

Abstract
Recovery from blood loss requires a greatly enhanced supply of iron to support expanded erythropoiesis. After hemorrhage, suppression of the iron-regulatory hormone hepcidin allows increased iron absorption and mobilization from stores. We identified a new hormone, erythroferrone (ERFE), which mediates hepcidin suppression during stress erythropoiesis. ERFE is produced by erythroblasts in response to erythropoietin. ERFE-deficient mice fail to suppress hepcidin rapidly after hemorrhage and exhibit a delay in recovery from blood loss. ERFE expression is greatly increased in murine HbbTh3/+ thalassemia intermedia where it contributes to the suppression of hepcidin and systemic iron overload characteristic of this disease.
INTRODUCTION
Iron is an essential component of heme and hemoglobin. The production of functional erythrocytes therefore requires the timely delivery of sufficient iron to erythroid precursors. The iron supply to the marrow comes under particular strain after hemorrhage, hemolysis and other conditions that trigger stress erythropoiesis. Both the absorption of dietary iron and the release of iron from stores are increased as erythropoiesis intensifies but the mechanism through which erythropoiesis modulates iron homeostasis, historically referred to as the “erythroid regulator”, is not understood1. After the loss of erythrocytes from hemorrhage or hemolysis, critical signals facilitating the provision of iron for restorative erythropoiesis would be expected to act within hours, as rapid recovery of red cell mass and oxygen carrying capacity confers obvious evolutionary advantages.
The liver-produced hormone hepcidin is the main circulating regulator of iron absorption and tissue distribution2. Hepcidin controls the major flows of iron into plasma: absorption of dietary iron in intestine, recycling of iron by macrophages which phagocytose old erythrocytes and other cells, and mobilization of stored iron from hepatocytes. Iron is exported from these tissues into plasma through ferroportin, the sole known cellular iron exporter and the hepcidin receptor. Hepcidin causes endocytosis and degradation of ferroportin, leading to the retention of iron in iron-exporting cells and decreased flow of iron into plasma. In turn, hepcidin production is transcriptionally regulated in response to changes in circulating iron concentration, iron stores, or the development of inflammation and iron-restricted erythropoiesis2.
In view of the central role of hepcidin in systemic iron homeostasis, the putative erythroid regulators could facilitate iron delivery to the marrow by decreasing blood hepcidin concentrations, and thereby allowing increased iron absorption and the release of iron from stores into blood plasma. Indeed, anemia induced by hemorrhage or hemolysis decreased hepcidin in mice3, and the suppressive effect of anemia on hepcidin depended on erythropoietin and functional bone marrow4, 5. In normal volunteers, the administration of erythropoietin was sufficient to lower serum hepcidin profoundly within less than 1 day in the absence of any significant changes in serum iron6.
In contrast to their adaptive role in the recovery from blood loss, erythroid regulators may act pathologically to mediate iron overload and its severe clinical complications in inherited anemias with ineffective erythropoiesis such as β-thalassemia and congenital dyserythropoietic anemia. This mechanism may be particularly prominent in untransfused patients in whom iron overload develops because of the pathological suppression of hepcidin synthesis and the resulting hyperabsorption of dietary iron7. Even in transfused patients, in whom ineffective erythropoietic activity is partially ameliorated, erythroid regulators and associated hepcidin suppression may contribute to the iron overload, particularly late during the intervals between transfusions. Growth differentiation factor 15 (GDF15) and twisted-gastrulation 1 (TWSG1) are bone morphogenetic protein family members that have been proposed as pathological suppressors of hepcidin in thalassemia 8, 9. However, the evidence for the pathogenic role of these mediators in iron-loading anemias is not definitive and recent studies argue against their contribution in physiological hepcidin suppression after hemorrhage10–12. In the current study, we describe the identification of a new erythroid regulator essential for early suppression of hepcidin after erythropoietic stimulation.
RESULTS
Suppression of hepcidin by erythropoietic stimulation
We first examined the time-course of hepatic hepcidin response to increased erythropoietic activity. Six week-old C57BL/6 males were phlebotomized 500 μL or treated with 200 units of erythropoietin (EPO). Hepcidin mRNA levels were reduced within 9 hours after erythropoietic stimulation, reached a nadir 10-fold suppression 15 hours after phlebotomy or EPO treatment and remained partially decreased even at 48 hours (Fig. 1A). Serum hepcidin concentrations were concordant with the mRNA data (Fig. 1B). The suppression occurred with a similar timing in mice as had been reported in human volunteers (between 9–24 hours after EPO injection) 6 thus we focused on the early mechanism of hepcidin suppression, active in the first 24 hours after hemorrhage. As was the case in the human study, no significant changes in serum iron concentration were observed after either stimulus (Supplementary Fig. 1).
Figure 1. Time-course of hepcidin mRNA and protein concentrations after phlebotomy (500 μL) or treatment with erythropoietin (EPO, 200 U).

(A) Hamp mRNA levels were reduced within 9 hours and maximally suppressed 12–15 hours after phlebotomy (solid line) or treatment with EPO (dashed line). (B) Serum hepcidin levels after phlebotomy and treatment with EPO reflect mRNA concentrations. Both graphs show means ± SEM. Log2 hepcidin mRNA concentration relative to that of a housekeeping gene Hprt, −ΔCt (i.e., Ct Hprt – Ct Hamp) is shown as measured by qRT-PCR and serum hepcidin values were obtained by enzyme-linked immunosorbent assay. Each time-point was compared to the control mice (t=0) by two-tailed Student t-test (n=4 mice per point). ***p<0.001, **p<0.01, *p<0.05 for phlebotomy, ###p<0.001, ##p<0.01, #p<0.05 for EPO treatment.
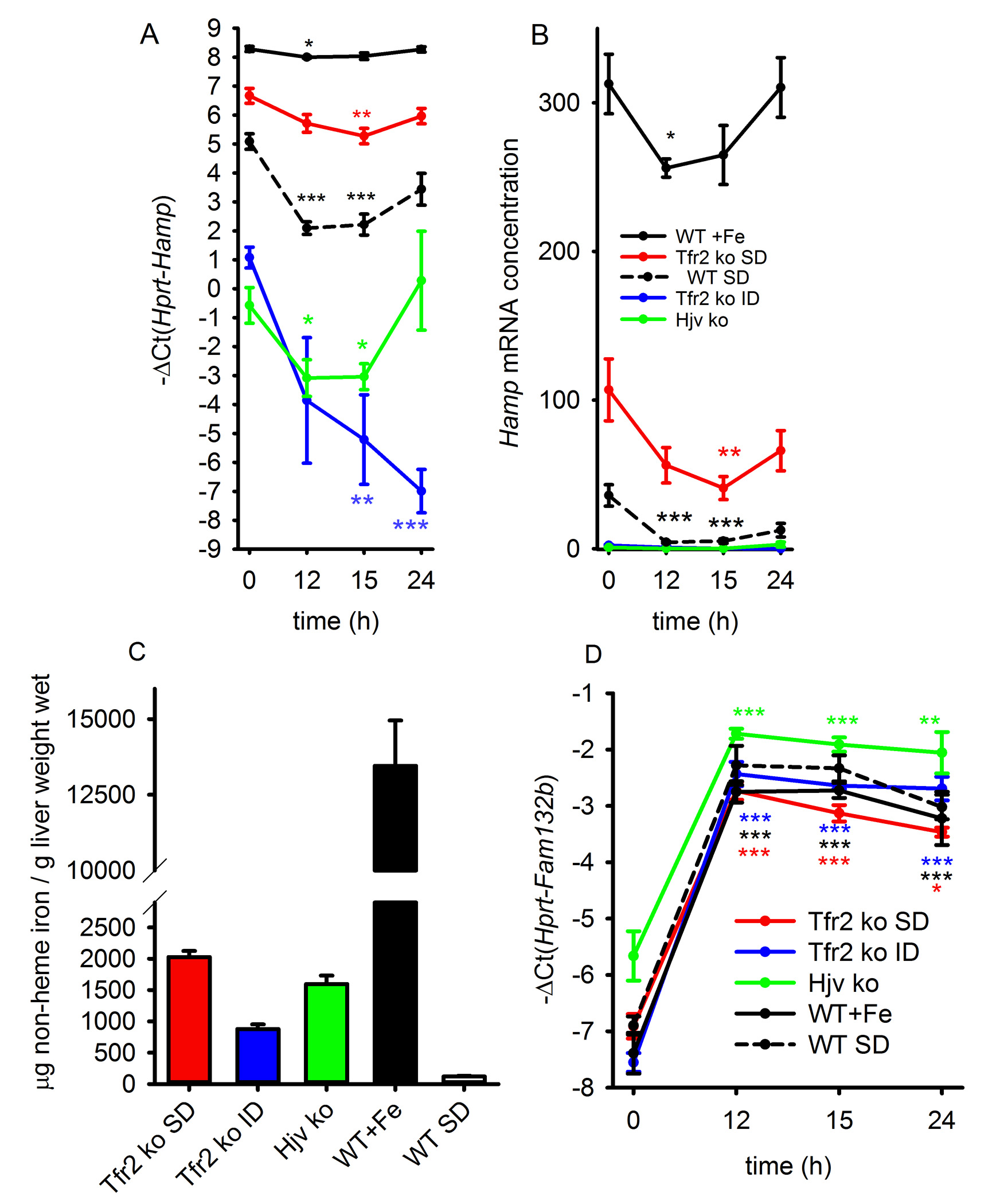
BMP/Smad signaling pathway is a major regulator of hepcidin transcription13, 14, and is involved in iron-dependent modulation of hepcidin expression. To assess if the hepatic BMP/Smad signaling is decreased in response to erythropoietic stimulation, we measured Smad5 phosphorylation in the livers of phlebotomized or EPO-treated mice 15 hours after the stimulus. Smad5 phosphorylation was not significantly decreased in either phlebotomized (Supplementary Fig. 2A, B) or EPO-treated mice (Supplementary Fig. 2C, D) compared to control mice. We also examined Id1 mRNA, a well-established marker of BMP/Smad activation15. In contrast to the profound suppression of hepcidin, only a mild decrease in Id1 mRNA levels was observed 9 and 15 hours after phlebotomy but no changes were observed 12 hours after bleeding or at any time after EPO treatment (Supplementary Fig. 2E). Together, these data suggest that hepcidin suppression by increased erythropoietic activity occurs independently of the canonical BMP/Smad signaling pathway. Moreover, using mice deficient in transferrin receptor 2 (TfR2) or hemojuvelin (HJV), important regulators of hepcidin response to iron and modulators of BMP signaling, we showed that neither TfR2 nor HJV were required for erythropoietic suppression of hepcidin (Supplementary Fig. 3A, B). Additionally, we stimulated the BMP pathway in WT mice by administering parenteral iron to cause extreme iron overload (liver iron content in Supplementary Fig. 3C). Even in these mice with very high hepcidin baseline, phlebotomy acutely suppressed hepcidin (Supplementary Fig. 3B). Thus, the erythropoietic suppression of hepcidin appears to act independently of the BMP pathway.
