

Abstract
Carcinoid tumors are slow growing and highly vascular neuroendocrine neoplasms that are increasing in incidence. Previously, we showed that carcinoid tumors express vascular endothelial growth factor receptor 2 (VEGFR-2) in the epithelial compartment of carcinoid tumor sections; yet, its role is not completely understood. The purpose of our study was to: (i) assess the expression of VEGFR-2 in the novel human carcinoid cell line BON, (ii) to determine the role of PI3K/Akt signaling on VEGFR-2 expression and (iii) to assess the effect of VEGFR-2 on BON cell invasion, migration and proliferation. We found that, although VEGFR-2 is expressed in BON cells, reduction in VEGFR-2 expression actually enhanced proliferation, invasion, and migration of the BON cell line. Also, expression of VEGFR-2 was inversely related to PI3K signaling. Carcinoid liver metastases in mice demonstrated decreased VEGFR-2 expression. Furthermore, the expression of a truncated, soluble form of VEGFR-2 (sVEGFR-2), a protein demonstrated to inhibit cell growth, was detected in BON cells. The presence of VEGFR-2 in the epithelial component of carcinoid tumors and in the BON cell line suggests an alternate role for VEGFR-2, in addition to its well-defined role in angiogenesis. The expression of sVEGFR-2 may explain the inverse relationship between VEGFR-2 expression and PI3K/Akt signaling and the inhibitory effect VEGFR-2 has on BON cell proliferation, migration and invasion.
Keywords: cell motility and migration, tumor markers and detection of metastasis, angiogenic factors and receptors
Carcinoid tumors are slow growing neuroendocrine neoplasms arising from the enterochromaffin cells of the gut.1,2 Because of their indolent nature, most carcinoids are not detected until metastases or the carcinoid syndrome has developed in patients.1,2 The carcinoid syndrome represents a constellation of symptoms, including flushing, diarrhea and cardiac valve fibrosis caused by the amine and peptide products secreted from carcinoid tumors.3 While carcinoid tumors are relatively rare, data from the Surveillance Epidemiology and End Results (SEER) database indicates an increasing incidence of carcinoid tumors since the 1970s.4 The increased frequency of carcinoids may be due to a greater awareness of the disease, as well as enhanced diagnostic techniques. Currently, surgery is the most effective treatment for this disease as carcinoid tumors do not respond well to standard chemotherapeutic agents.2,5
Because carcinoid tumors generally do not have altered expression or mutation of common tumor suppressors or oncogenes, the development of novel treatments for carcinoid tumors has focused on targeting growth factor receptors, such as vascular endothelial growth factor receptor 2 (VEGFR-2). Studies have demonstrated the efficacy of treating carcinoid disease with the VEGF-signaling inhibitors bevacizumab and sunitinib malate, as a unique aspect of neuroendocrine tumors is their highly vascular nature.6–8 VEGF signaling is well known to stimulate angiogenesis during tumor development. The VEGF protein binds to one of its receptors, VEGFR-1, -2 or -3, to promote either physiologic or pathologic angiogenesis or lymphangiogenesis, with VEGFR-2 and VEGFR-3 normally confined to endothelial cells lining blood vessels or lymphatic vessels, respectively.9,10 Previously, we detected the presence of VEGFR-2 in ∼48% of carcinoid tumors with the highest expression in foregut and hindgut carcinoids.11 VEGFR-2, also designated as kinase domain receptor (KDR), is a tyrosine kinase that can signal through the phosphatidylinositol-3-kinase/protein kinase B (PI3K/Akt) pathway.9,10 When VEGF binds to its receptor, it activates the PI3K protein, which is made up of a p85 regulatory subunit and a p110 catalytic subunit.12 Activated-PI3K results in the phosphorylation of Akt.12 Akt signaling can promote cellular metabolism, survival and proliferation.12 PI3K signaling can be inhibited with either the endogenous inhibitor phosphatase and tensin homolog deleted on chromosome ten (PTEN) or synthetic inhibitors such as wortmannin.13,14 PI3K/Akt signaling is upregulated in many types of cancer, including carcinoid tumors.15–17 This frequently occurs through either constitutive activation of the PI3K subunits or downregulation or mutation of PTEN. While the role of VEGF signaling in angiogenesis is well established, the mechanisms by which the expression of VEGFR is regulated are still unclear, and the role of VEGFR on carcinoid tumor cell proliferation and metastasis is not fully understood. Studies have demonstrated that VEGF signaling through the PI3K/Akt pathway regulates the expression of the transforming growth factor-β (TGF-β) and connective tissue growth factor (CTGF) genes; the protein products play a role in metastasis and fibrosis.18,19 Thus, it is now hypothesized that VEGF signaling has alternate roles, such as promotion of fibrosis,20,21 in addition to its contribution to angiogenesis. Furthermore, recent studies have indicated that VEGFR-2 has an inhibitory effect on cell proliferation22 and tumor metastasis.23,24 Albuquerque et al.22 demonstrated the expression of a soluble form of VEGFR-2 (sVEGFR-2), resulting from retention of intron 13 in the mature mRNA transcript, which acts as an inhibitor of lymphangiogenesis by sequestering VEGF-C and preventing it from binding and activating membrane bound VEGFR-3.
The cell line BON, derived from a carcinoid lymph node metastasis, was established and characterized in our laboratory and has served as a unique model to understand carcinoid cell biology.25,26 BON cells synthesize and secrete neurotensin, pancreastatin, chromogranin A (CgA) and serotonin.25,27–29 Various growth and cell signaling inhibitors are noted to inhibit BON cell growth in vitro and in vivo.26 Recently, we have shown that BON cells, when injected into the spleen of athymic nude mice, metastasize to the liver and produce symptoms consistent with carcinoid syndrome.6 Additionally, we demonstrated that treating mice with BON liver metastases with the VEGF antibody bevacizumab30 significantly decreased tumor growth and metastasis compared to vehicle-treated mice. The purpose of our current study was to assess the expression, regulation and function of VEGFR-2 in the BON human carcinoid cell line.
Material and Methods
Materials
Dulbecco's modified Eagle medium (DMEM)/F12K (50/50) media was purchased from Mediatech (Herndon, VA). Fetal bovine serum was purchased from Atlanta Biologicals (Lawrenceville, GA). An Envision+® System-HRP (DAB) kit for immunohistochemistry was purchased from Dako (Carpinteria, CA). An endocrine tumor tissue array, containing carcinoid sections, was obtained from U.S. Biomax (Rockville, MD). ECL™ Western blotting detection reagents were purchased from GE Healthcare (Buckinghamshire, United Kingdom). A Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Molecular Technologies (Rockville, MD). Antibody against chromogranin A was obtained from Abcam (Cambridge, MA). Antibodies against VEGF-C and VEGFR-3 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Other antibodies were purchased from Cell Signaling Technology (Danvers, MA). An RNeasy Mini kit was purchased from Qiagen (Valencia, CA), and a high-capacity cDNA reverse transcription kit was obtained from Applied Biosystems (Foster City, CA). Primers for qRT-PCR were purchased from Integrated DNA Technologies (Coralville, IA). VEGF-A was purchased from Sigma (St. Louis, MO). Recombinant human VEGF-C was obtained from R&D Systems (Minneapolis, MN). PTEN and VEGFR-2 shRNA were purchased from Open Biosystems (Huntsville, AL). For invasion assays, matrigel was obtained from BD Biosciences (San Jose, CA) and 4′,6-diamidino-2-phenylindole (DAPI) and glutaraldehyde were purchased from Sigma. A soluble VEGFR-2 enzyme-linked immunosorbent assay (ELISA) kit was purchased from R&D Systems (Minneapolis, MN). HMVEC-L cells were provided by Dr. Juan P. Olano (The University of Texas Medical Branch [UTMB], Galveston, TX).
