

Significance
New effective therapies are desperately needed for lung cancer because most current lung cancer treatments rarely prevent the metastatic disease that causes the majority of patient deaths. We discovered that the neurotrophin receptor TrkB is an important therapeutic target in metastatic lung cancer using a combination of cell line assays, conditional mouse models, and patient samples.
Keywords: TrkB/NTRK2, NSCLC
Abstract
Lung cancer is notorious for its ability to metastasize, but the pathways regulating lung cancer metastasis are largely unknown. An in vitro system designed to discover factors critical for lung cancer cell migration identified brain-derived neurotrophic factor, which stimulates cell migration through activation of tropomyosin-related kinase B (TrkB; also called NTRK2). Knockdown of TrkB in human lung cancer cell lines significantly decreased their migratory and metastatic ability in vitro and in vivo. In an autochthonous lung adenocarcinoma model driven by activated oncogenic Kras and p53 loss, TrkB deficiency significantly reduced metastasis. Hypoxia-inducible factor-1 directly regulated TrkB expression, and, in turn, TrkB activated Akt signaling in metastatic lung cancer cells. Finally, TrkB expression was correlated with metastasis in patient samples, and TrkB was detected more often in tumors that did not have Kras or epidermal growth factor receptor mutations. These studies demonstrate that TrkB is an important therapeutic target in metastatic lung adenocarcinoma.
Lung cancer remains the major cause of death from cancer worldwide. The two types of lung cancer are non-small-cell lung cancers (NSCLCs; 80% of all lung cancers), which include adenocarcinomas, squamous cell carcinomas, and large cell carcinomas, and small-cell lung cancers (20%, exhibiting neuroendocrine features). The average 5-y survival rate for NSCLC is only 16% (1). Most NSCLC patients have advanced disease at diagnosis; 22% have regional lymph node metastases, and 56% have distant metastases in the brain, bone, liver, or adrenal glands (1). Surgery or therapies that treat primary lung tumors rarely prevent metastases (1). Understanding the mechanisms of NSCLC metastasis is crucial for the development of new therapies to improve survival for lung cancer patients.
Tropomyosin-related kinase B (TrkB; also called NTRK2), a neurotrophin receptor, is important for neural development and is an independent prognostic marker in many tumor types (2). TrkB-expressing neural precursors migrate from their proliferative zone toward a gradient of the TrkB ligand, brain-derived neurotrophic factor (BDNF), which is made near the site of residence of the mature neurons (3). High levels of TrkB are correlated with poor patient prognosis in neuroblastomas and ovarian, pancreatic, colon, prostate, and gastric cancers (2). TrkB was overexpressed in ovarian adenocarcinoma metastases compared with primary lesions (4). TrkB is also a supressor of anoikis in cell lines (5). Finally, overexpression of TrkB and BDNF in nonmalignant rat intestinal epithelial cells promoted metastasis from s.c. injection sites to the lungs (6).
Despite these clues that TrkB regulates parts of the metastatic cascade in other tumor types, the role of TrkB in NSCLC metastasis remains unclear. TrkB immunoreactivity was correlated with advanced stage disease in lung squamous cell carcinomas, yet was also correlated with better survival (7). In contrast, TrkB expression was associated with worse survival in a panel of NSCLC samples containing squamous cell carcinomas, adenocarcinomas, and large-cell neuroendocrine carcinomas (8). Despite the evidence that TrkB is important for metastasis of cell lines, it has not been established that TrkB is required for NSCLC metastatic activity in endogenous tumors, and only xenograft models have been used to investigate TrkB function in other tumor types.
We developed an in vitro system to identify factors crucial for lung cancer metastasis. In this study, we determined that BDNF secreted by lymph node fibroblasts induces migration of lung cancer cells. TrkB was required for migration of lung adenocarcinoma cancer cell lines and their metastatic capacity in vivo. Loss of TrkB significantly reduced lymph node metastases in a lung adenocarcinoma model driven by activated oncogenic Kras and p53 loss. TrkB expression was regulated by hypoxia-inducible factor-1 (HIF-1), and TrkB was required for Akt activation during lung tumor cell migration. TrkB expression was correlated with a higher stage and with observed metastasis in lung cancer patient samples. Our work demonstrates that TrkB is crucial for the migratory steps in lung adenocarcinoma metastasis.
Results
Human Lung Cancer Cell Lines Migrate Toward BDNF Secreted from Lymph Node Fibroblasts.
We developed an in vitro assay to identify factors crucial for lung cancer cell migration. Because lung tumor cells routinely metastasize to the lymph nodes, we hypothesized that lymph node cells secrete proteins that stimulate tumor cell homing. To test this idea, we generated conditioned media (CM) from cultured primary human lymph node fibroblasts. CM robustly stimulated migration of A549, H322, H2030, and H2030-BrM3 human lung adenocarcinoma cell lines in transwell assays (Fig. 1A). To identify the secreted factors stimulating migration, serum-free control or lymph node fibroblast CM was incubated on antibody arrays, which allow for simultaneous detection of 507 human proteins including many cytokines. Although the positive control spot IgG levels were similar between the two types of media, BDNF was detected only in the conditioned media (Fig. 1B). These results identified BDNF as a candidate migration factor for lung cancer cells.
Fig. 1.