Bone marrow gene-expression profiling
Guided by previous studies4, 5 suggesting that the erythroid regulator may be secreted by the bone marrow after erythropoietic stimulation, we studied mRNA changes in the mouse marrow 0–48 hours after bleeding, using Affymetrix gene chip-based expression profiling. No changes were observed in Gdf15 and Twsg1 mRNAs, which encode previously proposed candidates for the erythroid regulator. We focused our attention on erythroid-specific transcripts encoding secreted proteins, with mRNA expression that was induced prior to, and was maintained at high levels during the suppression of hepatic hepcidin mRNA expression. A previously uncharacterized transcript, listed in the databases as Fam132b, fit these criteria: it was predicted to encode a secreted protein and was highly induced within 4 hours after bleeding, prior to hepcidin suppression. After its further characterization, we named this factor “erythroferrone” (ERFE), to denote its function as an erythroid hormone acting on iron metabolism. In the meantime, the protein encoded by Fam132b was also described by others as myonectin or CTRP1516, a member of the C1q/TNF-related protein family (CTRP). Pending definitive resolution of the nomenclature, we will refer to erythroferrone protein as ERFE and to the corresponding mRNA and gene as Fam132b.
Validation studies by qRT-PCR demonstrated that the Fam132b mRNA was induced in the bone marrow already within 4 hours after phlebotomy and reached maximal induction of over 30-fold by 9 hours (Fig. 2A) before decreasing toward baseline after 24–48 hours. Fam132b mRNA expression after phlebotomy mirrored serum erythropoietin concentrations (Supplementary Fig. 4). EPO treatment resulted in a similar (but more rapid) induction of Fam132b mRNA suggesting that the gene encoding ERFE is EPO-responsive (Fig. 2A). In the absence of any stimulation, Fam132b mRNA in the spleen was expressed at a very low level but was induced 8–10 fold after phlebotomy or EPO injection (Fig. 2B) supporting its role in stress erythropoiesis. The induction of Fam132b mRNA by phlebotomy was not affected by modulation of BMP signaling through iron loading or genetic ablation of TfR2 or hemojuvelin (Supplementary Figure 3D).
Figure 2. Induction of ERFE-encoding Fam132b mRNA after phlebotomy (500 μL) or treatment with erythropoietin (EPO, 200 U).

Fam132b mRNA in the bone marrow (A) and the spleen (B) of 6 week-old C57BL/6 wild-type males is rapidly increased 4–9 hours after phlebotomy (solid line) or EPO treatment (dashed line). Means of −ΔCt (i.e., Ct Hprt – Ct Fam132b) ± SEM are shown as measured by qRT-PCR, and compared for each time-point to the t=0 control by two-tailed Student t-test (n=4). ***p<0.001, **p<0.01, *p<0.05 for phlebotomy, ###p<0.001, ##p<0.01, #p<0.05 for EPO treatment.
ERFE is produced by erythroblasts
We next assessed tissue-specific expression of Fam132b mRNA from control and EPO-treated 6 week-old male wild-type mice. At baseline, the highest Fam132b expression was detected in the colon, the duodenum, the muscle, the bone marrow and the testis (Fig. 3A). After EPO stimulation, however, Fam132b induction was observed only in the bone marrow and the spleen, the erythropoietic organs of the adult mouse. Moreover, EPO-stimulated marrow became the organ with the highest expression of Fam132b mRNA supporting the idea that it may have erythroid-specific function. To examine Fam132b expression across the stages of erythroid differentiation, we sorted by flow cytometry the erythroblast population from the marrow of control or phlebotomized (15 hours time-point) wild-type mice. Erythroblasts populations were separated based on their size and the expression of the erythroid markers Ter119 and transferrin receptor 1 (TfR1) into pro-erythroblasts, basophilic, polychromatic and orthochromatic erythroblasts as previously described17. Fam132b mRNA was increased after phlebotomy in all four populations of erythroid precursors, with highest expression detected in basophilic and polychromatic erythroblasts (Fig. 3B).
Figure 3. ERFE is produced in erythroblasts and induced by erythropoietic stimuli through the Jak2/Stat5 signaling pathway.

(A) Tissue-specific expression of Fam132b mRNA. Fam132b mRNA expression was stimulated only in the bone marrow and in the spleen of 6 weeks-old C57BL/6 mice 15 hours after 200 U EPO treatment (n=6). (B) Fam132b mRNA expression across stages of erythroblast development from proerythroblasts (Pro-E) through basophilic (baso), polychromatic (poly) to orthochromatic (ortho) erythroblasts. Fam132b mRNA was measured by qRT-PCR in erythroid cells isolated from the bone marrows of three controls and three phlebotomized (15 h) 6 week-old C57BL/6 mice. Cells were sorted by size and anti-Ter119 and anti-CD71 staining18. (C) Fam132b induction by EPO is dependent on Stat5. Stimulation of Fam132b mRNA expression in erythroblasts by EPO treatment (15 h, 10 U/mL) was inhibited by Stat5 inhibitors pimozide (10 μM) or N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide (200 μM). Erythroferrone (Fam132b) mRNA levels were measured by qRT-PCR. Values shown are means ± SEM of −ΔCt (i.e., Ct Hprt or Rpl4 – Ct Fam132b). Means of −ΔCt values were compared between control and erythropoietic stimulation by two-tailed Student t-test (A, B). Bonferroni correction was applied to the multiple comparisons represented in panel A. (C) Expression ratio ± SEM of Fam132b transcripts in EPO-treated cells relative to controls and normalized to the gene Gypa mRNA was calculated using REST. Normalization by glycophorin A allowed the normalization of Fam132b mRNA expression per red cell. Results of 3 independent experiments with 15 hour treatments in duplicate are shown. Statistical significance was determined using randomization tests. ***p<0.001, **p<0.01
The cDNA sequence available for human FAM132B in public databases (GenBank XM_001714385.4) had been predicted from the genomic sequence and was not validated by mRNA sequencing. We performed 5′ RACE on total RNA from human fetal liver erythroblasts and cloned the dominant cDNA sequence which differed from the predicted sequence at the 5′ terminus, including the codons for the first 70 amino acids. With this change, the human and mouse ERFE protein sequences contain 354 and 340 amino acid residues respectively and are 71% identical. The C-terminal half of the predicted proteins shows even higher identity and encodes a TNF-α-like domain16. In human erythroblasts isolated from fetal liver or adult bone marrow, FAM132B mRNA expression was also strongly induced by erythropoietin (>16-fold) (Supplementary Fig. 5).