Cell culture and establishment of stable PTEN knockdown or VEGFR-2 knockdown cell lines
The BON cell line was last authenticated in October 2009 at the Johns Hopkins Genetic Resources Core Facility with short tandem repeat analysis using an Identifiler identification kit (Applied Biosystems). BON cells were maintained in DMEM/F12K (50/50) media, supplemented with 5% fetal bovine serum (FBS) in 5% CO2 at 37°C. To increase PI3K/Akt signaling, BON cells were transfected with GFP-tagged shRNA to PTEN, the natural inhibitor of PI3K/Akt, and selected in medium containing puromycin (2 μg/ml). To decrease VEGFR-2 signaling, BON cells were transfected with GFP-tagged shRNA to VEGFR-2 under the same conditions.
Cell proliferation assay
Cells were plated in 96-well plates (4,000 cells per 50 μl in each well). After 1 day (time - 0 hr), 50 μl of serum free DMEM/F12K (50/50) media or media containing VEGF-A, at a final concentration of either 25 or 100 ng/ml, was added to the wells. The media from all groups was supplemented with the VEGF-A cofactor heparin sulfate at a final concentration of 25 ng/ml. Cells treated with 5% FBS were used as a positive control. At 24, 48, 72, 96 and 120 hr, cell proliferation was measured with CCK-8 according to the manufacturer's protocol. The cell proliferation assay was performed with eight replicates.
Immunohistochemistry and immunocytochemistry
Tissue sections were incubated overnight in primary antibodies diluted in antibody diluent (Dako), as described previously.11 Briefly, the sections were stained and assessed, as positive or negative, in a blinded fashion by an experienced pathologist. Protein staining was performed using an Envision+® System-HRP (DAB) kit; samples were counter-stained with hematoxylin. For immunocytochemistry, BON cells were grown to ∼70% confluence and immunofluorescent staining was performed according to the protocol for VEGFR-2 antibody from Cell Signaling Technology® (Danvers, MA).
Western blot analysis
Total protein was resolved on NuPAGE® 4–12% Bis-Tris gels (Invitrogen) and transferred to Sequi-Blot™ PVDF membranes (Bio-rad). Membranes were incubated with specific primary antibodies. Following incubation with a horseradish peroxidase-conjugated secondary antibody, proteins were visualized using an enhanced chemiluminescence (ECL) detection system.
qRT-PCR
Total RNA was purified using an RNeasy kit including an optional DNase treatment, and cDNA was generated with a high-capacity cDNA reverse transcription kit, according to the manufacturer's protocol. Amplification of Vegfr2 was performed using the following primers: 5′-GAACATTTGGG AAATCTCTTGC-3′ (left) and 5′-CGGAAGAACAATGTA GTCTTTGC-3′ (right) and probe #18 from the Universal ProbeLibrary. To detect sVegfr2 mRNA, an sVegfr2 fragment covering the splicing site (exon 13–intron 13 junction) was amplified with the following primers: 5′-CTTGCTCAAGACA GGAAGACC-3′ (left) and 5′-GAATTGTCTCCCTACCTAG GACTG-3′ (right) and probe #27 from the Universal ProbeLibrary. cDNA was normalized to 18s rRNA and calibrated relative to control.
Migration assay
A monolayer scratch assay was used to compare the migratory ability of the BON VEGFR-2 shRNA cell line compared to BON cells transfected with NTC shRNA. VEGFR-2 and NTC shRNA-transfected cell lines were cultured to confluence, scratched and photographed using phase contrast microscopy at 0, 24 and 48 hr. The minimum distance in micrometers between the wound edges of the scratch area was analyzed using Adobe Photoshop 7.0. For transwell migration assays, the bottoms of the inserts were coated with collagen. BON cells were plated in the inserts in DMEM/F12K (50/50) media supplemented with 0.1% bovine serum albumin (BSA). BSA alone (0.1%) or BSA (0.1%) and VEGF-C (50 ng/ml) were used as chemoattractants. After 5 hr, cells were fixed in methanol and stained with crystal violet. Stained cells were counted in four different fields with an inverted microscope. All experiments were performed in triplicate.
Invasion assay
A modified Boyden chamber invasion assay with Matrigel-coated Transwell chambers was performed, as described previously. 31 Briefly, BON cells transfected with VEGFR-2 shRNA were compared to those transfected with NTC shRNA to evaluate invasiveness. Cells were grown in DMEM/F12K (50/50) media supplemented with 2% BSA, and complete BON media was used as a chemoattractant. After 24 and 48 hr, the cells were fixed with 3% glutaraldehyde and stained with DAPI fluorescent staining. DAPI-stained cells were counted in four different fields with an inverted fluorescent microscope. All experiments were performed in triplicate.
sVEGFR-2 ELISA
To quantitate the amount of sVEGFR-2 produced by BON cells, an ELISA was performed with BON cell lysates according to the manufacturer's directions. An equal amount of protein from BON cell lysates was added into each well, and the sVEGFR-2 concentration was calculated as pg/μg total protein. The ELISA was performed in quadruplicate.
Animal studies
Male athymic nude mice (4–6 weeks) were purchased from Harlan-Sprague-Dawley. Mice were anesthetized with isoflurane, and BON cells (1 × 107 per 100 μl) were injected into the pancreas with a 27-gauge needle. After 10 weeks, mice were sacrificed and primary and metastatic tumors were excised and harvested for assessment. All studies were approved by the Institutional Animal Care and Use Committee of UTMB.
Statistical analysis
Descriptive statistics including means and standard deviations were calculated and displayed in bar graphs to summarize cell counts, invasion, migration and qRT-PCR measurements across cell culture treatment and control groups. General linear models including analysis of variance and repeated measures models were used to test main effects as well as interaction between factors which includes cell culture groups, time points of measurement etc. Contrast statements were generated from the model to test for specific pairwise comparisons as well as test for trends over time. Two group comparisons (e.g., control shRNA vs. VEGFR-2 shRNA) were performed using two-sample t-tests. Assumptions on data normality and equality of variance were verified to determine validity of the statistical tests and log transformations were performed if necessary. The null hypothesis was rejected when p < 0.05. Data analysis was conducted using statistical software, SAS®, Release 9.2.
Results
VEGFR-2 is expressed in carcinoid tumors and the BON cell line
The progression of carcinoid tumors is thought to be due to the overexpression of growth factor receptors.32 Previously, we detected the presence of VEGFR-2 in the epithelial component of ∼48% (22 of 46) of carcinoid tumors.11 As VEGFR-2 expression is generally confined to endothelial cells surrounding blood vessels,9,10 the finding of VEGFR-2 in carcinoid tumors prompted us to assess the expression of VEGFR-2 in the BON carcinoid cell line. BON cells express VEGFR-2, as indicated by Western blot analysis and immunocytochemistry (Figs. 1a and 1b) and qRT-PCR (data not shown). Human microvascular endothelial cells of the lung (HMVEC-L) and HT29 colon cancer cells were used as positive and negative controls, respectively, for VEGFR-2 expression. Importantly, our results in the BON cell line corroborate our findings of VEGFR-2 expression in the carcinoid tumor samples from our previous study.
Figure 1.
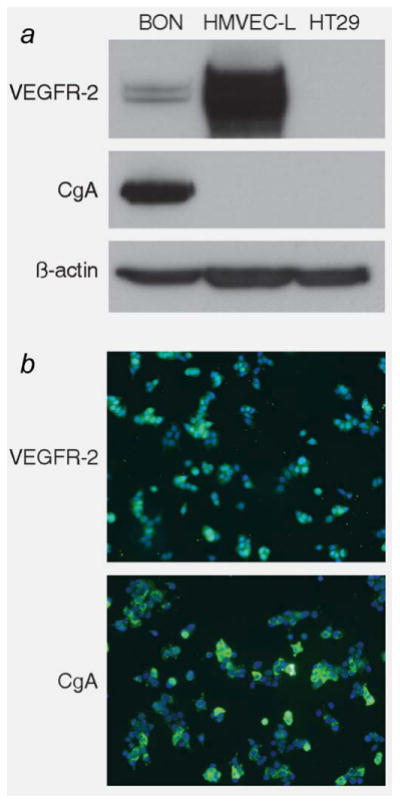
VEGFR-2 is expressed in the BON cell line. The expression of VEGFR-2 in the BON cell line was assessed with (a) Western blot and (b) immunocytochemistry. Human microvascular endothelial cells of the lung (HMVEC-L) were used as a positive control for VEGFR-2 expression, while the colon cancer cell line HT29 was used as a negative control. While VEGFR-2 is generally confined to endothelial cells lining blood vessels and involved in angiogenesis, these findings demonstrate that VEGFR-2 is expressed in the epithelial component, as well as the endothelial component of carcinoid tumors.