BDNF stimulates migration of human lung adenocarcinoma cell lines. (A) Human lung adenocarcinoma cell lines were plated into transwell assays with the indicated medias. n = 3. *P < 0.01. (B) Serum-free (SFM) and lymph node fibroblast conditioned media (CM) were incubated on protein array membranes. n = 1. (C) A549 transwell migration assays with control CM (Control IgG IP), BDNF-depleted CM (BDNF IP), or BDNF-depleted CM supplemented with BDNF (BDNF IP + Rescue). n = 3. *P < 0.01. (D) Quantitative PCR (qPCR) analysis showing BDNF expression of the indicated sorted cell populations from wild-type mouse lungs. n = 3. *P < 0.01. (E) ELISA showing BDNF protein levels in wild-type mouse tissues (n = 4). *P < 0.01 indicates a significant difference between that tissue and the amount of BDNF in the stomach. All error bars represent mean ± SEM.
To determine if BDNF was important in lung cancer cell migration, we assessed cell migration when BDNF was depleted from CM. ELISA confirmed BDNF depletion Fig. S1A). Transwell migration assays with A549 cells indicated that BDNF depletion from CM reduced lung cancer cell migration by 1.9-fold (Fig. 1C). A549 cell migration was rescued when BDNF was added back to the depleted CM (Fig. 1C). These studies indicate that the BDNF secreted by human lymph node fibroblasts is crucial for lung adenocarcinoma cell line migration.
We next wanted to determine the sites of BDNF expression in vivo. Although we demonstrated that human lymph node fibroblasts secrete BDNF, other cells in the lung and lymphatic system, including lymphocytes and macrophages, have also been shown to secrete BDNF (9). To examine the source of BDNF in the lung microenvironment, wild-type mouse lungs were dissociated, and macrophages (Mac1+/CD45+), hematopoietic cells (Mac1−/CD45+), endothelial cells (Mac1−CD45−CD31+), fibroblasts (Mac1−CD45−CD31−EpCAM−), and epithelial cells (Mac1−CD45−CD31−EpCAM+) were collected and confirmed with cell-type–specific markers (Fig. S1 B–G). BDNF transcript levels were detected only in the EpCAM− population, indicating that BDNF is primarily secreted by fibroblasts in the lung (Fig. 1D). We also isolated macrophages, lymphocytes, and fibroblasts from mouse lymph nodes and found that BDNF was expressed only in the EpCAM− fibroblast population (Fig. S1H). Using ELISA, we found that BDNF protein levels were highest in the lymph nodes, adrenal glands, kidney, and liver (Fig. 1E), all tissues to which mouse lung tumor cells most commonly metastasize (10, 11). These data suggested that a gradient of BDNF may promote the migration and homing of lung tumor cells to metastatic tissues.
Because it has been shown that neuron precursors migrate toward BDNF during development and secrete BDNF to amplify this migration gradient (12), we also investigated if an autocrine BDNF loop mediates lung tumor cell migration. All four lung cancer cell lines secreted BDNF into serum-free media (Fig. S1I). Interestingly, BDNF was also expressed in the EpCAM+ lung cell population of mice with lung tumors (Fig. S1J), indicating that lung tumor cells in this population may be secreting BDNF. Therefore, autocrine BDNF production may contribute to lung tumor cell migration.
TrkB Plays a Functional Role in Lung Cancer Cell Migration and Metastasis.
Because BDNF binds to TrkB and nerve growth factor receptor (NGFR; also called p75) (3), we next asked which receptor has a functional role in migration of lung cancer cells. Although knockdown of NGFR with three shRNA hairpins did not decrease migration of A549 cells (Fig. S2A), knockdown of TrkB with two different hairpins significantly decreased the migration of several lung adenocarcinoma cell lines. A 2.5- to 25-fold knockdown of TRKB transcript levels was achieved with an shRNA construct against TRKB (shTrkB) in the A549, H322, H2030, and H2030-BrM3 cells compared with the control shRNA to green fluorescent protein (shGFP) (Fig. S2B). Levels of TRKA, TRKC, and NGFR were not altered in the TrkB knockdown cells, indicating that other Trk family members did not compensate for the loss of TrkB expression (Fig. S2C). Migration assays with lymph node fibroblast CM showed that TrkB knockdown A549, H322, H2030, and H2030-BrM3 cells were 1.3- to 2-fold less migratory than their respective shGFP control cells (Fig. 2A). Importantly, proliferation was not different between the shGFP and shTrkB cells at 24 h (Fig. S2D), confirming that differences in migration rates were not attributable to altered proliferation. A second hairpin against TrkB (shTrkB-2) also significantly decreased the migration of H322 and H2030-BrM3 cells (Fig. S2E), without impairing proliferation (Fig. S2F). These data demonstrate that TrkB plays a functional role in lung adenocarcinoma cell line migration.
Fig. 2.

TrkB is required for human lung adenocarcinoma cell line migration and metastasis. (A) Transwell assays showing migration of the indicated cell lines expressing shTrkB toward conditioned media. n = 3. *P < 0.05. (B) Quantification of lung tumor burden of three to five mice per group i.v.-injected with H2030-BrM3 shGFP, shTrkB, or shTrkB-2 cells (n = 2–4). *P < 0.01. (C) Transwell assays showing migration of the indicated cell lines treated with ANA-12. n = 2–3. *P < 0.03. (D) Transwell assays showing migration of H23 cells overexpressing empty vector, wild-type TrkB (TrkB-OE), or kinase dead TrkB (TrkB-K571M). n = 2. *P < 0.02. (E) Representative pAkt and Akt immunoblots of the protein extracts from H2030-BrM3 shGFP and shTrkB cells treated with BDNF. n = 3. (F) Transwell assays showing migration of shGFP and shTrkB H2030-BrM3 cells expressing empty vector (EV) control or constitutively active Akt. n = 3. *P < 0.02. All error bars represent mean ± SEM.