Binding of EPO to its receptor is known to activate the Jak2-Stat5 signaling pathway necessary for stress erythropoiesis18. Because in silico analysis of the murine Fam132b promoter spanning 3000bp upstream of the transcription initiation site revealed multiple putative Stat5 binding elements (MatInspector, Genomatix Software) (Supplementary Table 1), we explored the involvement of this pathway in ERFE regulation. Treatment of total mouse bone marrow cells with two chemically unrelated Stat5 inhibitors (pimozide or N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide) prior to a 15 hours incubation with EPO inhibited Fam132b mRNA stimulation (Fig. 3C). Thus, EPO regulates Fam132b expression in erythroblasts through the stress erythropoiesis-related Jak2/Stat5 signaling pathway.
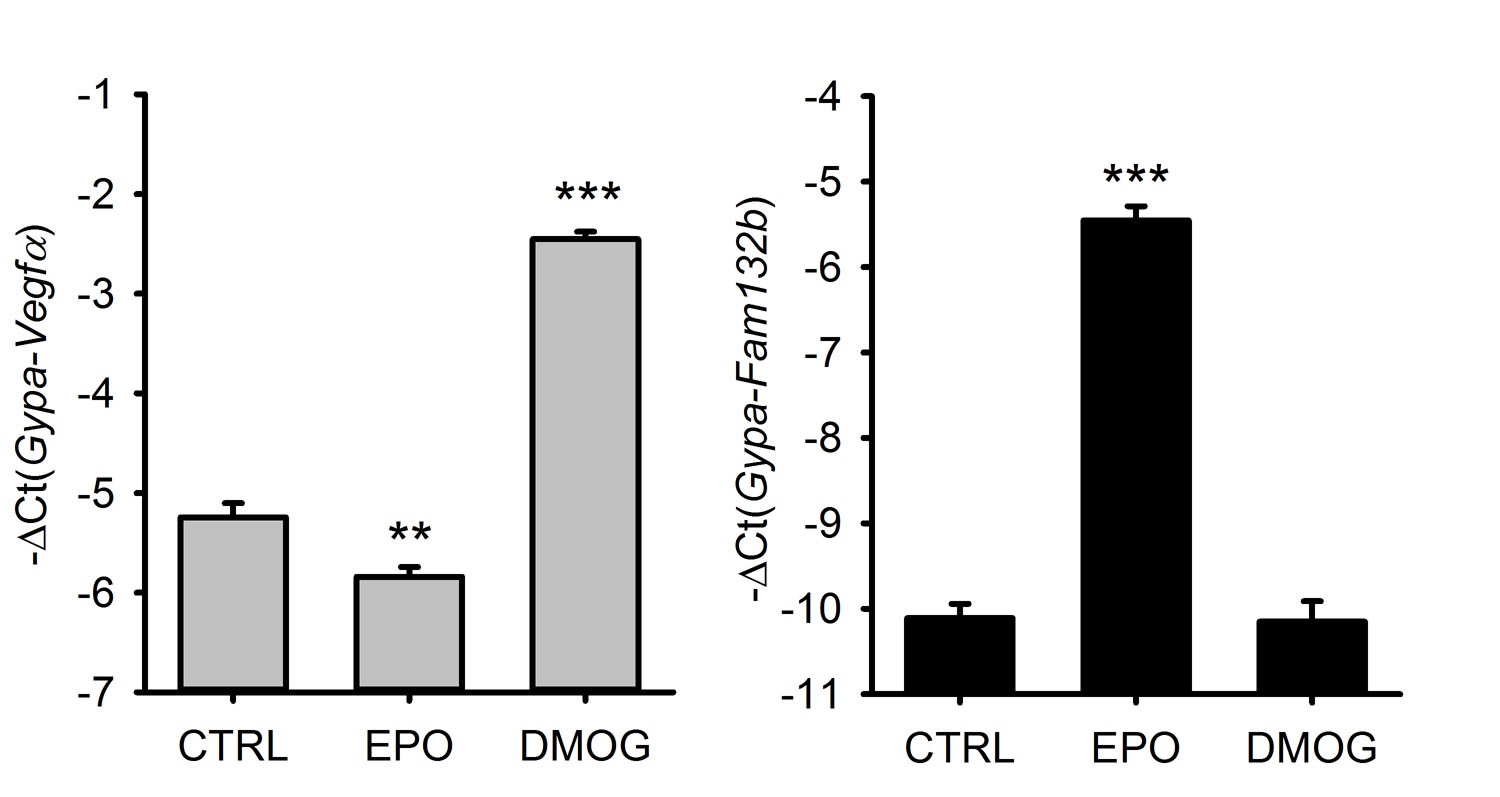
In contrast, we found no effect of hypoxia-related or systemic inflammatory signaling on Fam132b mRNA expression in the marrow. Treatment of mouse marrow cells in vitro with dimethyloxalylglycine (DMOG), an inhibitor of oxygen-sensing prolyl hydroxylases that mediate hypoxia-related signaling, had no effect on Fam132b mRNA expression even though it strongly induced Vegfα mRNA (Supplementary Fig. 6). Bone marrow Fam132b mRNA concentrations were also unresponsive to an acute inflammatory stimulus in vivo. Four hours after lipopolysaccharide injection into mice, TNFα, IL6 and hepcidin mRNAs were induced but Fam132b mRNA expression was unchanged (Supplementary Fig. 7).