VEGF signals through the PI3K/Akt pathway without significantly affecting BON cell growth
To determine if VEGFR-2 is functional in BON cells, cells were serum-starved for 24 hr and then treated with VEGF-A ligand (25 ng/ml) over a time course. VEGF-A treatment activated VEGFR-2, as indicated by autophosphorylation, and activated the Akt pathway, with a peak in phospho-Akt (Ser 473) at 5 min after addition of the VEGF-A ligand (Fig. 2a). Given that PI3K/Akt signaling promotes cell proliferation, BON cells were treated with VEGF-A over an extended time course to evaluate if VEGF can increase BON cell growth. While VEGF-A treatment transiently increased Akt signaling, VEGF-A (25 ng/ml and 100 ng/ml) did not affect BON cell proliferation during the 120-hr time course (Fig. 2b). Furthermore, VEGF-A treatment did not increase activation of extracellular signal-regulated kinases (ERK1/2) (data not shown).
Figure 2.
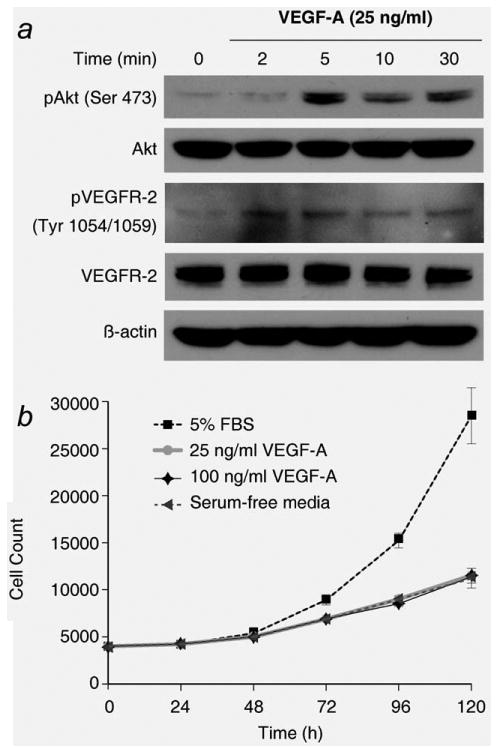
VEGFR-2 signaling has a limited effect on BON cell growth. (a) Incubation of BON cells with 25 ng/ml VEGF-A activates PI3K/Akt signaling, as demonstrated by Western blot. Maximal activation of Akt appears at 5 min following treatment with VEGF-A. (b) Treatment with VEGF-A (25 or 100 ng/ml) did not produce a significant change in BON cell proliferation, as assessed with a CCK-8 assay (p > 0.15 vs. serum-free media). A standard curve generated from a series of different cell counts was used to calculate cell number at each time point. The proliferation assay was performed with eight replicates.
The expression of VEGFR-2 in BON cells is inversely related to PI3K signaling
Interestingly, during the course of treating BON cells with either VEGFR-2 or PI3K inhibitors, we observed that PI3K inhibition increased VEGFR-2 expression. To investigate this phenomenon further, we altered PI3K signaling, by chemical and genetic means, and assessed the effect on VEGFR-2 expression. The organic compound wortmannin inhibits the PI3K-mediated conversion of PIP2 to PIP3, thereby preventing the phosphorylation of Akt.33 Treatment with wortmannin [500 nM in DMEM/F12K (50/50) media with 5% FBS] increased VEGFR-2 protein and mRNA expression over a time course (Figs. 3a and 3b; p < 0.0001). Akt Inhibitor VIII acts downstream of wortmannin, binding to the pleckstrin homology domain of Akt, thereby preventing Akt phosphorylation and activation at the cell membrane.34 As noted with wortmannin, Akt Inhibitor VIII [500 nM in DMEM/F12K (50/50) media with 5% FBS] also increased VEGFR-2 protein and mRNA expression (Figs. 3c and 3d; p = 0.0021). The effect of PI3K/Akt inhibition is specific to VEFGR-2 expression, as little to no change was detected in the expression of EGF and IGF-1β receptors (Figs. 3a and 3c). Treatment with vehicle control (DMSO) for 0 and 24 hr (Figs. 3a–3d; right) did not increase either VEGFR-2 mRNA or protein expression, indicating that the effect on VEGFR-2 expression is due to PI3K/Akt inhibition. Conversely, BON cells were transfected with shRNA to PTEN, the natural inhibitor of PI3K. Increased PI3K signaling, via a reduction in PTEN, specifically decreased VEGFR-2 protein and mRNA expression (Figs. 3e and 3f; p = 0.0004).
Figure 3.
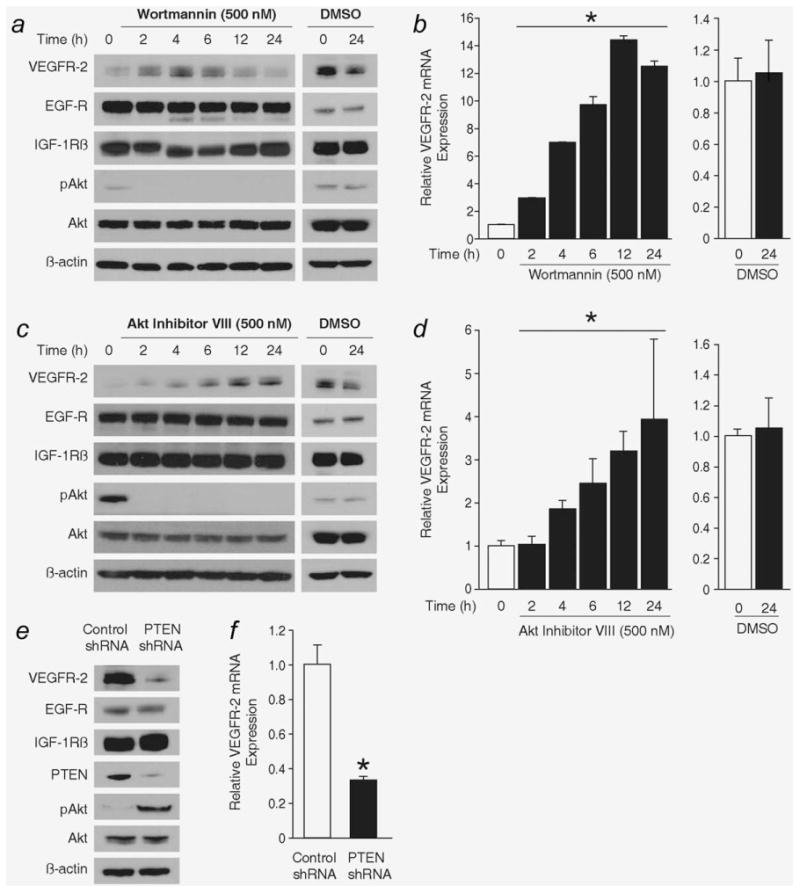
Altering PI3K signaling adjusts VEGFR-2 expression in BON cells. Inhibition of PI3K signaling with the chemical wortmannin increases VEGFR-2 protein and mRNA expression as assessed by (a) Western blot and (b) qRT-PCR, respectively (*, p < 0.0001 test for trend over time with wortmannin treatment). The inhibitory effect of wortmannin is demonstrated by the absence of Akt phosphorylation following wortmannin treatment. Akt Inhibitor VIII also increases VEGFR-2 (c) protein and (d) mRNA expression (*, p = 0.002 test for trend over time with Akt Inhibitor VIII treatment). (a–d), right) Treatment with the vehicle control (DMSO) had no effect on VEGFR-2 expression. Small hairpin RNA (shRNA) was used to stably reduce the expression of PTEN in the BON cell line. Increasing PI3K signaling decreased VEGFR-2 expression, as demonstrated by (e) Western blot and (f) qRT-PCR (*, p = 0.0004 vs. treatment with Control shRNA).