We next determined that loss of TrkB affected lung adenocarcinoma cancer cell line metastasis in vivo. We used s.c. injection assays, which allow for assessment of cell proliferation and tumor growth, and i.v. tail-vein injections, which measure the ability of cells to successfully navigate several steps of the metastatic cascade, including survival during circulation and colonization of secondary sites. The growth of s.c. tumors from H2030 shTrkB cells, H2030-BrM3 shTrkB cells, and their matched shGFP derivatives was not statistically significant, indicating that TrkB may not be crucial for tumor cell proliferation in vivo (Fig. S3A). After i.v. injection, H2030 shTrkB cells and H2030-BrM3 shTrkB cells formed fewer lung tumors and showed decreased tumor burden compared with matched shGFP cells (Fig. S3 B–D). Again using two different hairpins for TrkB, we found that both H2030-BrM3 shTrkB cells and H2030-BrM3 shTrkB-2 cells exhibited reduced lung tumor burden (Fig. 2B). Notably, the degree of knockdown correlated with the effect on tumor burden; shTrkB cells and shTrkB-2 cells had 5.2- and 1.9-fold less tumor burden, attributable to 7.7- and 2.6-fold less TrkB expression, respectively (Fig. S3E). Similar effects were seen for tumor number; H2030-BrM3 shTrkB cells produced 2.5-fold fewer tumors than shGFP cells, whereas shTrkB-2 cells did not show decreased tumor number (Fig. S3F). Notably, cultured H2030 shTrkB cells had a fivefold reduction in TrkB expression, whereas shTrkB cells isolated from lung tumors that formed after i.v. injection only had a twofold reduction (Fig. S3G). Thus, some shTrkB cells may escape TrkB knockdown in vivo and produce lesions. Taken together, these results illustrate that TrkB is required for human lung cancer cell line metastasis.
We also treated a panel of cell lines with the TrkB-specific inhibitor, ANA-12 (13). Migration assays showed that A549, H322, H2030, and H2030-BrM3 cells were 2- to 5.9-fold less migratory at 30 µM and 4.8- to 8.3-fold less migratory at 50 µM ANA-12 than their respective DMSO-treated control cells (Fig. 2C). Cells treated with 30 or 50 µM ANA-12 were 1.1- to 1.3-fold less proliferative than DMSO-treated control cells (Fig. S3H); only a small change in proliferation accompanied the observed decrease in migration. These results demonstrate that ANA-12 treatment significantly reduces migration of lung adenocarcinoma cells.
We next investigated if the kinase activity of TrkB is required for lung cancer cell migration. Wild-type TrkB (TrkB-OE) or kinase dead TrkB (TrkB-K571M) was overexpressed in H23 cells, which have undetectable levels of TrkB expression (Fig. S4A). H23 TrkB-OE cells were 1.7-fold more migratory than empty vector control cells (Fig. 2D). However, TrkB-K571M did not change migration ability (Fig. 2D). As in other experiments above, a minor change in proliferation (1.1-fold increase) was seen in TrkB-OE cells (Fig. S4B). These data show that TrkB kinase activity is crucial for lung adenocarcinoma cell migration.
To determine the downstream mechanism of TrkB activation in lung adenocarcinoma metastasis, the protein lysates of shGFP and shTrkB H2030-BrM3 cells treated with 50 ng/mL BDNF were incubated on a human phospho-kinase array, which allows for simultaneous detection of relative site-specific phosphorylation of 43 kinases. Levels of phospho-Akt (pAkt), part of the PI3K-signaling pathway that promotes lung tumor progression via increased growth, motility, and invasion (14), were greatly reduced in the TrkB knockdown cells (Fig. S4C). Western blots showed that BDNF treatment increased pAkt levels in control cells and confirmed that pAkt levels were greatly reduced in H2030-BrM3 shTrkB and shTrkB-2 knockdown cells (Fig. 2E and Fig. S4D). Notably, levels of another TrkB target, pERK, were not reduced in TrkB knockdown cells (Fig. S4 C and E). Treatment of H2030-BrM3 cells with the pan-Trk inhibitor K252A also decreased pAkt levels compared with control DMSO-treated cells (Fig. S4F). Finally, expression of myristoylated, constitutively active Akt (15) rescued the migration defect of the TrkB knockdown cells. As expected, shTrkB-empty vector cells were 1.4-fold less migratory than shGFP-empty vector cells, whereas cells with constitutive Akt (shTrkB-Akt cells and shGFP-Akt cells) had comparable migration (Fig. 2F and Fig. S4 G and H). These results demonstrate that pAkt is downstream of TrkB signaling during lung adenocarcinoma cell migration.
TrkB Is Required for Endogenous Lung Tumor Metastasis.
We also determined that TrkB is crucial for metastasis of endogenous murine lung tumors that accurately mimic human lung adenocarcinomas. Adenomas and early stage adenocarcinomas develop in mice expressing oncogenic Kras from the endogenous locus, yet none of these Kras mice develop metastases (10, 16, 17). In contrast, advanced stage lung adenocarcinomas develop in Lox-Stop-Lox-KrasG12D;p53flox/flox (Kras;p53) animals after adenovirus expressing Cre recombinase (AdCre) is delivered to the lungs. Fifty percent of Kras;p53 mice also develop metastases, most commonly in the mediastinal lymph nodes (10). Interestingly, two cell lines derived from Kras primary lung tumors (LKR10 and LKR13) expressed no TrkB transcript, whereas two cell lines from primary Kras;p53 lung tumors (CK1750 and SC241) expressed significantly higher levels of TrkB (Fig. S5A). In addition, cell lines derived from Kras;p53 lung tumors that have metastatic capacity (TMet-1 and TMet-2) expressed 11.8-fold greater levels of TrkB transcript than those that lacked metastatic behavior (TnonMet-1 and TnonMet-3) (11) (Fig. S5B). Thus, higher TrkB expression was correlated with increased metastatic potential in murine lung tumors.
