DT insertion in V5. Model building suggests a possible salt bridge between D-loop Asp279 and UCA light-chain Lys53 in FR3 and reduced steric clash from the D-loop V281A/G mutation. For I2, the D-loop mutations improved affinity for gp120 with three- and five-residue V5 loop insertions, which occurred only after fixation of the D-loop double mutation. In our study, the D-loop mutations alone did not enhance affinity of gp120 core for any of the CH103 lineage antibodies. Thus, the timing probably resolves any apparent paradox that escape from neutralization by whatever antibody lineage drove the D-loop changes would have enhanced affinity for antibodies in the CH103 lineage.
Discussion
The most striking conclusion from our analysis is that a critical feature of affinity maturation in the CH103 lineage is a small but significant shift in the relative orientations of the VH and VL domains. We do not know of any similar cases with naturally occurring antibodies described in the literature to date. Other reported pathways leading to bnAbs involve direct or indirect modification of the CDRH3 loop. In antibodies directed against the V1V2 loop region of the HIV Env, for example, the size and charge of the CDRH3 loop have changed (17). In a lineage of antibodies directed against the influenza virus hemagglutinin receptor site, the CDRH3 loop rigidifies during affinity maturation although its amino acid sequence does not change and the relevant mutations lie outside the antibody−antigen interface (18). A recent study of in vitro antibody evolution has shown that VH−VL interface residues are usually conserved to maintain interaction with a given antigen (19, 20). The domain interface therefore should be considered a locus for affinity maturation because sequence changes there can produce differences in packing that alter the geometry of the CDR surface.
The somatically mutated residues at the domain interface that correlate with the observed orientation shift (Fig. 5) are at positions conventionally designated as “framework.” Somatic mutations appear more often in CDRs than in framework residues (21, 22), consistent with the reasonable assumption that framework changes are more likely than CDR changes to destabilize the entire variable-region structure. The abnormal extent of somatic mutation in many HIV bnAbs has prompted the inference that framework-region mutations might be necessary to generate breadth (23). Restructuring the variable module in response to a viral escape mutation, by mutation at the domain interface or in any other determinant of overall conformation, is a mechanism for overcoming broad classes of escape mutation (V5 insertions of varying length, sequence, and possibly conformation in the present example), whereas point mutations in the CDRs are more likely to enhance affinity for a specific target configuration or to respond by reciprocal mutation to a defined amino acid substitution in the antigen. The former mechanism—alteration of long-range structure—is more likely to enable rapid development of breadth than the more prevalent mutation of local amino acid residue identity, because many point mutations in the CDRs may be needed to achieve the same effect.
The selective advantage of the VH−VL shift in CH103 would have been to restore affinity for the envelope of viruses that had acquired insertion mutations in the gp120 V5 loop, allowing them to escape neutralization by the progenitor antibody of the lineage (the UCA). That is, we attribute the rearrangement of chains in the variable module as closure of one cycle in the virus−antibody arms race. We can assign to the light chain the principal mutations that produced the change, because the two synthetic intermediates, I3.2 and I3.1, which share a heavy chain, have configurations similar to those of the UCA and the mature antibodies, respectively. The VH−VL configuration in the more mature antibodies can accommodate some longer V5 loops, not only in the CH505 virus but also in others such as ZM176.66 and 92ug037.8, both of which have loops two residues longer than does the CH505 T/F. Thus, the domain shift might account both for neutralization of autologous viruses by I2 and for the increased heterologous neutralization breadth that distinguishes I2 from I3. There appears to have been no further change in the relative orientations of VH and VL beyond I2, presumably because the even longer V5 loops from weeks 100 and later did not interfere with antibody binding.