Metastatic carcinoid cells have reduced VEGFR-2 expression
A decrease in PTEN enhances the metastatic potential of colorectal cancer cells.35,36 Increased PI3K activity has also been noted in carcinoid tumors.15,17 Based on our finding that reduced PTEN activity decreases the expression of VEGFR-2 in BON cells, we hypothesized that VEGFR-2 expression is decreased in metastatic carcinoid cells. During metastasis, cells undergo epithelial-to-mesenchymal transition (EMT), which allows the cells to break away from the primary tumor, invade the blood stream and migrate to a new location.37,38 During EMT, the expression of several genes is altered to provide cells with an enhanced migratory and invasive potential. For example, TGF-β and Snail are increased in cells undergoing EMT, as these proteins signal to decrease the expression of the cell adhesion marker E-cadherin.37,38 BON cells with stably reduced PTEN expression displayed increased Snail and TGF-β and decreased E-cadherin expression compared to control BON cells (Fig. 4a).
Figure 4.
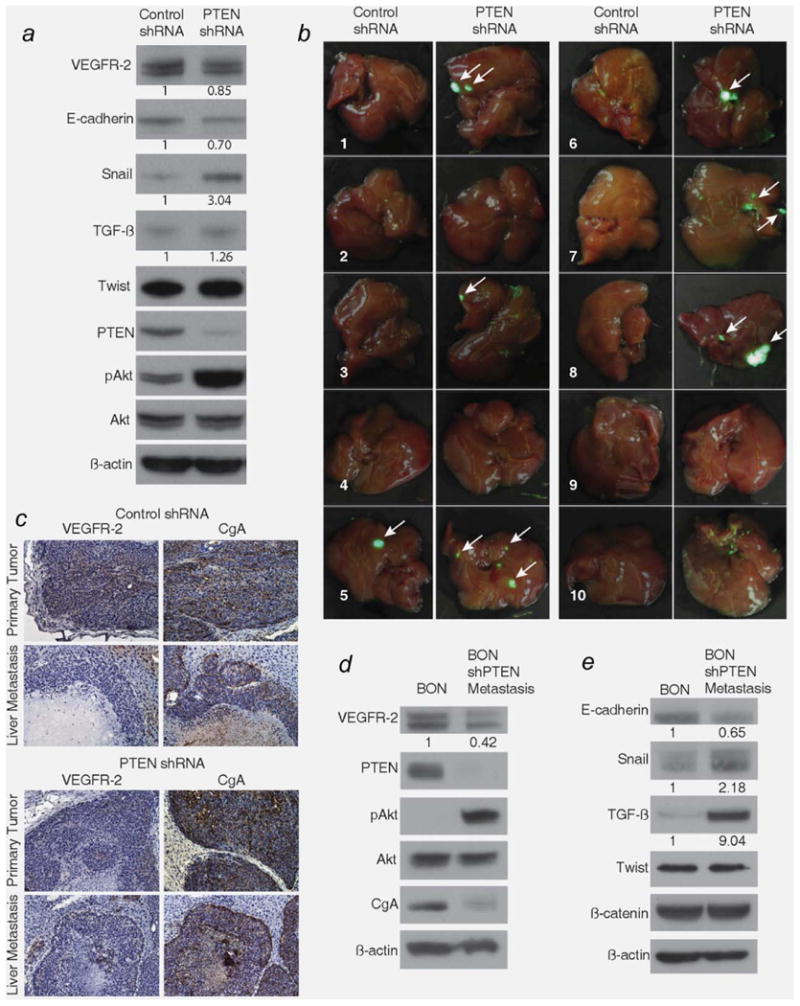
VEGFR-2 expression is decreased in metastatic carcinoid cells. (a) BON shPTEN cells were analyzed for changes in the expression of markers known to be involved in metastasis and were injected into the pancreas of athymic nude mice. Following sacrifice, liver metastases were noted (b) grossly and (c) histologically (×200). Only 10% of mice injected with BON shControl cells formed liver metastases, and VEGFR-2 was expressed in the primary tumor but not in the liver metastasis (c, top left). 60% of mice injected with BON shPTEN cells formed liver metastases compared to 10% injected with BON shControl cells (p = 0.057), and VEGFR-2 expression was reduced in both primary and metastatic tumors (c, bottom left). The liver metastatic cells from a BON shPTEN-injected mouse were harvested and cultured and displayed persistent reduction in (d) PTEN and VEGFR-2 compared to parental BON cells and E) alterations in markers known to be involved in metastasis, such as E-cadherin, snail and TGF-β. Densitometric analysis was performed to compare the expression of VEGFR-2, E-cadherin, Snail, and TGF-β in BON shPTEN and BON shPTEN metastasis cells to that in control BON cells; numbers under the lanes represent relative densitometric values.
To evaluate the expression of VEGFR-2 in carcinoid metastases, we injected BON cells (1 × 107) stably transfected with small hairpin RNA (shRNA), containing a puromycin resistance marker, directed to PTEN or non-targeting control into the pancreas of athymic nude mice. Previously, we demonstrated that injection of BON cells into the spleen of athymic nude mice resulted in liver metastases6; however, it is uncertain if the tumors in the liver are actual metastases or primary tumors, resulting from direct seeding to the liver immediately after injection. BON cells were derived from a metastatic pancreatic carcinoid tumor, so injection into the pancreas provides a better orthotopic model to analyze metastasis.
Only one of ten mice injected with non-targeting control shRNA BON cells developed liver metastases, while 60% of mice (six of ten) injected with PTEN shRNA BON cells developed one to three liver metastases (p = 0.057), noted grossly, via GFP fluorescence, (Fig. 4b) and histologically (Fig. 4c). Liver metastases derived from both control shRNA BON cells and PTEN shRNA BON cells (Fig. 4c) had reduced expression of VEGFR-2 compared to the corresponding primary tumors in the pancreas. As expected, primary tumors derived from PTEN shRNA BON cells had reduced expression of VEGFR-2 compared to primary tumors derived from control shRNA BON cells. To assess the expression of VEGFR-2 in liver metastases, cells from a liver metastasis (derived from PTEN shRNA BON cells) were harvested and cultured in vitro for six passages and treated with 2 μg/ml puromycin to select for BON cells. BON metastatic cells (designated as BON shPTEN metastasis) demonstrated reduced expression of VEGFR-2 and PTEN compared to parental BON cells (Fig. 4d). The expression of CgA, hypothesized to function partly as an adhesive marker in carcinoid tumors,39,40 was also reduced in the BON shPTEN metastatic cells. As with the in vitro analysis of EMT markers (Fig. 4a), the expressions of Snail and TGF-β were increased, while the expression of E-cadherin was decreased in BON shPTEN metastatic cells compared to parental BON cells (Fig. 4e). The concomitant reduction of VEGFR-2 expression with a decrease in PTEN expression suggests that VEGFR-2 inhibits EMT and, hence, metastasis of carcinoid tumor cells.
Decreasing VEGFR-2 expression enhances migration, invasion and proliferation of BON cells
As VEGFR-2 is decreased in BON cells with enhanced PI3K signaling and in BON metastatic cells, we assessed the effect of VEGFR-2 on BON cell migration, invasion and proliferation. To determine if VEGFR-2 affects the expression of EMT markers, BON cells were transfected with GFP-tagged shRNA to VEGFR-2. A reduction in VEGFR-2 resulted in upregulation of TGF-β and reduced expression of E-cadherin (Fig. 5a). However, Snail and Twist expressions were not altered, suggesting that the effect of VEGFR-2 on E-cadherin is independent of these pathways. Consistent with the adhesion properties of E-cadherin, VEGFR-2 knockdown, resulting in decreased E-cadherin expression, increased the migratory and invasive potential of BON cells at 24 and 48 hr, as indicated by scratch and Boyden chamber assays, respectively (Figs. 5b, p = 0.0002 and 5c, p < 0.0001). Furthermore, knockdown of VEGFR-2 increased the proliferative potential of the BON cell line at 48 and 72 hr (p < 0.05) (Fig. 5d). At 24 hr following plating, there was no significant difference in the proliferation of shPTEN and shVEGFR-2 BON cells compared to control shRNA BON cells (data not shown), which is consistent with the observation that the doubling time of parental BON cells is ∼60 hr.25 The lack of proliferative differences at 24 hr provides evidence that the observed increases in migration and invasion of VEGFR-2 shRNA BON cells are not the result of enhanced proliferation.