To directly test if loss of TrkB in endogeneous lung tumors would lead to decreased lung tumor metastatic rates, we bred Kras;p53 animals with mice containing conditional TrkB knockout alleles (TrkBflox/flox) (18) to generate Kras;p53;TrkB mice. Kras;p53 and Kras;p53;TrkB mice were euthanized 17–18 wk after AdCre delivery, when the majority of the Kras;p53 mice appeared sick. Both sets of animals developed grade 2, 3, and 4 lung adenocarcinomas with similar lung tumor burdens and number of lung tumors (Fig. 3 A and B; Fig. S5C). In addition, there was not a signficant difference between genotypes in the percentage of lung tumors of each grade or the number of grade IV invasive lung tumors, defined as having invaded into the stroma, bronchus, lymphatic vessels, or blood vessels (19) (Fig. S5 D and E). Lung tumors from both groups of animals had similar histology, percentages of Ki67+ cells, and CD31+ blood vessels (Fig. S5 C, F, and G), indicating that proliferation and angiogenesis rates were similar. However, Kras;p53 animals had significantly more lymph node metastases (Fig. S5C). Strikingly, whereas 57% of Kras;p53 mice had metastases, only 17% of Kras;p53;TrkB mice developed lymph node metastases; loss of TrkB decreased metastatic rates 3.3-fold (Fig. 3C). Kras;p53;TrkB mice also had significantly fewer metastases per mouse than Kras;p53 animals (Fig. 3D). These results showed that TrkB is critical for lung adenocarcinoma metastasis.
Fig. 3.

TrkB is required for metastasis in a murine model of lung cancer. (A) Quantification of lung tumor burden of Kras;p53 (n = 14) and Kras;p53;TrkB (n = 18) mice. (B) Average number of lung tumors of the mice analyzed in A. (C) Number of mice analyzed in A with lymph node metastases. (D) The average number of metastases per mouse analyzed in A. *P < 0.01. All error bars represent mean ± SEM.
TrkB Is Regulated by HIF-1α.
We next investigated the possibility that TrkB is regulated by hypoxia in lung adenocarcinoma cells. TRKB has 12 hypoxia response elements (HREs) 2 kb upstream of its start site, and a luciferase reporter containing the TrkB promoter was activated by hypoxia in neuroblastoma cells (20). During low-oxygen conditions, HIF-1α, the labile subunit of HIF-1, is stabilized and promotes transcription of genes with HREs to overcome hypoxic stress and to promote tumor progression and metastasis (21). Gene expression profiling of individual human lung tumors using the Oncomine cancer database (www.oncomine.org) revealed that TRKB was significantly correlated with HIF-1α (22) (Fig. S6A). To confirm that TrkB expression is increased by HIF-1α in lung cancers, we incubated A549, H322, H2030, and H2030-BrM3 cell lines in normoxic (21% O2) or hypoxic (1% O2) conditions. As expected, cell line HIF-1α protein levels greatly increased after incubation in 1% oxygen for 16 h (Fig. S6B), whereas HIF-2α levels were unchanged (Fig. S6C). TRKB transcript levels also increased 2.1- to 4-fold during hypoxic incubation (Fig. 4A), demonstrating that hypoxia significantly increased TrkB expression. Two shRNA hairpins (shHIF-1α, shHIF-1α-2) were used to successfully knock down HIF-1α protein levels in H2030-BrM3 cells, which express detectable levels of HIF-1α protein in normoxic conditions (Fig. S6D). TrkB transcript and protein levels were significantly decreased 1.8- to 2.2-fold and 3.9- to 10-fold in the HIF-1α knockdown cells compared with shGFP cells, respectively (Fig. 4B and Fig. S6E).
Fig. 4.
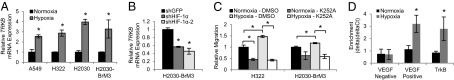
HIF-1α induces TrkB expression in human lung adenocarcinoma cell lines. (A) qPCR analysis of TRKB expression of the indicated cell lines cultured in hypoxia or normoxia. n = 3–4. *P < 0.04. (B) qPCR analysis of TRKB expression of H2030-BrM3 cells expressing the indicated hairpins. n = 3. *P < 0.01. (C) Transwell assays showing migration of the indicated cell lines cultured in normoxia or hypoxia toward media with DMSO or K252A. n = 3. *P < 0.03. (D) qPCR using VEGF negative or positive control or TrkB promoter primers and DNA purified from HIF-1α ChIP in A549 cells incubated in normoxia or hypoxia. n = 3. The enrichment relative to β-actin and the input is shown. *P < 0.01. All error bars represent mean ± SEM.
Because hypoxia has been demonstrated to increase tumor cell migration and metastasis (21), we next wanted to test if upregulation of TrkB in hypoxic cells enhanced migration. After confirming that H322 and H2030-BrM3 lung cancer cell lines were significantly more migratory when incubated in hypoxic conditions (Fig. 4C), cells were treated with the pan-Trk inhibitor K252a. K252a treatment significantly decreased migration of hypoxic cells 2- to 3.5-fold compared with DMSO vehicle-treated control cells (Fig. 4C), indicating that the enhanced ability of lung tumor cells to migrate during hypoxia is at least partially due to increased TrkB signaling.
Finally, to determine if HIF-1α directly regulates TrkB expression, chromatin immunoprecipitation (ChIP) with a HIF-1α antibody on A549 cells cultured in normoxic or hypoxic conditions was performed. As expected, a negative control region 5 kb upstream of the VEGF promoter did not show enrichment in the hypoxic cells (23), but a positive control region containing several HREs in the VEGF promoter was enriched 2.8-fold (24) (Fig. 4D). The TrkB promoter was similarly enriched 3.2-fold in the cells incubated in hypoxia compared with those cultured in normoxia (Fig. 4D). These results clearly demonstrated that HIF-1α directly regulates TrkB expression in human lung cancer cells.