The relative orientation of VH and VL in I3.1, I2, and CH103 lies outside the range found in other variable modules from human Fabs. A comparison with a set of 10 diverse human Fabs, having naturally paired heavy and light chains and directed against HIV Env or influenza hemagglutinin (16, 18, 24–30), shows that most resemble the UCA and I3.2 (Fig. S6). An exception is VRC01, whose light-chain variable domain appears to shift away from the heavy chain even more than in I2 (Fig. S6). The VL of CH31, a VRC01-class antibody, also shifts away from the “average” orientation, but not to the same extent as VRC01. The VRC01-class antibodies interact with the V5 and D loops of gp120, although with contacts quite different from those made by CH103. To accommodate diversity in this region of the virus envelope, VRC01-class antibodies have CDRL1 loops that, by deletion or reconfiguration during affinity maturation, avoid a clash with gp120 (29). A significantly shortened CDRL3 loop may contribute to the reconfiguration of the CDRL1 loop in the case of VRC01 (16, 31). In the CH103 lineage antibodies, the CDRL1 loop does not vary greatly in length or conformation (except for CH103 itself, which, unlike CH104 and CH106 with comparable affinities for gp120, has a three-residue deletion in CDRL1). Thus, the orientational shift of VL appears to have been the principal response to an augmented V5 loop during the first 20–30 wk of infection.
The CDRH3 loop, encoded principally by the D and J segments with somatic diversity at the V-D and D-J junctions, is the most variable of the CDRs (32). Within this lineage, however, both its sequence and length are conserved. Its tip appears to be quite flexible, even disordered, in the UCA, but less so in I3.2, in which the local configuration of the entire CDR is essentially the same as in the mature antibody, except for a shift of about 2 Å relative to the rest of the domain. Despite this shift, the heavy chain could associate with the CD4 binding loop of gp120 essentially as in the published complex, conserving contacts with all three CDRs, including the critical salt bridge between Arg97H of the antibody and Asp368 of gp120, thereby explaining the I3.2 affinities in Table 2. We note, however, that an intermediate such as I3.2 would never have been made if (as seems likely) the Tyr32LAsn mutation, already present in I2, had been introduced into the light chain by the time the I3 heavy chain appeared.
Mutations in regions of the CH505 envelope other than the V5 and D loops might in principle have influenced binding of antibodies in the CH103 lineage. Insertions in the V1 loop and accompanying variations in potential glycosylation sites occurred at relatively early time points after infection. A superposition of the CH103/gp120 complex (or of the corresponding model with any of the intermediates described here) on the structure of the SOSIP gp140 trimer, determined by cryoEM and X-ray crystallography (33–35), indicates that outside the V5/D loop region, amino acid residues that mutated during the first 96 wk of infection do not contact the CH103 antibody. Moreover, parts of the V1 and V3 loops that might contact the antibody have no mutations during this time period.
Recent proposals for vaccine development focus on designing immunogens with high affinity for the unmutated ancestor of a particular broadly neutralizing antibody (12). The UCA of the CH103 lineage binds tightly to the T/F virus, suggesting that a suitably modified CH505 T/F gp120 core might itself be such an immunogen. If so, it would be interesting to examine the effect of boosting with a variant with insertions in the V5 loop.
Methods
All proteins were expressed in 293T or 293F cells, and purified on Ni-NTA resin (Qiagen) followed by size exclusion chromatography. VRC01 IgG was tagless and therefore purified using protein A agarose resin (Pierce), cleaved, and further purified by gel filtration. Fabs were crystallized by hanging drop vapor diffusion and structures determined by molecular replacement with CH103 from the CH103/gp120 outer domain complex structure (PDB ID 4JAN) as the starting model. All graphical representations with protein crystal structures were made in PyMol. Binding measurements by biolayer interferometry were performed according to manufacturer’s protocols using the Octet QKe (ForteBio) and anti-Human Fab-CH1 biosensors. More detailed experimental procedures are provided in SI Methods. Coordinates and diffraction data have been deposited in the PDB (PDB ID codes: 4QHK, 4QHL, 4QHM, and 4QHN for UCA, I3.2, I3.1, and I2, respectively).