Figure 5.
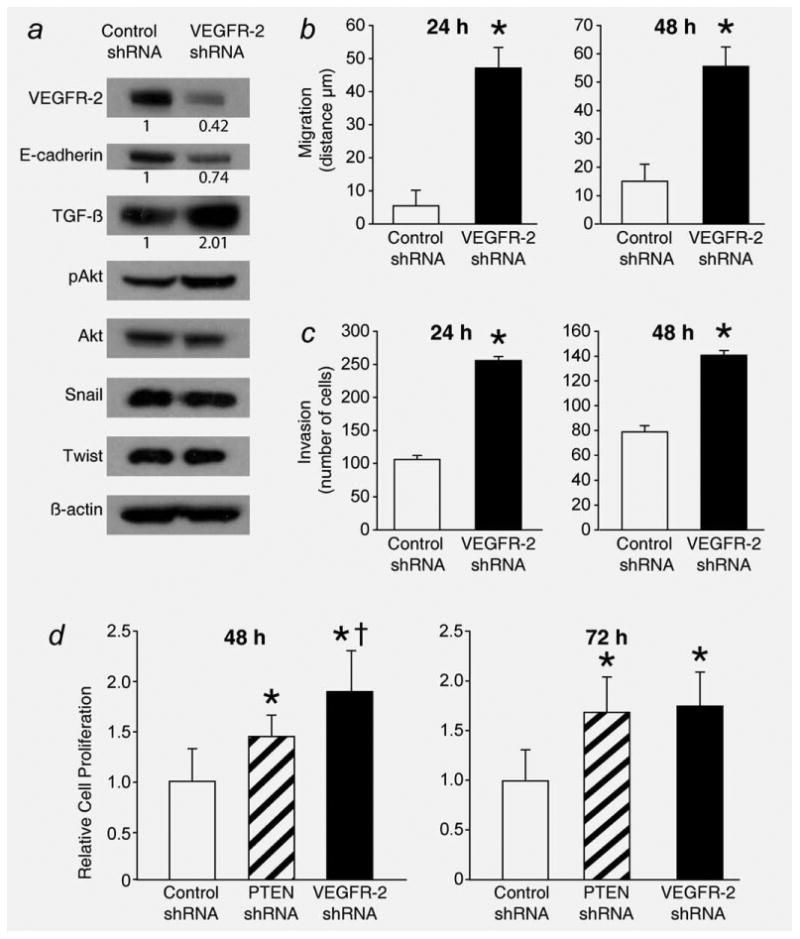
BON cell invasion and migration is increased with stable reduction in VEGFR-2. (a) shRNA against VEGFR-2 results in stable reduction in VEGFR-2 and E-cadherin expression and increased expression of TGF-β but no significant change in downstream EMT markers, such as snail or twist. Consistent with VEGFR-2 regulated expression of E-cadherin, BON cells with stable reduction in VEGFR-2 have an increased migratory and invasive potential (b and c) at both 24 and 48 hr incubation times (*, p = 0.0002 and p < 0.0001 vs. treatment with Control shRNA). (d) VEGFR-2 reduction also increases BON cell proliferation at 48 and 72 hr, as assessed by CCK-8 assay (*, p < 0.05 vs. treatment with Control shRNA; †, p < 0.05 vs. treatment with PTEN shRNA). The CCK-8 proliferation assay was performed with 16 replicates. Densitometric analysis was performed to compare the expression of VEGFR-2, E-cadherin, and TGF-β in BON shVEGFR-2 cells to that in control BON cells; numbers under the lanes represent relative densitometric values.
BON cells express soluble (s)VEGFR-2
To better understand our observations that VEGFR-2 inhibits cellular proliferation, migration and invasion, we next analyzed BON cells for the expression of sVEGFR-2, a truncated form resulting from retention of intron 13, that endogenously inhibits lymphangiogenesis by sequestering VEGF-C from VEGFR-3.22 BON cells express ∼0.37 pg sVEGFR-2 per μg of total protein as determined by an ELISA specific for sVEGFR-2 (Fig. 6a, top). To further verify the expression of sVEGFR-2 in BON cells, qRT-PCR was performed. Following the methods of Albuquerque et al.,22 we developed primers targeting the exon 13–intron 13 junction of sVegfr2 to quantify sVegfr2 mRNA expression. sVegfr2 mRNA was present in BON and HMVEC-L but not in HT29 cells (p < 0.05 vs. BON; Fig. 6a, bottom). We also verified through ELISA and qRT-PCR that VEGFR-2 shRNA reduces sVEGFR-2 expression, in addition to reducing membrane-bound VEGFR-2 expression (p = 0.045) (Fig. 6b). Furthermore, stable reduction in PTEN decreases sVEGFR-2 expression (Fig. 6c), in addition to membrane bound VEGFR-2, as shown in Figures 3e and 3f. To evaluate the possibility that sVEGFR-2 inhibits carcinoid metastasis through the same mechanism as lymphangiogenesis inhibition,22 we assessed the expression of VEGF-C and VEGFR-3 in the BON cell line and in carcinoid sections from an endocrine tumor tissue array. Both VEGF-C and VEGFR-3 are expressed in BON cells (Fig. 6d) and carcinoid tumors (Fig. 6e). Additionally, a 5-hr treatment with VEGF-C (50 ng/ml) promotes BON cell migration, as demonstrated by a transwell migration assay (p = 0.0046 vs. no treatment with VEGF-C) (Fig. 6f). Taken together, our results demonstrate that BON cells produce sVEGFR-2 and suggest that, by reducing sVEGFR-2 expression, more VEGF-C is available to bind VEGFR-3 and the metastatic potential of this carcinoid cell line is enhanced (Fig. 6g).
Figure 6.
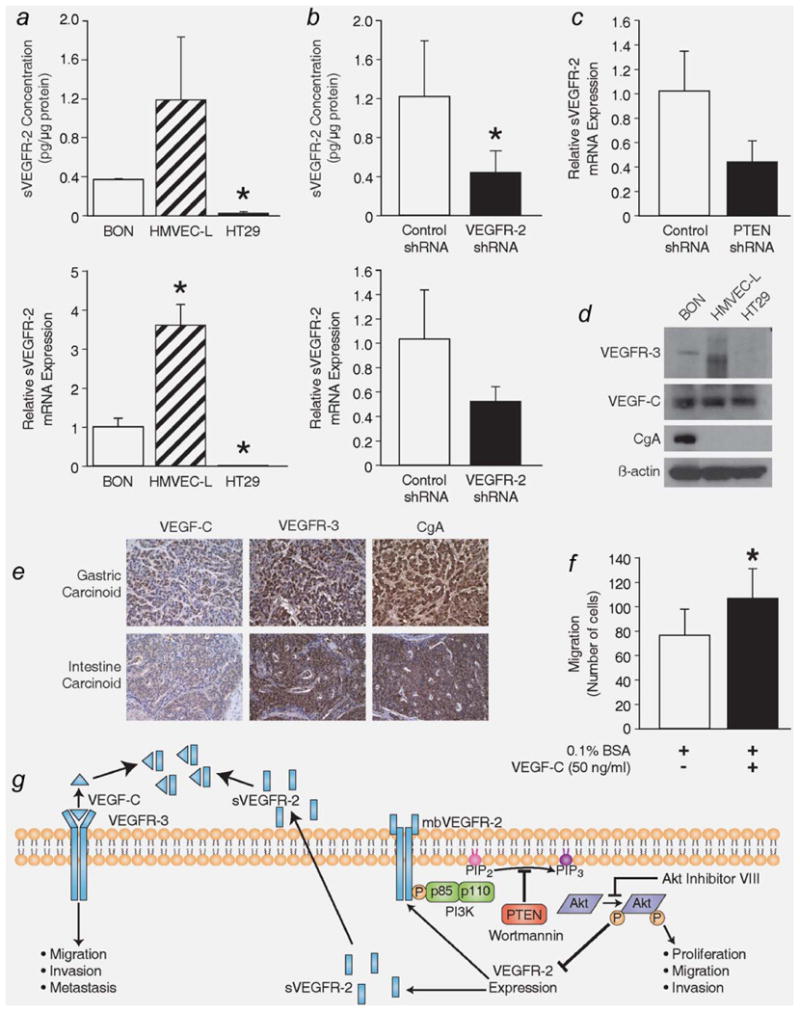
BON cells express sVEGFR-2. (a, top) BON cells express sVEGFR-2, as assessed by ELISA. HMVEC-L and HT29 cells were used as positive and negative controls, respectively, of sVEGFR-2 expression. (a, bottom) qRT-PCR further confirms the expression of sVEGFR-2 in the BON cell line. Primers targeting the exon13-intron 13 junction in Vegfr2 were used to specifically detect sVegfr2 transcripts (*, p < 0.05 vs. BON). (b) ELISA and qRT-PCR verify that a reduction in VEGFR-2 expression, via VEGFR-2 shRNA, also decreases the expression of sVEGFR-2 (*, p = 0.045 vs. treatment with Control shRNA). (c) BON cells with stably reduced PTEN expression have decreased expression of sVEGFR-2. Both VEGFR-3 and its ligand VEGF-C, which can also bind sVEGFR-2, are expressed in (d) BON cells and in (e) gastric and intestine carcinoid sections (200X). (f) Treatment with VEGF-C (50 ng/ml) for 5 h enhances BON cell migration (*, p = 0.0046 vs. no treatment with VEGF-C). (g) This schematic diagram summarizes our findings demonstrating an inverse relationship between PI3K/Akt signaling and VEGFR-2 and sVEGFR-2 expression and the possible relationship between sVEGFR-2 and VEGF-C/VEGFR-3 signaling in BON cells.