TrkB Expression Is Correlated with Aggressive Human Lung Tumors.
To determine the relevance of TrkB expression in human lung cancers, we evaluated TrkB expression in six human lung adenocarcinoma gene expression data sets available in the Oncomine cancer database (www.oncomine.org). TRKB expression was 3.9-fold higher in the lung tumors of patients who died 1 y after treatment than those who survived (25) (Fig. S7A). Our analysis of these data also revealed that higher TRKB expression was significantly correlated with more aggressive disease using four different criteria for clinical relevance among six microarray studies: tumor expression, tumor stage, metastasis status, and survival (Fig. S7B). To verify that TrkB expression is increased in more aggressive human lung tumors, we analyzed TrkB transcript and protein levels in 22 primary human lung adenocarcinoma samples. Strikingly, TrkB transcript and protein levels were 5.4- and 3.3-fold higher, respectively, in more advanced stage IIIB-IV tumors compared with earlier stage IA-IIIA lesions (Fig. 5A and Fig. S7C). Human lung tumor staging takes into account the size of the primary tumor, lymph node involvement, and presence of distant metastases. HIF-1α transcript levels were also significantly higher in these stage IIIB-IV tumor samples (Fig. S7D), and there was a significant correlation between HIF-1α and TRKB in these specimens (Fig. S7E). pTrkB protein levels were also 4.1-fold higher in poorly differentiated tumors compared with more differentiated tumors (Fig. S7F).
Fig. 5.

High TrkB expression is correlated with poor lung adenocarcinoma patient prognosis. (A) qPCR analysis showing TRKB expression in stage IA-IIIA tumors (n = 18) relative to stage IIIB-IV human lung adenocarcinoma tumors (n = 4). *P < 0.01. (B) Presence of TrkB expression in genotyped human lung adenocarcinomas. (C) Presence of distant metastases and smoking status in patients at the time of diagnosis or following treatment. All error bars represent mean ± SEM.
To further examine the spectrum of TrkB-expressing human lung tumors, a set of lung adenocarcinomas with known oncogenic mutations was stained for TrkB. TrkB staining specific to the cellular membrane and cytoplasm (Fig. S7G) was scored as “no expression” (0), “low expression” (1), or “high expression” (2) (Fig. S7H). Interestingly, tumors with TrkB expression (level 1 or 2 staining) were significantly more likely to be wild-type for KRAS and epidermal growth factor receptor (EGFR) (Fig. 5B). Thus, upregulation of TrkB may be important in particular subsets of lung cancer patients, depending on their tumor genotype.
To determine if TrkB protein levels in patient lung tumor samples predict a greater rate of metastasis, we stained a well-characterized tissue microarray containing triplicate core lung adenocarcinoma sections from 89 patients (26). TrkB expression was correlated with the presence of distant metastases, supporting our findings that TrkB is crucial for lung adenocarcinoma metastasis; patients with tumors expressing TrkB (level 1 or 2 staining) were 2.2-fold more likely to develop distant metastases than patients with tumors expressing no TrkB (level 0 staining) (Fig. 5C). In addition, patients with tumors expressing TrkB smoked an average of 22 pack years more than patients with tumors expressing no TrkB (Fig. 5C). Adenocarcinomas with TrkB expression also tended to be a later pathological stage and less differentiated than those with no TrkB expression (Table S1), although these differences were not significant. Overall, these findings clearly illustrate that high TrkB expression in human lung adenocarcinoma samples is correlated with metastasis.
Discussion
Our data highlight several mechanisms by which lung adenocarcinoma cells can become metastatic and how they may be therapeutically targeted (Fig. S8). We show for the first time, to our knowledge, that TrkB is crucial for lung cancer metastasis of endogenous tumors in vivo and specifically point to a role for TrkB in the migratory steps of the metastatic cascade. Conditional loss of TrkB signaling significantly reduced the incidence of metastases in the Kras;p53 lung adenocarcinoma model, and, importantly, Kras;p53 and Kras;p53;TrkB mice had similar lung tumor numbers, size, and histological grade, showing that TrkB functions specifically during metastasis.
In lung cancer cell lines, there is some evidence that TrkB signaling may contribute to other steps of metastasis, including proliferation and invasion. Transient knockdown of TrkB in the lung adenocarcinoma cell line A549 inhibited cell invasion in culture (27), and treatment of A549 cells with the pan-Trk inhibitor K252a induced cell death and reduced the ability of the cells to form colonies in soft agar (28). Reduction of TrkB signaling similarly decreased invasion, soft agar colony formation, and tumor xenograft growth of small-cell lung cancer neuroendocrine cell lines (29). TrkB was originally identified as a supressor of anoikis (6), yet TrkB knockdown did not alter anoikis in our studies (Fig. S9A), arguing against a role for this pathway in metastatic lung cancer cell survival. Overexpression of TrkB in nonmalignant rat intestinal epithelial or endometrial carcinoma cells led to an epithelial-mesenchymal transition (EMT)-like state dependent on the Twist-Snail axis and Zeb1 (30–32), and treatment of pleural effusion cells with high concentrations of the pan-Trk inhibitor K252A or knockdown of TrkB decreased expression of SLUG, TWIST, SNAIL, and VIMENTIN, indicating that cells with lower levels of TrkB were in less of an EMT-like state (33). Our shTrkB cell lines did not express significantly different levels of ECADHERIN, TWIST, or SNAIL mRNA transcript compared with shGFP control cells (Fig. S9 B and C), nor did they exhibit EMT-related morphological changes. Although we cannot rule out that TrkB signaling may be important in other steps of the lung cancer metastatic cascade, our data clearly show a role for TrkB in lung cancer cell migration.