Supplementary Material
Acknowledgments
We thank Donald Raymond, James Kovacs, Jia Chen, Gary Frey, and Bing Chen for advice and assistance; Tongqing Zhou and Peter Kwong for sending coordinates of the CH103:gp120 complex before publication; Peter Kwong for comments; and beam-line staff at Advanced Photon Source 24-ID-E and Advanced Light Source 8.2.2. Research reported in this publication was supported by National Institute of Allergy and Infectious Diseases Grant AI100645 (Center for HIV/AIDS Vaccine Immunology-Immunogen Discovery). S.C.H. is an investigator of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
Data deposition: The atomic coordinates, crystallography, and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4QHK, 4QHL, 4QHM, and 4QHN).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1409954111/-/DCSupplemental.
References
- 1.Stamatatos L, Morris L, Burton DR, Mascola JR. Neutralizing antibodies generated during natural HIV-1 infection: Good news for an HIV-1 vaccine? Nat Med. 2009;15(8):866–870. doi: 10.1038/nm.1949. [DOI] [PubMed] [Google Scholar]
- 2.Doria-Rose NA, et al. Frequency and phenotype of human immunodeficiency virus envelope-specific B cells from patients with broadly cross-neutralizing antibodies. J Virol. 2009;83(1):188–199. doi: 10.1128/JVI.01583-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simek MD, et al. Human immunodeficiency virus type 1 elite neutralizers: Individuals with broad and potent neutralizing activity identified by using a high-throughput neutralization assay together with an analytical selection algorithm. J Virol. 2009;83(14):7337–7348. doi: 10.1128/JVI.00110-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kwong PD, Mascola JR, Nabel GJ. Broadly neutralizing antibodies and the search for an HIV-1 vaccine: The end of the beginning. Nat Rev Immunol. 2013;13(9):693–701. doi: 10.1038/nri3516. [DOI] [PubMed] [Google Scholar]
- 5.Julien JP, Lee PS, Wilson IA. Structural insights into key sites of vulnerability on HIV-1 Env and influenza HA. Immunol Rev. 2012;250(1):180–198. doi: 10.1111/imr.12005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisen HN, Siskind GW. Variations in affinities of antibodies during the immune response. Biochemistry. 1964;3:996–1008. doi: 10.1021/bi00895a027. [DOI] [PubMed] [Google Scholar]
- 7.McKean D, et al. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81(10):3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacob J, Kelsoe G, Rajewsky K, Weiss U. Intraclonal generation of antibody mutants in germinal centres. Nature. 1991;354(6352):389–392. doi: 10.1038/354389a0. [DOI] [PubMed] [Google Scholar]
- 9.Liao HX, et al. High-throughput isolation of immunoglobulin genes from single human B cells and expression as monoclonal antibodies. J Virol Methods. 2009;158(1-2):171–179. doi: 10.1016/j.jviromet.2009.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wardemann H, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–1377. doi: 10.1126/science.1086907. [DOI] [PubMed] [Google Scholar]
- 11.Wrammert J, et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature. 2008;453(7195):667–671. doi: 10.1038/nature06890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Haynes BF, Kelsoe G, Harrison SC, Kepler TB. B-cell-lineage immunogen design in vaccine development with HIV-1 as a case study. Nat Biotechnol. 2012;30(5):423–433. doi: 10.1038/nbt.2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao HX, et al. NISC Comparative Sequencing Program Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature. 2013;496(7446):469–476. doi: 10.1038/nature12053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kepler TB. 2013. Reconstructing a B-cell clonal lineage. I. Statistical inference of unobserved ancestors [v1; ref status: indexed, http://f1000r.es/z6]. F1000Research 2:103.