ERFE-deficient mice have a transient hemoglobin deficit
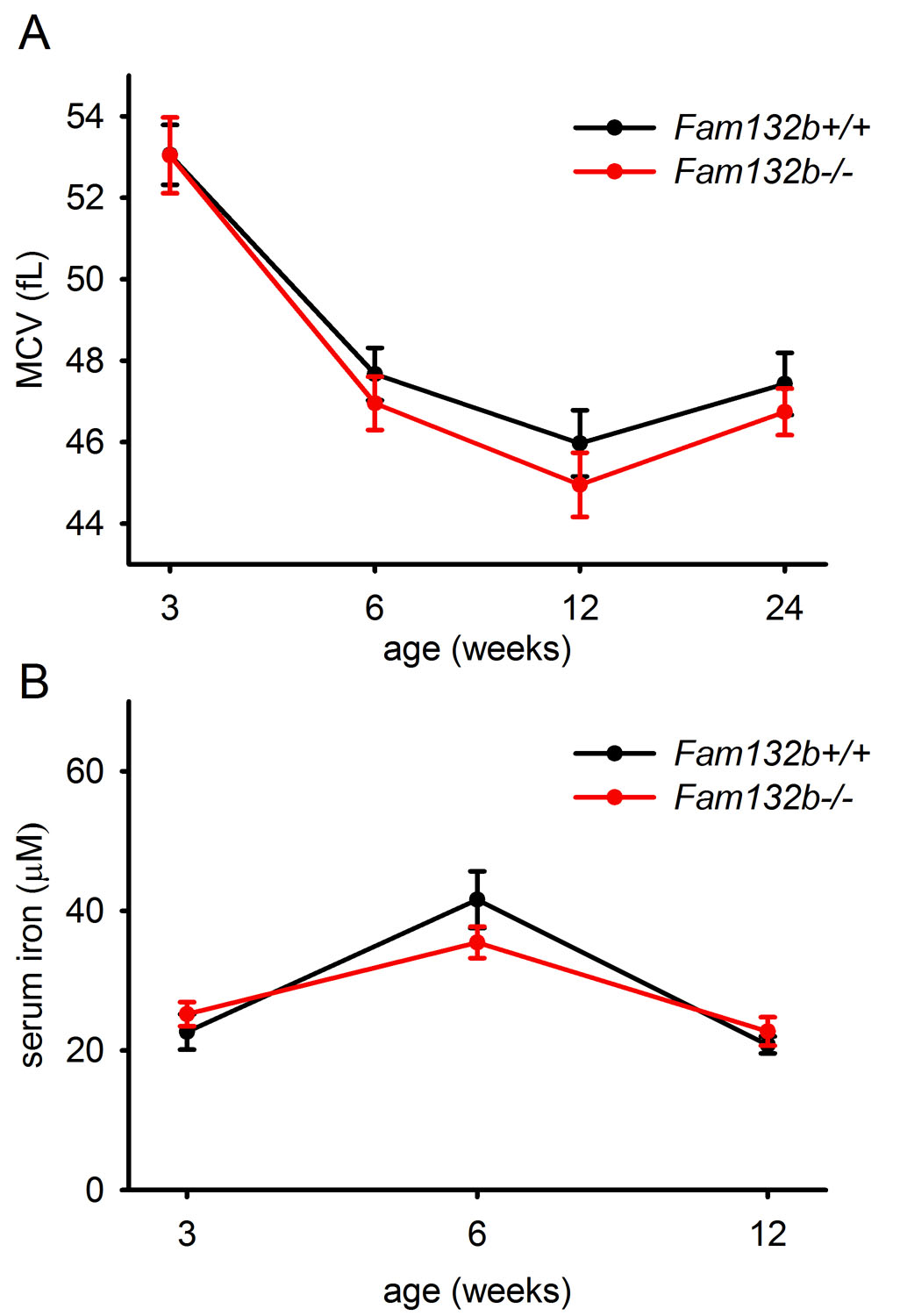
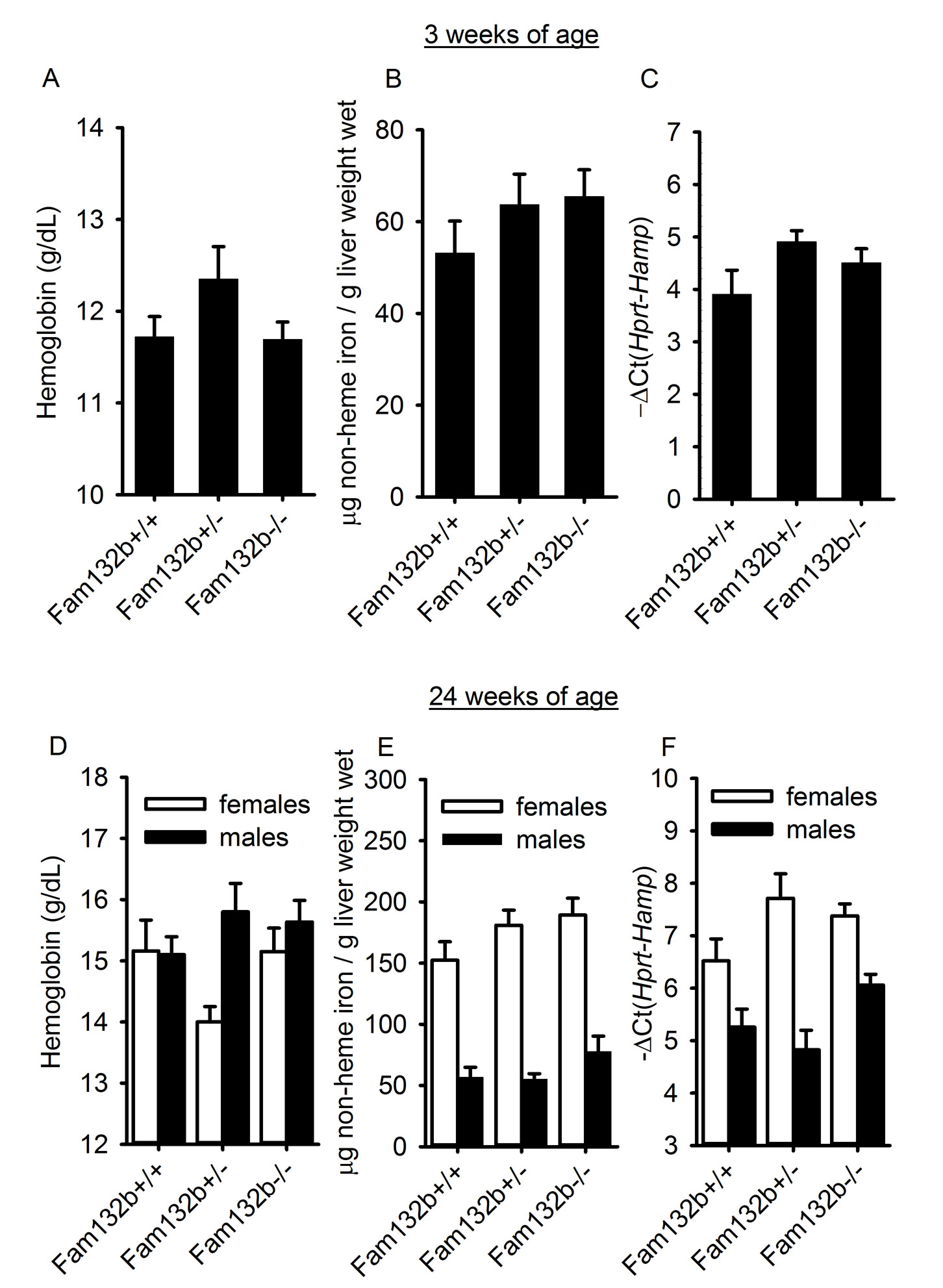
Fam132b−/− mice were previously generated as part of a large-scale mouse knockout library19 and were reported to be viable and fertile without any abnormal phenotypic features. In our more detailed examination, Fam132b-deficient mice had mildly lower hemoglobin and mean corpuscular hemoglobin concentrations than wild-type mice at 6 weeks of age (Fig. 4A, B), a time of rapid growth, erythropoietic expansion and high iron demand. Six week-old Fam132b+/− heterozygotes also exhibited slightly decreased hemoglobin levels compared to WT animals (Supplementary Fig. 8A). In contrast, 3, 12 and 24 week-old Fam132b−/− mice had hematological parameters similar to those of wild-type mice. Fam132b−/− mice exhibited a trend toward lower mean corpuscular volume at 6, 12 and 24 weeks, and lower serum iron level at 6 weeks, but the differences were not statistically significant (Supplementary Fig. 9)
Figure 4. At 6 weeks of age, ERFE-deficient mice have lower hemoglobin and mean corpuscular hemoglobin (MCH) levels than wild type mice.

Hemoglobin and MCH were compared between Fam132b+/+ and Fam132b−/− littermates at the ages of 3, 6, 12, and 24 weeks. Values shown are means of hemoglobin or MCH ± SEM. Means were compared between genotypes by two-tailed Student t-test (n=8 to 16 mice per time point and genotype). ***p<0.001, **p<0.01, *p<0.05.
We did not observe any baseline difference in liver iron or hepcidin between Fam132b+/+, Fam132b+/− and Fam132b−/− littermates at the ages examined (3, 6 and 24 weeks) (Supplementary Fig. 8 and 10). Spleen iron content was measured at 6 weeks of age and was also similar between wild-type and Fam132b−/− mice (Supplementary Fig. 8D).