Our data suggest that lung adenocarcinoma cells become metastatic via increased expression of wild-type TrkB rather than via oncogenic mutation of TrkB. Interestingly, ∼3% of lung adenocarcinomas harbor TrkB exon missense mutations (9/230, 3.9%, https://tcga-data.nci.nih.gov/tcga/; 23/848, 2.7%, www.sanger.ac.uk/genetics/CGP/cosmic/), yet the impact of these mutations on TrkB function is not understood. Notably, none of the TrkB mutations in lung cancer that have been investigated have shown any demonstratable gain-of-function activity; known mutations were not sufficient for transformation and did not impact migration (34). Our data illustrate the important point that metastatic drivers need not be mutated in lung cancer but can be activated by posttranscriptional, posttranslational, or epigenetic means, similar to what has been described in prostate cancer (35). Our data linking TrkB expression to metastasis in patient samples are supported by a study that showed that TrkB expression was higher in NSCLCs with lymph node metastases than in NSCLCs with no lymph node metastases (27). Finally, the correlation of TrkB expression with smoking history is interesting, as it is known that smoking increases lung alveolar hypoxia (36). This supports our discovery that hypoxia induces TrkB expression in lung cancer cells and that TrkB and HIF-1α are correlated in lung tumor specimens.
Our data identify both up- and downstream mechanisms by which TrkB drives lung adenocarcinoma metastasis. We show that HIF-1α directly upregulates TrkB expression. In lung cancer, intratumoral hypoxia correlates with a decreased overall survival (37). Although loss of HIF-1α in the Kras lung adenocarcinoma mouse model did not impact tumorigenesis (38), HIF-1α has not been examined in the metastatic Kras;53 model. We also show that the kinase activity of TrkB is crucial for lung cancer cell migration. Although we cannot rule out the possibility that other signaling pathways may also be involved, our data suggest that TrkB drives migration at least partially by activating Akt signaling. PI3K signaling is amplified in lung cancers and their metastases, and overexpression of pAkt has been reported as a poor prognostic factor for NSCLC patients with stage I primary tumors (14). Studies with Akt inhibitors would help clarify if, and to what extent, TrkB-stimulated migration relies on Akt activation. Similarly, MEK inhibitors could be used to determine if ERK signaling is not crucial as our results suggest. We found that lung tumors expressing TrkB were more often wild type for KRAS and EGFR, suggesting that overexpression of TrkB may be an alternative way for tumors to enhance PI3K signaling. Targeting TrkB may be a useful strategy for patient subsets for whom there is no currently available targeted therapy.
These studies demonstrate the crucial role of signals produced by the local and metastatic microenvironment in lung adenocarcinoma progression. Our data suggest that fibroblasts in the lung tumor microenvironment secrete BDNF and induce the metastatic behavior of TrkB-expressing tumor cells by promoting migration into nearby tissue. Once tumor cells have escaped the primary tumor and have entered the blood or lymphatics, they may migrate (or extravasate) toward a gradient of BDNF secreted by metastatic sites such as the lymph nodes. Supporting this hypothesis, we found that the tissues to which Kras;p53 mouse lung tumor cells routinely metastasize, including the adrenal glands, kidney, and liver, expressed the highest levels of BDNF. Whereas it is known that lymphatic endothelial cells can secrete protein gradients that attract tumor cells (39), our studies demonstrate that lymph node fibroblasts may also provide these homing signals. Although a role for other cell types cannot be ruled out, these studies demonstrate that fibroblasts in the lung and lymph node microenvironment are the main source of BDNF. We have also shown that lung tumor cells secrete BDNF. Similar to the autocrine BDNF mechanism that neuron precursors use to amplify the migration gradient during development (12), an autocrine BDNF loop may also be important for tumor cell migration. Our findings support the idea that metastatic cancer cells harness pathways that are critical for normal developmental processes such as migration.
Taken together, our studies suggest that TrkB is an important therapeutic target in metastatic lung adenocarcinoma. Because therapeutically inhibiting transcription factors like HIF-1α is challenging and the tumor specificity of HIF-1α inhibition remains unclear (21), inhibition of the hypoxia-induced target genes crucial for metastasis (21), such as TrkB, are more amenable targets. Because pAkt is downstream of TrkB activation in lung cancer migration, targeting TrkB may be another way to therapeutically dampen PI3K signaling in certain lung tumors (14). Finally, we demonstrate that the TrkB-specific inhibitor ANA-12 inhibits lung adenocarcinoma cell line migration. Our studies indicate that it may be beneficial to target TrkB or the BDNF gradient secreted by the tumor microenvironment to limit the dissemination of lung cancer cells and improve patient survival rates.
Materials and Methods
Generation of Cell Lines.
Human lymph node fibroblasts and cell lines were purchased from ScienCell (2530) or the American Type Culture Collection. See SI Materials and Methods for details.
In Vitro Assays.
Migration assays were performed with transwell plates according to the manufacturer’s instructions (Corning). See SI Materials and Methods for details.
Antibody Arrays and Immunoprecipitations.
Medias were incubated on antibody array I membranes (AAH-BLM-1–2, RayBiotech). See SI Materials and Methods for details.
Quantitative RT-PCR Gene Expression Analysis.
RNA and cDNA were prepared using standard methods, and Taqman probes, including GAPDH as an endogenous control, were used with a StepOnePlus Real-Time PCR System (Applied Biosystems). See SI Materials and Methods for details.