Discussion
Carcinoid tumors are highly vascular neuroendocrine tumors that are increasing in incidence. Recently, the proangiogenic protein VEGFR-2 has been described as a target for novel treatment regimens of carcinoid tumors.7,8 Our results demonstrate that VEGFR-2 is expressed in the epithelial component of carcinoid tumors and in the BON carcinoid cell line. Paradoxically, we noted that VEGFR-2 is inversely regulated by PI3K/Akt signaling and has an inhibitory effect on BON cell invasion, migration and proliferation, which is hypothesized to be partly due to the concurrent expression of sVEGFR-2 in BON cells. Our in vivo studies further suggest that VEGFR-2 has an antimetastatic effect in the BON carcinoid cell line, as its expression is reduced in the liver metastases compared to the primary tumors.
The finding of VEGFR-2 expression in the epithelial component of carcinoid tumors and in BON carcinoid cells led us to hypothesize that VEGF-A can directly promote carcinoid cancer cell growth, in addition to stimulating angiogenesis. Consistent with the findings of others,9,10 VEGF-A signaling transiently activated the PI3K/Akt pathway. However, VEGF-A treatment did not significantly increase BON cell proliferation, possibly due to the growth factors secreted by BON cells which can sustain cell growth.25 An unexpected finding was that VEGFR-2 expression was inversely proportional to PI3K signaling. While the role of VEGFR-2 in angiogenesis is well established, the mechanisms regulating its expression are still under investigation. Recently, it has been determined that the nuclear receptor peroxisome proliferators-activated receptor alpha (PPARα) inhibits VEGFR-2 expression,41 while the Rac1 GTPase promotes VEGFR-2 expression.42 Our findings of the relationship between VEGFR-2 expression and PI3K signaling are consistent with PPARα-induced inhibition of VEGFR-2, as PPARα activates PI3K/Akt signaling in endothelial cells lining the blood vessels of the myocardium as a defense mechanism against ischemia-reperfusion injury.43 Because VEGFR-2 mRNA expression is increased following inhibition of PI3K/Akt signaling, it is likely that PI3K/Akt negatively regulates the transcription of the Vegfr2 gene, rather than exerting a post-translational effect on VEGFR-2 protein expression.
Previous studies in our laboratory have demonstrated that injection of BON cells into the spleen of athymic nude mice result in liver metastases and manifestations of carcinoid syndrome.6 Additionally, we have detected increased PI3K/Akt signaling in colorectal cancer metastases.35,36 Specifically, we determined that PTEN deficiency enhances the metastatic phenotype of colorectal cancer.35,36 Because a reduction in PTEN decreases VEGFR-2 expression in BON cells, we assessed the role of VEGFR-2 in carcinoid metastasis. When injected into the pancreas of athymic nude mice, PTEN-deficient BON cells formed liver metastases; cells harvested from the liver metastases displayed increased PI3K/Akt signaling and altered expression of EMT markers. Additionally, VEGFR-2 expression remained decreased in the BON metastatic cells. The development of metastases from BON cells displaying increased PI3K/Akt signaling, due to decreased expression of PTEN, supports the hypothesis that this pathway contributes to carcinoid metastasis. Interestingly, the reduced expression of VEGFR-2 in BON shPTEN and BON shPTEN metastatic cells suggests that VEGFR-2 may actually inhibit metastasis.
E-cadherin is a cell adhesion protein that is downregulated during EMT, thus providing cancer cells with an enhanced migratory potential.37,38 In endothelial cells, activated-VEGFR-2 induces expression of the transcription factor Ets1, which transactivates the vascular endothelial (VE)-cadherin promoter.44,45 Several studies have suggested that VEGFR-2 interacts with VE-cadherin to promote endothelial cell actin remodeling, vascular permeability and angiogenesis.46,47 Consistent with these observations, our results indicate that VEGFR-2 in BON cells promotes E-cadherin expression, possibly by downregulating TGF-β, since reduction of VEGFR-2 in BON cells produces an upregulation of TGF-β and a decrease in E-cadherin. While VE-cadherin associates with VEGFR-2 to induce angiogenesis, E-cadherin acts as an epithelial cell adhesion protein and limits migratory potential. Thus, the relationship between VEGFR-2 and E-cadherin expression in BON cells suggests that VEGFR-2 inhibits migration and invasion in carcinoid cells.
Our finding of sVEGFR-2 expression in BON cells provides a potential explanation for the paradoxical effects of VEGFR-2. Recent reports have demonstrated an increase in tumor metastasis following treatment with angiogenesis inhibitors.23,24 While we previously demonstrated a decrease in tumor growth following antiangiogenic therapy,6 we treated mice with bevacizumab, an antibody to VEGF that prevents it from binding to the receptor.30 In contrast, Ebos et al.23 and Paez-Ribes et al.24 specifically targeted VEGFR-2 with either anti-VEGFR-2 antibodies or the small molecule VEGFR-2 inhibitor sunitinib.48 Also, there has been a growing interest in the function of sVEGFR-2, with recent studies demonstrating that sVEGFR-2 inhibits lymphatic vessel growth22 and neuroblastoma progression.49 sVEGFR-2 exerts its inhibitory effect by binding and sequestering the VEGF-C ligand in the extracellular environment. VEGF-C normally binds to VEGFR-3 to promote lymphangiogenesis22 and cancer cell migration and invasion.50 Su, et al.50 demonstrated that VEGF-C and VEGFR-3 are expressed by cancer cells and promote cancer cell invasion and metastasis in an autocrine manner. In a preliminary approach to determine the mechanism of sVEGFR-2 inhibition of BON cell migration and invasion, we detected the expression of VEGFR-3 and VEGF-C in BON cells and determined that VEGF-C promotes BON cell migration. Based on the recently identified role of sVEGFR-2, reducing sVEGFR-2 may increase the proliferative and metastatic potential of BON cells by permitting the binding of more VEGF-C to VEGFR-3. The schematic in Figure 6g summarizes the proposed relationship between PI3K/Akt signaling, VEGFR-2 expression and VEGF-C/VEGF-3 signaling. Future studies will better define the roles of VEGFR-2, sVEGFR-2 and VEGF-C/VEGFR-3 signaling in carcinoid tumor growth and metastasis.