ERFE is required for hepcidin suppression after blood loss
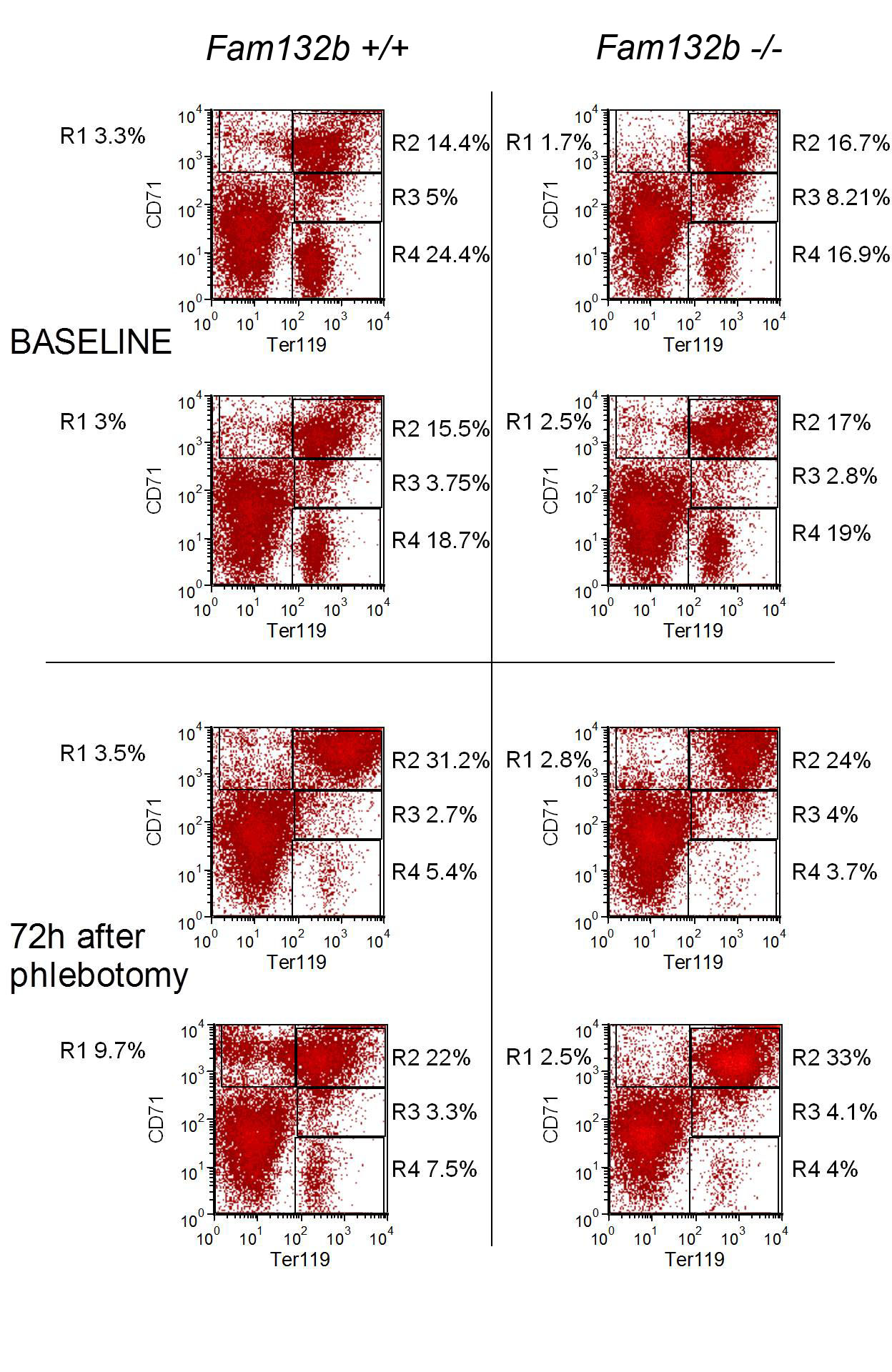
To determine whether ERFE is an erythroid regulator necessary for hepcidin regulation by increased erythropoietic activity, we studied hepatic hepcidin response to phlebotomy in 6 week-old Fam132b+/+, Fam132b+/− and Fam132b−/− littermate mice. In contrast to the rapid and profound suppression of hepcidin mRNA in Fam132b+/+ mice (10-fold suppression at 15 hours after phlebotomy), Fam132b-deficient mice failed to mount this response (Fig. 5A). Even Fam132b+/− mice showed a blunted suppression of hepcidin (~4-fold) compared to their wild-type counterparts, consistent with lower levels of Fam132b mRNA expression in the bone marrow (Fig. 5B). After phlebotomy, serum iron concentrations in Fam132b−/− mice (but not in Fam132b+/+) were at all times significantly lower than baseline, consistent with the lack of suppression of hepcidin and a homeostatic defect in iron regulation after hemorrhage (Supplementary Fig. 11). These results support our hypothesis that ERFE is required for the regulation of hepcidin by increased erythropoietic demand. Flow cytometry analysis of bone marrow erythroblasts from Fam132b+/+ and Fam132b−/− littermate mice 72 hours after phlebotomy and in control mice did not reveal any difference in erythroblast differentiation (Supplementary Fig. 12) suggesting that ERFE is not necessary for erythroblast differentiation during baseline and stress erythropoiesis.
Figure 5. Fam132b-deficient mice do not suppress hepcidin in response to phlebotomy and show delayed recovery from anemia.

(A) After phlebotomy, hepatic Hamp mRNA expression was measured in Fam132b+/+ (dashed line), Fam132b+/− (dotted line) and Fam132b−/− (solid line) littermates (n=6 to 14 per genotype and time-point). Hepcidin is suppressed after hemorrhage in wild-type mice but not in Fam132b-deficient mice, with Fam132b+/− presenting an intermediate response between the wild-types and knockouts. (B) After phlebotomy, both Fam132b+/+ (dashed line) and Fam132b+/− (dotted line) mice increase Fam132b mRNA expression in the bone marrow, although the maximal levels were about twice as high in Fam132b+/+ compared to Fam132b+/−. (C and D) Phlebotomized Fam132b-deficient mice (solid line) compared to wild-type mice (dashed line) showed delayed recovery of hemoglobin and lower mean corpuscular hemoglobin (MCH). Hepcidin (Hamp) (A) and Fam132b (B) mRNA levels were measured by qRT-PCR and shown as −ΔCt (i.e., Ct Hprt – Ct Hamp or Fam132b). For each genotype, means of −ΔCt values were compared at each time-point to the respective control mice t=0 by two-tailed Student t-test. Hematological parameters (C, D) were compared for each measurement between WT (n=17) and KO (n=15) by Student t-test. All graphs show means ± SEM. In the absence of gender differences, the genders were combined for each parameter. ***p<0.001, **p<0.01, *p<0.05
We next investigated whether ERFE facilitates compensatory iron acquisition during recovery from hemorrhage. Twelve week-old Fam132b+/+ and Fam132b−/− were phlebotomized and their hematological parameters monitored for 9 days. Throughout this period, ERFE-deficient mice exhibited lower hemoglobin, mean corpuscular hemoglobin, mean corpuscular volume and hematocrit levels compared to wild-type controls (Fig. 5C, D and Supplementary Fig. 13) demonstrating that the recovery of ERFE-deficient mice from hemorrhage-induced anemia was delayed by several days.
ERFE acts on the liver to suppress hepcidin
To test whether secreted erythroferrone protein is a suppressor of hepcidin, we injected C57BL/6 male mice with murine recombinant ERFE (2 μg/g body weight). We found that hepcidin mRNA and serum protein levels were significantly reduced 15 hours after ERFE treatment compared to saline-injected mice (Fig. 6A, B). despite a substantial inflammatory response (as shown by increased expression of Saa1 mRNA, Supplementary Fig. 14A), which would be expected to increase hepcidin. Using high performance gel-permeation chromatography under nondenaturing conditions, we confirmed a previous report16 that the purified recombinant protein had an effective molecular mass of 500–600 kDa suggesting that it was highly aggregated, a tendency shared by several other members of the TNFα superfamily. Measures that decrease aggregation may decrease the inflammatory effects of the recombinant protein and further increase its potency.
Figure 6. ERFE acts directly on the liver to suppress hepcidin.