Mice and Histology.
All animal studies were approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee. Lox-Stop-Lox-KrasG12D (Kras), Lox-Stop-Lox-KrasG12D;p53flox/flox (Kras;p53), and TrkBflox/flox (TrkB) mice have been described (10, 16, 18). See SI Materials and Methods for details on AdCre infections, histology, and transplantation assays.
Phospho-Kinase Arrays and Immunoblots.
BDNF-treated cell lysates were incubated on Proteome Profiler Human Phospho-Kinase Arrays (R&D Systems) or immunoblots using standard procedures. See SI Materials and Methods for details.
ChIP.
ChIP in normoxic and hypoxic conditions was performed as previously described (23). See SI Materials and Methods for details.
Statistics.
Unpaired two-tailed Student t tests or ANOVA were performed unless otherwise noted. See SI Materials and Methods for details.
Supplementary Material
Acknowledgments
We thank L. Parada for the TrkB conditional knockout mice; S. Grande for tail-vein technical assistance; J. Zhao for the Akt plasmids; J. Massague for cell lines; A. Kung, G. Qing, and C. Simon for ChIP advice; the Cancer Genome Atlas Research Network for use of the lung adenocarcinoma TCGA dataset; and K. Cichowski, R. Segal, B. Zetter, L. Zon, and members of the C.F.K. laboratory for critical reading of the manuscript and helpful discussions. This work was supported by American Cancer Society Postdoctoral Fellowship PF-09-121-01-DDC; a Harvard Stem Cell Institute National Institutes of Health (NIH) Training Grant; a Free to Breathe (formerly National Lung Cancer Partnership) 2012 Young Investigator Research Grant (to K.W.S.); the Howard Hughes Medical Institute Medical Research Fellowship Program (J.L.); the Department of Defense Air Force Office of Scientific Research, National Defense Science and Engineering Graduate Fellowships, 32 Code of Federal Regulations 168a (to A.N.L.); the Dana–Farber Harvard Cancer Center Lung Cancer Specialized Programs of Research Excellence Grants P50 CA090578, R01 AG2400401, R01 CA122794, and R01 CA140594 (to K.-K.W.); the Children’s Hospital Stem Cell Program (T.M.S.); the Glenn Foundation for Medical Research, American Cancer Society New Scholar Award (to M.C.H.); the V Foundation for Cancer Research; American Cancer Society Research Scholar Grant RSG-08-082-01-MGO; the Freeman Trust; the Harvard Stem Cell Institute; NIH/National Heart, Lung, and Blood Institute U01 HL100402 and RO1 HL090136; the Thoracic Foundation; Joan’s Legacy; and the Boston Children’s Hospital Faculty Career Development Fellowship (to C.F.K.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1404399111/-/DCSupplemental.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63(1):11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Desmet CJ, Peeper DS. The neurotrophic receptor TrkB: A drug target in anti-cancer therapy? Cell Mol Life Sci. 2006;63(7-8):755–759. doi: 10.1007/s00018-005-5490-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thiele CJ, Li Z, McKee AE. On Trk: The TrkB signal transduction pathway is an increasingly important target in cancer biology. Clin Cancer Res. 2009;15(19):5962–5967. doi: 10.1158/1078-0432.CCR-08-0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zheng W, et al. Overexpression of tyrosine kinase receptor B promotes metastasis of ovarian serous adenocarcinoma by lymphangiogenesis. Tumori. 2011;97(6):756–761. doi: 10.1177/030089161109700613. [DOI] [PubMed] [Google Scholar]
- 5.Geiger TR, Peeper DS. Critical role for TrkB kinase function in anoikis suppression, tumorigenesis, and metastasis. Cancer Res. 2007;67(13):6221–6229. doi: 10.1158/0008-5472.CAN-07-0121. [DOI] [PubMed] [Google Scholar]
- 6.Douma S, et al. Suppression of anoikis and induction of metastasis by the neurotrophic receptor TrkB. Nature. 2004;430(7003):1034–1039. doi: 10.1038/nature02765. [DOI] [PubMed] [Google Scholar]
- 7.Terry J, et al. Immunohistochemical expression of neurotrophic tyrosine kinase receptors 1 and 2 in lung carcinoma: Potential discriminators between squamous and nonsquamous subtypes. Arch Pathol Lab Med. 2011;135(4):433–439. doi: 10.5858/2010-0038-OA.1. [DOI] [PubMed] [Google Scholar]
- 8.Okamura K, et al. Expression of TrkB and BDNF is associated with poor prognosis in non-small cell lung cancer. Lung Cancer. 2012;78(1):100–106. doi: 10.1016/j.lungcan.2012.07.011. [DOI] [PubMed] [Google Scholar]
- 9.Braun A, et al. Cellular sources of enhanced brain-derived neurotrophic factor production in a mouse model of allergic inflammation. Am J Respir Cell Mol Biol. 1999;21(4):537–546. doi: 10.1165/ajrcmb.21.4.3670. [DOI] [PubMed] [Google Scholar]
- 10.Jackson EL, et al. The differential effects of mutant p53 alleles on advanced murine lung cancer. Cancer Res. 2005;65(22):10280–10288. doi: 10.1158/0008-5472.CAN-05-2193. [DOI] [PubMed] [Google Scholar]
- 11.Winslow MM, et al. Suppression of lung adenocarcinoma progression by Nkx2-1. Nature. 2011;473(7345):101–104. doi: 10.1038/nature09881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou P, et al. Polarized signaling endosomes coordinate BDNF-induced chemotaxis of cerebellar precursors. Neuron. 2007;55(1):53–68. doi: 10.1016/j.neuron.2007.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cazorla M, et al. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J Clin Invest. 2011;121(5):1846–1857. doi: 10.1172/JCI43992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wojtalla A, Arcaro A. Targeting phosphoinositide 3-kinase signalling in lung cancer. Crit Rev Oncol Hematol. 2011;80(2):278–290. doi: 10.1016/j.critrevonc.2011.01.007. [DOI] [PubMed] [Google Scholar]