In summary, we have identified the unique expression of VEGFR-2 and sVEGFR-2 in the epithelial component of carcinoid tumors and in the BON carcinoid cell line. Our findings reveal that inhibiting VEGFR-2 increases the proliferative, migratory and invasive capacities of BON cells. Furthermore, the expression of VEGFR-2 is inversely related to PI3K/Akt signaling. With the evolving complexity of VEGFR-2 regulation and function, we have identified a soluble form of VEGFR-2 produced in BON cells, which may act as an inhibitor of carcinoid cell growth and metastasis. Thus, our findings suggest the need for a multimodal approach in treating carcinoid disease, as targeting VEGFR-2 alone in carcinoid tumors may limit angiogenesis and decrease the growth of the primary tumor. Conversely, targeting VEGFR-2 alone may enhance the metastatic potential of surviving cancer cells.
Acknowledgments
The authors would like to thank Mrs. Karen Martin, Mrs. Donna Gilbreath, and Dr. Nathan L. Vanderford for manuscript preparation; Mr. Tatsuo Uchida for statistical analysis; and Drs. Kathleen O'Connor, Tianyan Gao, and Jayakrishna Ambati for thoughtful suggestions and review of the manuscript.
Grant sponsor: National Institutes of Health; Grant numbers: R37 AG10885, R01 DK48489, T32DK07639, R01 CA104748
References
- 1.Modlin IM, Kidd M, Latich I, Zikusoka MN, Shapiro MD. Current status of gastrointestinal carcinoids. Gastroenterology. 2005;128:1717–51. doi: 10.1053/j.gastro.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 2.Woodside KJ, Townsend CM, Jr, Mark Evers B. Current management of gastrointestinal carcinoid tumors. J Gastrointest Surg. 2004;8:742–56. doi: 10.1016/j.gassur.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Druce M, Rockall A, Grossman AB. Fibrosis and carcinoid syndrome: from causation to future therapy. Nat Rev Endocrinol. 2009;5:276–83. doi: 10.1038/nrendo.2009.51. [DOI] [PubMed] [Google Scholar]
- 4.Modlin IM, Oberg K, Chung DC, Jensen RT, de Herder WW, Thakker RV, Caplin M, Delle Fave G, Kaltsas GA, Krenning EP, Moss SF, Nilsson O, et al. Gastroenteropancreatic neuroendocrine tumours. Lancet Oncol. 2008;9:61–72. doi: 10.1016/S1470-2045(07)70410-2. [DOI] [PubMed] [Google Scholar]
- 5.Modlin IM, Kidd M, Drozdov I, Siddique ZL, Gustafsson BI. Pharmacotherapy of neuroendocrine cancers. Expert Opin Pharmacother. 2008;9:2617–26. doi: 10.1517/14656566.9.15.2617. [DOI] [PubMed] [Google Scholar]
- 6.Jackson LN, Chen LA, Larson SD, Silva SR, Rychahou PG, Boor PJ, Li J, Defreitas G, Stafford WL, Townsend CM, Jr, Evers BM. Development and characterization of a novel in vivo model of carcinoid syndrome. Clin Cancer Res. 2009;15:2747–55. doi: 10.1158/1078-0432.CCR-08-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kulke MH, Lenz HJ, Meropol NJ, Posey J, Ryan DP, Picus J, Bergsland E, Stuart K, Tye L, Huang X, Li JZ, Baum CM, et al. Activity of sunitinib in patients with advanced neuroendocrine tumors. J Clin Oncol. 2008;26:3403–10. doi: 10.1200/JCO.2007.15.9020. [DOI] [PubMed] [Google Scholar]
- 8.Yao JC, Phan A, Hoff PM, Chen HX, Charnsangavej C, Yeung SC, Hess K, Ng C, Abbruzzese JL, Ajani JA. Targeting vascular endothelial growth factor in advanced carcinoid tumor: a random assignment phase II study of depot octreotide with bevacizumab and pegylated interferon alpha-2b. J Clin Oncol. 2008;26:1316–23. doi: 10.1200/JCO.2007.13.6374. [DOI] [PubMed] [Google Scholar]
- 9.Holmes K, Roberts OL, Thomas AM, Cross MJ. Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal. 2007;19:2003–12. doi: 10.1016/j.cellsig.2007.05.013. [DOI] [PubMed] [Google Scholar]
- 10.Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–71. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- 11.Bowen KA, Silva SR, Johnson JN, Doan HQ, Jackson LN, Gulhati P, Qiu S, Riall TS, Evers BM. An analysis of trends and growth factor receptor expression of GI carcinoid tumors. J Gastrointest Surg. 2009;13:1773–80. doi: 10.1007/s11605-009-0958-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Franke TF. PI3K/Akt: getting it right matters. Oncogene. 2008;27:6473–88. doi: 10.1038/onc.2008.313. [DOI] [PubMed] [Google Scholar]
- 13.Liu W, Zhou Y, Reske SN, Shen C. PTEN mutation: many birds with one stone in tumorigenesis. Anticancer Res. 2008;28:3613–9. [PubMed] [Google Scholar]
- 14.Vogt PK, Gymnopoulos M, Hart JR. PI 3-kinase and cancer: changing accents. Curr Opin Genet Dev. 2009;19:12–7. doi: 10.1016/j.gde.2008.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pitt SC, Chen H, Kunnimalaiyaan M. Phosphatidylinositol 3-kinase-Akt signaling in pulmonary carcinoid cells. J Am Coll Surg. 2009;209:82–8. doi: 10.1016/j.jamcollsurg.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 17.Wang L, Ignat A, Axiotis CA. Differential expression of the PTEN tumor suppressor protein in fetal and adult neuroendocrine tissues and tumors: progressive loss of PTEN expression in poorly differentiated neuroendocrine neoplasms. Appl Immunohistochem Mol Morphol. 2002;10:139–46. doi: 10.1097/00129039-200206000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Lee KS, Park SJ, Kim SR, Min KH, Lee KY, Choe YH, Hong SH, Lee YR, Kim JS, Hong SJ, Lee YC. Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur Respir J. 2008;31:523–31. doi: 10.1183/09031936.00125007. [DOI] [PubMed] [Google Scholar]
- 19.Suzuma K, Naruse K, Suzuma I, Takahara N, Ueki K, Aiello LP, King GL. Vascular endothelial growth factor induces expression of connective tissue growth factor via KDR. Flt1, and phosphatidylinositol 3-kinase-akt-dependent pathways in retinal vascular cells. J Biol Chem. 2000;275:40725–31. doi: 10.1074/jbc.M006509200. [DOI] [PubMed] [Google Scholar]
- 20.Ardura JA, Berruguete R, Ramila D, Alvarez-Arroyo MV, Esbrit P. Parathyroid hormone-related protein interacts with vascular endothelial growth factor to promote fibrogenesis in the obstructed mouse kidney. Am J Physiol Renal Physiol. 2008;295:F415–25. doi: 10.1152/ajprenal.00018.2008. [DOI] [PubMed] [Google Scholar]
- 21.Malmstrom NK, Kallio EA, Rintala JM, Nykanen AI, Raisanen-Sokolowski AK, Paavonen T, Lemstrom KB, Koskinen PK. Vascular endothelial growth factor in chronic rat allograft nephropathy. Transpl Immunol. 2008;19:136–44. doi: 10.1016/j.trim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- 22.Albuquerque RJ, Hayashi T, Cho WG, Kleinman ME, Dridi S, Takeda A, Baffi JZ, Yamada K, Kaneko H, Green MG, Chappell J, Wilting J, et al. Alternatively spliced vascular endothelial growth factor receptor-2 is an essential endogenous inhibitor of lymphatic vessel growth. Nat Med. 2009;15:1023–30. doi: 10.1038/nm.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ebos JM, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–9. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Vinals F, Inoue M, Bergers G, Hanahan D, Casanovas O. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell. 2009;15:220–31. doi: 10.1016/j.ccr.2009.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evers BM, Ishizuka J, Townsend CM, Jr, Thompson JC. The human carcinoid cell line. BON. A model system for the study of carcinoid tumors. Ann N Y Acad Sci. 1994;733:393–406. doi: 10.1111/j.1749-6632.1994.tb17289.x. [DOI] [PubMed] [Google Scholar]