(A, B, C) Six C57BL/6 male mice were treated intraperitoneally with either mouse recombinant ERFE (2 μg/g) or saline and analyzed 15 hours later. Hepatic hepcidin mRNA (A) and serum hepcidin (B) levels were significantly suppressed by ERFE treatment. (C, D, E) Seven week-old C57BL/6 males were transduced with a lentivirus encoding GFP or mouse ERFE (n=8 per group) and analyzed after 3 weeks. (C) Fam132b mRNA expression was not detectable (ND) in the liver of control mice, and was moderately increased in the liver of mice transduced with the ERFE lentivirus although only 2-fold higher than the baseline bone marrow levels. However, this increase was sufficient to significantly reduce hepatic hepcidin mRNA (D) and serum hepcidin (E) levels. (F) Recombinant mouse ERFE is secreted by HEK293T cells infected with the ERFE-lentivirus, as detected by Western blot with an anti-FLAG antibody. (G) Treatment of mouse primary hepatocytes with supernatants (50% v/v) from HEK293T control cells or HEK293T cells overexpressing ERFE indicated that ERFE acts directly on the liver to suppress hepcidin mRNA expression. Hepcidin (Hamp), serum amyloid 1 (Saa1) and ERFE (Fam132b) mRNA levels were measured by qRT-PCR and are shown as −ΔCt (i.e., Ct Hprt – Ct Hamp, Saa1 or Fam132b). Graphs show means± SEM. Mean values were compared between treated mice and control mice or between ERFE-treated to control samples by two-tailed Student t-test. ***p<0.001, **p<0.01, *p<0.05
To avoid the acute inflammatory effect of the recombinant ERFE preparation, C57BL/6 male mice were treated with a lentivirus-based vector encoding ERFE or a control vector (expressing GFP). Three weeks later, although we only achieved a modest increase in Fam132b mRNA in the liver (but not the bone marrow) (Fig. 6C), hepcidin mRNA and serum levels were reduced 30-fold and 10-fold respectively (Fig. 6D, E) suggesting that even a small amount of ERFE in the liver was sufficient to exert its inhibitory effect on hepcidin transcription. Saa1 mRNA was not increased in these mice (Supplementary Fig. 14B). Neither recombinant ERFE nor chronic lentiviral expression of ERFE affected the BMP pathway, as measured by the expression of its canonical target Id1 mRNA (Supplementary Fig. 14C,D). We then tested whether ERFE could act directly on the liver to regulate hepcidin. To circumvent recombinant ERFE aggregation and loss of potency during the purification process, mouse primary hepatocytes were treated for 15 hours with 50% (v/v) supernatants from control HEK293T cells or HEK293T cells overexpressing ERFE (Fig. 6F), resulting in a 4-fold decrease in hepcidin mRNA levels in ERFE-treated cells (Fig. 6G). Taken altogether, these results show that ERFE is a potent suppressor of hepcidin that can act directly on the liver to repress hepcidin mRNA expression.
Mice with β-thalassemia intermedia have high levels of ERFE
HbbTh3/+ mice are an established model of β-thalassemia intermedia, and exhibit anemia, ineffective erythropoiesis and splenomegaly20. Their hepatic hepcidin expression is inappropriately low, leading to hyperabsorption of iron and hepatic iron overload20. We assayed Fam132b mRNA in the marrow and spleen of 6-week-old HbbTh3/+ mice. As expected, the mice were anemic and had very high serum EPO concentrations (Fig. 7A, B). Compared to their WT littermates, Fam132b mRNA levels in HbbTh3/+ mice were increased 10-fold in the bone marrow and 16-fold in the spleen (Fig. 7C) suggesting that ERFE may be involved in hepcidin dysregulation in β-thalassemia. Ablation of ERFE in β-thalassemic mice (Fam132b−/−HbbTh3/+) raised hepcidin levels (Fig. 7D) and decreased hepatic iron overload compared to HbbTh3/+ mice (Fig. 7E). Consistent with the expected effect of increased hepcidin levels in Fam132b−/−HbbTh3/+ mice, serum iron concentrations and erythrocyte MCV and MCH were also decreased compared to HbbTh3/+ mice (Supplementary Fig. 15). Thus, ERFE contributes to hepcidin suppression and iron overload in this model of β-thalassemia intermedia.
Figure 7. In a mouse model of β-thalassemia intermedia, erythroferrone is markedly increased, suppresses hepcidin and contributes to iron overload.

Six week-old HbbTh3/+ mice had (A) lower hemoglobin and (B) greatly increased serum Epo concentrations compared to their wild-type littermate mice (n=7 mice per genotype). Fam132b mRNA expression (C) was highly increased in the bone marrow and the spleen of HbbTh3/+ mice compared to wild-type controls. (D and E) Comparing six week-old littermates WT (n=12), HbbTh3/+ (n=12) and Fam132b−/−HbbTh3/+ (n=18), ablation of ERFE reversed hepcidin suppression (D) and significantly decreased hepatic iron overload (E). Values shown are means ± SEM. Statistical comparisons were performed by two-tailed Student t-test for normally distributed variables and Mann-Whitney rank sum test otherwise. ***p<0.001, **p<0.01, *p<0.05.
DISCUSSION
The search for the erythroid regulator of iron homeostasis that assures adequate iron supply for increased erythropoietic activity has been ongoing for many years1. Discovery of the iron regulatory hormone hepcidin and its suppression in anemia suggested a mechanism. It was proposed that hepcidin suppression in anemias may result from direct hepcidin regulation by hepatocellular hypoxia3, 21 or erythropoietin3, 22. However, later studies provided convincing evidence against a biologically-important direct effect of hypoxia on hepcidin transcription23. The lack of hepcidin suppression after bleeding or EPO injections in mice with ablated bone marrow 4, 5, 23 also argues against a direct effect of hypoxia or EPO. Rather, these studies suggest that high levels of EPO cause hepcidin suppression indirectly, by inducing the secretion of erythroid regulators from the bone marrow which, in turn, act on the liver to suppress hepcidin expression and increase iron delivery from dietary absorption and stores. We therefore initiated a search by a gene-expression microarray for a potential hepcidin suppressor, highly induced in the bone marrow prior to hepcidin suppression in the liver (Fig. 1). We focused on secreted proteins whose mRNA expression was rapidly increased 4 hours after phlebotomy or EPO injection, and identified the orphan TNF-α superfamily member Fam132b as a new erythroid regulator secreted by differentiating erythroblasts (Fig. 2, 3). We named this factor erythroferrone as our results suggest it functions as a hormone linking erythropoiesis and iron metabolism (Fig. 8). In gene expression databases, FAM132B was highly expressed in the bone marrow and fetal liver, and an in vitro study of differentiated human erythroblasts24 reported that FAM132B expression was highest in intermediate erythroblasts. We documented the high expression of Fam132b mRNA in mouse and human erythroblasts and established that it is greatly induced by erythropoietic stimulation (Fig. 3 and Supplementary Fig. 5) but not directly affected by hypoxia-related or inflammatory stimuli (Supplementary Fig. 6 and 7). In the meantime, another study reported that Fam132b (also termed myonectin or CTRP15) is generated predominantly in skeletal muscle, is suppressed by fasting and induced by refeeding, and participates in the regulation of systemic lipid metabolism16. The study, however, did not include analysis of the bone marrow. Further work will be needed to understand how these distinct activities coexist and to assess their respective biological and pathological relevance.
Figure 8. Proposed role of the erythroid factor erythroferrone (ERFE).

After erythropoietic stimulation, differentiating erythroblasts in the bone marrow and spleen rapidly increase ERFE production in an EPO-Stat5 dependent manner. ERFE is secreted into the circulation and acts directly on the liver to repress hepcidin. ERFE-mediated hepcidin suppression in turn increases iron availability for new red blood cells synthesis.
In the absence of ERFE, the mechanism suppressing hepcidin early after erythropoietic stimulation is abrogated and Fam132b-deficient mice exhibit several days delay in recovery from hemorrhage compared to wild-type animals (Fig. 5). This confirms that ERFE is a biologically important regulator of hepcidin expression and is required for the rapid compensatory response to hemorrhage. Nevertheless, Fam132b-deficient mice do recover from hemorrhage suggesting that other mechanisms eventually increase iron availability for new erythrocyte synthesis. This is not surprising as recovery from hemorrhage is subject to strong evolutionary pressure, and likely mediated by multiple mechanisms. Even if hepcidin is not suppressed rapidly, increased iron demand from accelerated erythropoiesis could eventually cause transient iron deficiency (i.e. decreased plasma iron concentration) which would be expected to decrease hepcidin levels and therefore increase iron absorption and release from the stores. Interestingly, mature Fam132b-deficient mice at baseline do not exhibit any detectable abnormalities as their iron parameters and hepcidin expression are similar to wild-type controls (Supplementary Fig. 8, 9, 10 and 12). Moreover, the induction of Fam132b mRNA after blood loss was not modulated by the BMP/Smad pathway or iron (Supplementary Fig. 3D). Thus, ERFE is a stress erythropoiesis-specific regulator, and does not play a major role in baseline erythropoiesis. Consistent with this, ERFE ablation in mice only causes a transient deficit in hemoglobin synthesis as erythropoiesis intensifies during rapid growth between 3 and 6 weeks of age (Fig. 4). The strong dependence of Fam132b induction by EPO on the Stat5 pathway (Fig. 3C) is also consistent with a role in stress erythropoiesis.