- 15.Gao F, et al. Cooperation of B cell lineages in induction of HIV broad neutralizing antibodies. Cell. 2014 doi: 10.1016/j.cell.2014.06.022. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou T, et al. Structural basis for broad and potent neutralization of HIV-1 by antibody VRC01. Science. 2010;329(5993):811–817. doi: 10.1126/science.1192819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doria-Rose NA, et al. NISC Comparative Sequencing Program Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 2014;509(7498):55–62. doi: 10.1038/nature13036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt AG, et al. Preconfiguration of the antigen-binding site during affinity maturation of a broadly neutralizing influenza virus antibody. Proc Natl Acad Sci USA. 2013;110(1):264–269. doi: 10.1073/pnas.1218256109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsu HJ, et al. Antibody variable domain interface and framework sequence requirements for stability and function by high-throughput experiments. Structure. 2014;22(1):22–34. doi: 10.1016/j.str.2013.10.006. [DOI] [PubMed] [Google Scholar]
- 20.Narayanan A, Sellers BD, Jacobson MP. Energy-based analysis and prediction of the orientation between light- and heavy-chain antibody variable domains. J Mol Biol. 2009;388(5):941–953. doi: 10.1016/j.jmb.2009.03.043. [DOI] [PubMed] [Google Scholar]
- 21.Wu TT, Kabat EA. An analysis of the sequences of the variable regions of Bence Jones proteins and myeloma light chains and their implications for antibody complementarity. J Exp Med. 1970;132(2):211–250. doi: 10.1084/jem.132.2.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wagner SD, Milstein C, Neuberger MS. Codon bias targets mutation. Nature. 1995;376(6543):732. doi: 10.1038/376732a0. [DOI] [PubMed] [Google Scholar]
- 23.Klein F, et al. Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell. 2013;153(1):126–138. doi: 10.1016/j.cell.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabin C, et al. Crystal structure and size-dependent neutralization properties of HK20, a human monoclonal antibody binding to the highly conserved heptad repeat 1 of gp41. PLoS Pathog. 2010;6(11):e1001195. doi: 10.1371/journal.ppat.1001195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corti D, et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science. 2011;333(6044):850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 26.Pancera M, et al. Structural basis for diverse N-glycan recognition by HIV-1-neutralizing V1-V2-directed antibody PG16. Nat Struct Mol Biol. 2013;20(7):804–813. doi: 10.1038/nsmb.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kong L, et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat Struct Mol Biol. 2013;20(7):796–803. doi: 10.1038/nsmb.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong M, et al. Antibody recognition of the pandemic H1N1 Influenza virus hemagglutinin receptor binding site. J Virol. 2013;87(22):12471–12480. doi: 10.1128/JVI.01388-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou T, et al. NISC Comparative Sequencing Program Multidonor analysis reveals structural elements, genetic determinants, and maturation pathway for HIV-1 neutralization by VRC01-class antibodies. Immunity. 2013;39(2):245–258. doi: 10.1016/j.immuni.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao HX, et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity. 2013;38(1):176–186. doi: 10.1016/j.immuni.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.West AP, Jr, Diskin R, Nussenzweig MC, Bjorkman PJ. Structural basis for germ-line gene usage of a potent class of antibodies targeting the CD4-binding site of HIV-1 gp120. Proc Natl Acad Sci USA. 2012;109(30):E2083–E2090. doi: 10.1073/pnas.1208984109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morea V, Tramontano A, Rustici M, Chothia C, Lesk AM. Conformations of the third hypervariable region in the VH domain of immunoglobulins. J Mol Biol. 1998;275(2):269–294. doi: 10.1006/jmbi.1997.1442. [DOI] [PubMed] [Google Scholar]
- 33.Lyumkis D, et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science. 2013;342(6165):1484–1490. doi: 10.1126/science.1245627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Julien JP, et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science. 2013;342(6165):1477–1483. doi: 10.1126/science.1245625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bartesaghi A, Merk A, Borgnia MJ, Milne JL, Subramaniam S. Prefusion structure of trimeric HIV-1 envelope glycoprotein determined by cryo-electron microscopy. Nat Struct Mol Biol. 2013;20(12):1352–1357. doi: 10.1038/nsmb.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.