- 26.Evers BM, Townsend CM, Jr, Upp JR, Allen E, Hurlbut SC, Kim SW, Rajaraman S, Singh P, Reubi JC, Thompson JC. Establishment and characterization of a human carcinoid in nude mice and effect of various agents on tumor growth. Gastroenterology. 1991;101:303–11. doi: 10.1016/0016-5085(91)90004-5. [DOI] [PubMed] [Google Scholar]
- 27.Beauchamp RD, Coffey RJ, Jr, Lyons RM, Perkett EA, Townsend CM, Jr, Moses HL. Human carcinoid cell production of paracrine growth factors that can stimulate fibroblast and endothelial cell growth. Cancer Res. 1991;51:5253–60. [PubMed] [Google Scholar]
- 28.Evers BM, Ishizuka J, Townsend CM, Jr, Rajaraman S, Thompson JC. Expression of neurotensin messenger RNA in a human carcinoid tumor. Ann Surg. 1991;214:448–54. doi: 10.1097/00000658-199110000-00009. discussion 54–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jeng YJ, Townsend CM, Jr, Nagasawa S, Chuo S, Kern K, Yanaihara N, Ferrar RS, Hill FL, Thompson JC, Greeley GH., Jr Regulation of pancreastatin release from a human pancreatic carcinoid cell line in vitro. Endocrinology. 1991;128:220–5. doi: 10.1210/endo-128-1-220. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara N, Hillan KJ, Novotny W. Bevacizumab (Avastin), a humanized anti-VEGF monoclonal antibody for cancer therapy. Biochem Biophys Res Commun. 2005;333:328–35. doi: 10.1016/j.bbrc.2005.05.132. [DOI] [PubMed] [Google Scholar]
- 31.Bowen KA, Doan HQ, Zhou BP, Wang Q, Zhou Y, Rychahou PG, Evers BM. PTEN loss induces epithelial–mesenchymal transition in human colon cancer cells. Anticancer Res. 2009;29:4439–49. [PMC free article] [PubMed] [Google Scholar]
- 32.Duerr EM, Chung DC. Molecular genetics of neuroendocrine tumors. Best Pract Res Clin Endocrinol Metab. 2007;21:1–14. doi: 10.1016/j.beem.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 33.Kong D, Yamori T. Phosphatidylinositol 3-kinase inhibitors: promising drug candidates for cancer therapy. Cancer Sci. 2008;99:1734–40. doi: 10.1111/j.1349-7006.2008.00891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calleja V, Laguerre M, Parker PJ, Larijani B. Role of a novel PH-kinase domain interface in PKB/Akt regulation: structural mechanism for allosteric inhibition. PLoS Biol. 2009;7:e17. doi: 10.1371/journal.pbio.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rychahou PG, Jackson LN, Silva SR, Rajaraman S, Evers BM. Targeted molecular therapy of the PI3K pathway: therapeutic significance of PI3K subunit targeting in colorectal carcinoma. Ann Surg. 2006;243:833–42. doi: 10.1097/01.sla.0000220040.66012.a9. discussion 43–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rychahou PG, Kang J, Gulhati P, Doan HQ, Chen LA, Xiao SY, Chung DH, Evers BM. Akt2 overexpression plays a critical role in the establishment of colorectal cancer metastasis. Proc Natl Acad Sci USA. 2008;105:20315–20. doi: 10.1073/pnas.0810715105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 38.Wu Y, Zhou BP. New insights of epithelial-mesenchymal transition in cancer metastasis. Acta Biochim Biophys Sin (Shanghai) 2008;40:643–50. doi: 10.1111/j.1745-7270.2008.00443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Montero-Hadjadje M, Vaingankar S, Elias S, Tostivint H, Mahata SK, Anouar Y. Chromogranins A and B and secretogranin II: evolutionary and functional aspects. Acta Physiol (Oxf) 2008;192:309–24. doi: 10.1111/j.1748-1716.2007.01806.x. [DOI] [PubMed] [Google Scholar]
- 40.Ratti S, Curnis F, Longhi R, Colombo B, Gasparri A, Magni F, Manera E, Metz-Boutigue MH, Corti A. Structure-activity relationships of chromogranin A in cell adhesion. Identification of an adhesion site for fibroblasts and smooth muscle cells. J Biol Chem. 2000;275:29257–63. doi: 10.1074/jbc.M003796200. [DOI] [PubMed] [Google Scholar]
- 41.Meissner M, Stein M, Urbich C, Reisinger K, Suske G, Staels B, Kaufmann R, Gille J. PPARalpha activators inhibit vascular endothelial growth factor receptor-2 expression by repressing Sp1-dependent DNA binding and transactivation. Circ Res. 2004;94:324–32. doi: 10.1161/01.RES.0000113781.08139.81. [DOI] [PubMed] [Google Scholar]
- 42.Meissner M, Michailidou D, Stein M, Hrgovic I, Kaufmann R, Gille J. Inhibition of Rac1 GTPase downregulates vascular endothelial growth factor receptor-2 expression by suppressing Sp1-dependent DNA binding in human endothelial cells. Exp Dermatol. 2009;18:863–9. doi: 10.1111/j.1600-0625.2009.00867.x. [DOI] [PubMed] [Google Scholar]
- 43.Bulhak AA, Jung C, Ostenson CG, Lundberg JO, Sjoquist PO, Pernow J. PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-Kinase/Akt and NO pathway. Am J Physiol Heart Circ Physiol. 2009;296:H719–27. doi: 10.1152/ajpheart.00394.2008. [DOI] [PubMed] [Google Scholar]
- 44.Lelievre E, Mattot V, Huber P, Vandenbunder B, Soncin F. ETS1 lowers capillary endothelial cell density at confluence and induces the expression of VE-cadherin. Oncogene. 2000;19:2438–46. doi: 10.1038/sj.onc.1203563. [DOI] [PubMed] [Google Scholar]
- 45.Sato Y, Kanno S, Oda N, Abe M, Ito M, Shitara K, Shibuya M. Properties of two VEGF receptors. Flt-1 and. KDR, in signal transduction. Ann N Y Acad Sci. 2000;902:201–5. doi: 10.1111/j.1749-6632.2000.tb06314.x. discussion 5–7. [DOI] [PubMed] [Google Scholar]
- 46.Kouklis P, Konstantoulaki M, Malik AB. VE-cadherin-induced Cdc42 signaling regulates formation of membrane protrusions in endothelial cells. J Biol Chem. 2003;278:16230–6. doi: 10.1074/jbc.M212591200. [DOI] [PubMed] [Google Scholar]
- 47.Liao F, Doody JF, Overholser J, Finnerty B, Bassi R, Wu Y, Dejana E, Kussie P, Bohlen P, Hicklin DJ. Selective targeting of angiogenic tumor vasculature by vascular endothelial-cadherin antibody inhibits tumor growth without affecting vascular permeability. Cancer Res. 2002;62:2567–75. [PubMed] [Google Scholar]
- 48.Mendel DB, Laird AD, Xin X, Louie SG, Christensen JG, Li G, Schreck RE, Abrams TJ, Ngai TJ, Lee LB, Murray LJ, Carver J, et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: determination of a pharmacokinetic/pharmacodynamic relationship. Clin Cancer Res. 2003;9:327–37. [PubMed] [Google Scholar]
- 49.Becker J, Pavlakovic H, Ludewig F, Wilting F, Weich HA, Albuquerque R, Ambati J, Wilting J. Neuroblastoma progression correlates with downregulation of the lymphangiogenesis inhibitor sVEGFR-2. Clin Cancer Res. 2010;16:1431–41. doi: 10.1158/1078-0432.CCR-09-1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Su JL, Yang PC, Shih JY, Yang CY, Wei LH, Hsieh CY, Chou CH, Jeng YM, Wang MY, Chang KJ, Hung MC, Kuo ML. The VEGF-C/Flt-4 axis promotes invasion and metastasis of cancer cells. Cancer Cell. 2006;9:209–23. doi: 10.1016/j.ccr.2006.02.018. [DOI] [PubMed] [Google Scholar]