In mice, the injection of recombinant ERFE, or ERFE over-expression using lentiviral vectors confirmed that ERFE is an effective suppressor of hepcidin in vivo (Fig. 6). Treatment of primary hepatocytes with ERFE demonstrated that the hormone exerts its hepcidin-suppressive activity directly on hepatocytes (Fig. 6G). Further work is needed to identify ERFE’s receptor(s) and the downstream effectors controlling hepcidin expression. The BMP/Smad signaling pathway, the principal pathway mediating iron-dependent regulation of hepcidin, may not be involved in ERFE-mediated hepcidin regulation as neither HJV nor TfR2 were necessary for rapid hepcidin suppression after blood loss (Supplementary Fig. 3), Smad5 phosphorylation was not decreased (Supplementary Fig. 2A–D), and Id1, a canonical target of the BMP pathway, was not appreciably affected by erythropoietic stimulation (Supplementary Fig. 2E) or ERFE treatment (Supplementary Fig. 14). Thus, although both the iron-regulated BMP/Smad pathway and erythropoietin-regulated ERFE pathway converge on hepcidin, we saw no significant evidence of crosstalk between them. Hemorrhage/ERFE-mediated suppression of hepcidin mRNA was observed over a large range of iron loads and baseline hepcidin mRNA concentrations, and the ERFE signal appeared to act on the hepcidin gene independently from the iron-regulated BMP/Smad pathway. A recent study of hepcidin regulation by iron in an Epo-overproducing mouse model25 also supports the existence of independent pathways regulating hepcidin in response to hepatic iron load and erythropoietic activity.
Hepcidin suppression by increased erythropoietic activity underlies the development of iron overload in inherited anemias with ineffective erythropoiesis. This is particularly evident in untransfused patients26, 27 where hyperabsorption of dietary iron is the sole source of iron overload. Studies of human patients will be necessary to establish whether ERFE plays a role in hepcidin suppression and iron overload in anemias with ineffective erythropoiesis. The production of ERFE would be expected to increase in iron-loading anemias with ineffective erythropoiesis, considering that these patients have high EPO levels and massively expanded erythroblast populations28. Indeed, our measurements in the mouse model of β-thalassemia intermedia (Fig. 7C) document a large increase in Fam132b mRNA expression in the marrow and spleen. Ablation of Fam132b in a mouse model of β-thalassemia intermedia reverses the hepcidin defect, and decreases hepatic iron overload (Fig. 7D and E), serum iron concentrations and erythrocyte MCV and MCH (Supplementary Fig. 15). Thus, erythroferrone is a strong candidate for a pathological hepcidin suppressor in inherited anemias with ineffective erythropoiesis, where hepcidin levels are inversely correlated with EPO concentrations and erythropoietic activity29, 30. Although the changes induced by ERFE ablation are smaller and do not affect blood hemoglobin concentrations, the pattern is otherwise similar to the effect of pharmacologic increases of hepcidin expression in β-thalassemic mice31–33. If the contribution of ERFE to hepcidin suppression and iron overload in iron-loading anemias is confirmed in clinical studies, ERFE neutralization would be a rational and apparently specific therapeutic strategy. At the other end of the disease spectrum, the ability of ERFE to suppress hepcidin could be useful in the treatment of iron-restrictive anemias characterized by increased hepcidin, including anemia of inflammation, anemia of chronic kidney diseases and iron-refractory iron-deficiency anemia. 34
METHODS
Mice
Experiments were conducted in accordance with guidelines by the National Research Council and were approved by the University of California, Los Angeles. Wild type (WT) C57BL/6J males were purchased from The Jackson Laboratory (Bar Harbor, Me). Tfr2Y245X/Y245X (FVB background35, from Dr. Robert Fleming, Saint Louis University, MO) and Hjv−/−(129S6/SvEvTac background36, from Dr. Paul Schmidt, Children’s Hospital Boston, MA) mutant mice were maintained at UCLA. Fam132b+/− mice on a mixed Sv129/C57BL/6 background (Fam132btm1Lex) were generated by Lexicon Pharmaceuticals19 by deletion of 3204bp of the genomic sequence (exons 2–7) and insertion of a neomycin cassette and were obtained from the Mutant Mouse Regional Resource Center (MMRRC) at UC Davis (strain B6;129S5-Fam132btm1Lex/Mmucd, ID MMRRC:032289-UCD). Fam132b+/− mice were crossed to generate Fam132b+/+, Fam132b+/− and Fam132b−/− littermates that were maintained at UCLA. Hbbth3/+ mice were obtained from The Jackson Laboratory (strain name B6;129P-Hbb-b1tm1Unc Hbb-b2tm1Unc/J) and maintained at UCLA in heterozygosity by breeding with C57BL/6 mice. Unless otherwise specified, mice received a standard diet (NIH 31 rodent diet, 336 ppm iron, Harlan Teklad, Indianapolis, IN) and were analyzed between 6 and 8 weeks of age. A set of Tfr2Y245X/Y245X (Tfr2 ko ID) were fed an iron-deficient diet containing 4 ppm iron (Harlan Teklad, TD 80396) at weaning and were studied at 6 weeks of age. Fam132b+/+, Fam132b+/− and Fam132b−/− mice were analyzed at 3, 6, 12 and 24 weeks of age. To provide an erythropoietic stimulus, mice were phlebotomized by retro-orbital puncture (500 μL) or treated with a single dose of 200 units human erythropoietin (Epogen, Amgen), and analyzed within 24–48 hours. For the studies of recovery from anemia, Fam132b+/+ and Fam132b−/− mice were monitored for 9 days after a 500 μL phlebotomy and 25–50 μL of blood were collected by retro-orbital puncture at each time-point for hematological parameters assessment. In some experiments, C57BL/6 males were given 2 doses of 25 mg iron-dextran (Sigma-Aldrich) in two consecutive weeks and were phlebotomized and analyzed a week later at the age of 7 weeks. HbbTh3/+ mice were mated with Fam132b−/− to generate Fam132b+/−, HbbTh3/+ mice and subsequent breeding of these mice to Fam132b+/− mice generated littermates wild-type, HbbTh3/+ and Fam132b−/−, HbbTh3/+ (referred to as Fam132b−/−Th3/+) that were maintained at UCLA on an iron-balanced diet containing 50 ppm diet (Harlan Teklad, TD 06015) and studied at the age of 6 weeks.
Measurement of iron and hematologic parameters
Serum iron, spleen and liver non-heme iron concentrations were determined as previously described37, using acid treatment followed by a colorimetric assay for iron quantitation (Sekisui Diagnostics, Charlottetown, Canada). Complete blood counts were obtained with a HemaVet blood analyzer (Drew Scientific).
Microarray analysis
Total RNA from mouse bone marrow was extracted using Trizol (Invitrogen) and purified using RNeasy Micro kit (Qiagen). RNA quality was checked on RNA 6000 Nano chips using a Bioanalyzer 2100 (Agilent Technologies). Gene-level expression profiling of the bone marrow after phlebotomy was performed with the GeneChip® Gene 1.0 ST Array System for mouse (Affymetrix). Expression data were analyzed using the GeneSpring software (Agilent) by comparing control mice (n=3) to mice at the time-points 4h (n=1 to indicate stress-related variations of transcription), 9h (n=3), 12h (n=2), 15h (n=2) and 48h (n=2).