- 15.Boehm JS, et al. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129(6):1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 16.Jackson EL, et al. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001;15(24):3243–3248. doi: 10.1101/gad.943001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnson L, et al. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410(6832):1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- 18.Luikart BW, et al. TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci. 2005;25(15):3774–3786. doi: 10.1523/JNEUROSCI.0041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DuPage M, Dooley AL, Jacks T. Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat Protoc. 2009;4(7):1064–1072. doi: 10.1038/nprot.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martens LK, Kirschner KM, Warnecke C, Scholz H. Hypoxia-inducible factor-1 (HIF-1) is a transcriptional activator of the TrkB neurotrophin receptor gene. J Biol Chem. 2007;282(19):14379–14388. doi: 10.1074/jbc.M609857200. [DOI] [PubMed] [Google Scholar]
- 21.Bennewith KL, Dedhar S. Targeting hypoxic tumour cells to overcome metastasis. BMC Cancer. 2011;11:504. doi: 10.1186/1471-2407-11-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wachi S, Yoneda K, Wu R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics. 2005;21(23):4205–4208. doi: 10.1093/bioinformatics/bti688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xia X, et al. Integrative analysis of HIF binding and transactivation reveals its role in maintaining histone methylation homeostasis. Proc Natl Acad Sci USA. 2009;106(11):4260–4265. doi: 10.1073/pnas.0810067106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang MH, et al. Direct regulation of TWIST by HIF-1α promotes metastasis. Nat Cell Biol. 2008;10(3):295–305. doi: 10.1038/ncb1691. [DOI] [PubMed] [Google Scholar]
- 25.Beer DG, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8(8):816–824. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 26.Barletta JA, et al. Clinical significance of TTF-1 protein expression and TTF-1 gene amplification in lung adenocarcinoma. J Cell Mol Med. 2009;13(8B):1977–1986. doi: 10.1111/j.1582-4934.2008.00594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, et al. TrkB is highly expressed in NSCLC and mediates BDNF-induced the activation of Pyk2 signaling and the invasion of A549 cells. BMC Cancer. 2010;10:43. doi: 10.1186/1471-2407-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perez-Pinera P, et al. The Trk tyrosine kinase inhibitor K252a regulates growth of lung adenocarcinomas. Mol Cell Biochem. 2007;295(1-2):19–26. doi: 10.1007/s11010-006-9267-7. [DOI] [PubMed] [Google Scholar]
- 29.Osborne JK, et al. NeuroD1 regulates survival and migration of neuroendocrine lung carcinomas via signaling molecules TrkB and NCAM. Proc Natl Acad Sci USA. 2013;110(16):6524–6529. doi: 10.1073/pnas.1303932110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smit MA, Geiger TR, Song J-Y, Gitelman I, Peeper DS. A Twist-Snail axis critical for TrkB-induced epithelial-mesenchymal transition-like transformation, anoikis resistance, and metastasis. Mol Cell Biol. 2009;29(13):3722–3737. doi: 10.1128/MCB.01164-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smit MA, Peeper DS. Zeb1 is required for TrkB-induced epithelial-mesenchymal transition, anoikis resistance and metastasis. Oncogene. 2011;30(35):3735–3744. doi: 10.1038/onc.2011.96. [DOI] [PubMed] [Google Scholar]
- 32.Bao W, et al. Upregulation of TrkB promotes epithelial-mesenchymal transition and anoikis resistance in endometrial carcinoma. PLoS ONE. 2013;8(7):e70616. doi: 10.1371/journal.pone.0070616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ricci A, et al. TrkB is responsible for EMT transition in malignant pleural effusions derived cultures from adenocarcinoma of the lung. Cell Cycle. 2013;12(11):1696–1703. doi: 10.4161/cc.24759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harada T, et al. Role and relevance of TrkB mutations and expression in non-small cell lung cancer. Clin Cancer Res. 2011;17(9):2638–2645. doi: 10.1158/1078-0432.CCR-10-3034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Min J, et al. An oncogene-tumor suppressor cascade drives metastatic prostate cancer by coordinately activating Ras and nuclear factor-kappaB. Nat Med. 2010;16(3):286–294. doi: 10.1038/nm.2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kent BD, Mitchell PD, McNicholas WT. Hypoxemia in patients with COPD: Cause, effects, and disease progression. Int J Chron Obstruct Pulmon Dis. 2011;6:199–208. doi: 10.2147/COPD.S10611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Swinson DE, et al. Carbonic anhydrase IX expression, a novel surrogate marker of tumor hypoxia, is associated with a poor prognosis in non-small-cell lung cancer. J Clin Oncol. 2003;21(3):473–482. doi: 10.1200/JCO.2003.11.132. [DOI] [PubMed] [Google Scholar]
- 38.Mazumdar J, et al. HIF-2α deletion promotes Kras-driven lung tumor development. Proc Natl Acad Sci USA. 2010;107(32):14182–14187. doi: 10.1073/pnas.1001296107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Issa A, Le TX, Shoushtari AN, Shields JD, Swartz MA. Vascular endothelial growth factor-C and C-C chemokine receptor 7 in tumor cell-lymphatic cross-talk promote invasive phenotype. Cancer Res. 2009;69(1):349–357. doi: 10.1158/0008-5472.CAN-08-1875. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.