Quantitation of mRNA levels
Total RNA from mouse tissues was extracted using Trizol (Invitrogen). Total RNA from muscle, heart and brain was extracted using RNeasy fibrous tissue mini kit (Qiagen). cDNA was synthesized using iScript (Biorad). Quantitative PCR reactions were prepared with Sso advanced Sybr Green supermix (Biorad) and primers indicated in Supplementary Table 2, and run in duplicate on a CFXconnect Instrument (Biorad). Hamp, Fam132b, Id1, Saa1 and Gypa mRNA transcript abundance was normalized to the reference genes Hprt or Rpl4. Results are expressed as −ΔCt ± standard error of the mean (i.e., the cycle threshold differences between reference and target genes within each group of mice). Expression ratio and SEM values of Hamp transcripts in ERFE-treated cells relative to controls and normalized to the reference gene Hprt mRNA was calculated using REST38. Statistical significance was determined using randomization tests.
Isolation of human erythroblasts
CD34−/CD235+ cells from fetal livers were sorted by Novogenix Laboratories, LLC as previously described39 from de-identified specimens following informed consent. CD34−/CD235+ human bone marrow cells were a kind gift of Dr. Gay Crooks (Departments of Pathology & Laboratory Medicine and Pediatrics in the David Geffen School of Medicine, UCLA).
Rapid Amplification of cDNA Ends (RACE)
Human fetal liver erythroblasts obtained from Novogenix Laboratories, LLC., were resuspended in IMDM medium (GIBCO) supplemented with 20% FBS and 2mM L-Glutamine (Invitrogen) and treated 15 hours with 10U/mL of erythropoietin (Epogen, Amgen). Total RNA was extracted using Trizol (Invitrogen) and purified using RNeasy Micro kit (Qiagen). 5′ RACE was performed using Generacer (Invitrogen) according to the manufacturer’s instructions.
Mouse bone marrow erythroblasts isolation and treatment
Freshly isolated total bone marrow cells from C57BL/6 mice femurs were suspended in IMDM medium (GIBCO) supplemented with 20% FBS and 2mM L-Glutamine (Invitrogen). Cells were pre-treated for 2 hours with two different Stat5 inhibitors: 10 μM pimozide (Sigma) or 200 μM N′-((4-Oxo-4H-chromen-3-yl)methylene)nicotinohydrazide (Calbiochem) and incubated overnight (15h) with 10 U/mL of human recombinant erythropoietin (Epogen, Amgen). Treatments were done in duplicates in three independent experiments. To test the effect of hypoxia, mouse erythroblasts were treated overnight (15h) with 1mM DMOG (Dimethyloxalylglycine, N-(Methoxyoxoacetyl)-glycine methyl ester, SIGMA).
Fluorescence-Activated Cell Sorting
To isolate erythroblasts at different stages of maturation by cell sorting, >15 × 106 bone marrow cells from six 7 week-old Sv129/C57BL/6 Fam132b+/+ littermate mouse femurs (three controls and three 15 hours after phlebotomy) were filtered through a 30 μm filter and resuspended in 1 mL PBS/5% FCS. Cells were blocked with rat anti-mouse CD16/CD32 (0.25 μL/106 cells, BD Biosciences no. 553142) for 15 minutes and subsequently stained with FITC rat anti-mouse TER-119 (0.5 μL/106 cells, BD Biosciences no. 557915), and PE rat anti-mouse CD71 (0.5 μL/106 cells, BD Biosciences no. 553267) and incubated on ice for 30 minutes in the dark. Cells were washed twice with 1 mL PBS and resuspended in 2 mL PBS/5%FCS. Sorting was performed on a FACSAriaII High-Speed Cell Sorter (Becton Dickinson). Erythroblasts population were differentiated as previously described17 into four populations: pro-erythroblasts (pro-E, Ter119med CD71high FSChigh), basophilic erythroblasts (Ter119high CD71high FSChigh), polychromic erythroblasts (Ter119high CD71high FSClow) and orthochromic erythroblasts (Ter119high CD71low FSClow).
Construction of the ERFE-secreting cell line
Lentivirus-based vectors pRRL-sin-cPPT-hCMV-MCS-IRES-emdGFP encoding ERFE mouse cDNA ORF (accession number NM_173395) fused to a C-terminal 3xFLAG tag and empty control vector were produced by the UCLA Vector Core facility. Internal ribosome entry site ensured reinitiation and translation of Green Fluorescent Protein (GFP). Vector titers were determined by p24 ELISA (Perkin Elmer). HEK293T cells (ATCC® CRL-3216™) were transduced using 4 μg/ml Polybrene (Sigma, H9268). Insertion of the virus was confirmed by GFP detection by fluorescence microscopy. ERFE overexpression and secretion was monitored by western blot.
Recombinant protein production and purification
Supernatants from HEK293T overexpressing FLAG-tagged ERFE were harvested after 5 days, supplemented with protease inhibitor cocktail and purified using an anti-FLAG affinity gel according to the manufacturer’s protocol (Sigma) and eluted with 150 μg/ml FLAG peptide (Sigma). FLAG peptide was then eliminated by filtration through Amicon Ultra 30K device (Millipore) and recombinant ERFE was resuspended in saline (0.9% NaCl). Protein concentration was determined by Coomassie Imperial Protein Stain (Pierce) and bicinchoninic acid protein assay (Pierce).
Western blot analysis
Supernatants from ERFE-secreting HEK293T cells were subjected to SDS-PAGE and electroblotted to PVDF membranes (Biorad). Membranes were incubated with an anti-DDK monoclonal antibody (4C5, Origene) which recognizes DYKDDDDK epitope (FLAG-Tag) using the ONE-HOUR Western™ Detection System (Genscript) following manufacturer’s instructions. For Smad5 analysis, membranes were incubated with phospho-Smad5 (Ser463/465, Abcam, ab92698) and Smad5 (Epitomics, 1682-1) antibodies, as previously detailed40 and with GAPDH (Cell Signaling Technology, 14C10). Western blots were visualized by chemiluminescence using the ChemiDoc XRS+ imaging system with Image Lab software (Bio-Rad).
Enzyme-linked immunosorbent assays
Mouse hepcidin-1 monoclonal antibodies, Ab2B10 (capture), AB2H4-HRP (detection) and synthetic mouse hepcidin-25, were a generous gift from Amgen (Thousand Oaks, CA) 41. High binding 96-well EIA plates (Corning) were coated overnight at room temperature with 50 μL/well of 3.6 μg/mL Ab2B10 in 0.2 M carbonate-bicarbonate buffer pH 9.4 (Pierce). Plates were washed twice with wash buffer (PBS, 0.5% Tween-20) and blocked for 45 minutes with 200 μL/well blocking buffer (PBS, 1% BSA, 1% normal goat serum, 0.5% Tween-20). Samples and standards were added and incubated 1 hour at room temperature. After four washes, plates were incubated an hour with 50 μL/well of 130 ng/mL Ab2H4-HRP, washed 4 times then developed with 100 μL/well Ultra-TMB substrate (Thermo Scientific) for 30 minutes in the dark at room temperature. The reaction was stopped by adding 50 μL 2M sulfuric acid and the absorbance was measured at 450 nm. Serum Epo was measured using mouse Epo quantikine set (R&D Systems), according to the manufacturer’s instructions.
Recombinant protein injection
Recombinant ERFE (2 μg/g of body weight) or the equivalent volume of saline solution (0.9% NaCl) was injected intraperitoneally into 8 week-old C57BL/6 mice (n=6). To prevent any interference by dietary iron overload, mice were placed on iron-deficient diet (4ppm Fe) 10 days prior to the injection. Mice were analyzed 15 hours after treatment.
Mouse primary hepatocyte isolation and treatment
Hepatocytes were isolated from wild-type C57BL/6 mice by a two-step portal vein collagenase perfusion method as previously described42 and incubated overnight (15 hours) in fresh Williams E Medium (Sigma Aldrich) supplemented with 5% FBS and 200 μM L-glutamine (Invitrogen) and 50% (v/v) supernatants from control HEK293T cells or HEK293T cells overexpressing ERFE (also maintained in Williams E Medium/5%FBS/200 μM L-glutamine). Three independent experiments with 15 hour treatments in triplicate were performed.
Injection of lentiviral vector
Lentiviral vector supernatants were thawed and diluted in 0.9% saline. Six week-old C57BL/6 mice were injected intravenously with 1 × 108 IU of a control lentiviral vector or the lentiviral vector encoding ERFE (200 μL injection volume). Mice were analyzed three weeks after injection.
Statistical analysis
The statistical significance of differences between groups was evaluated using Sigmaplot 11.0 package (Systat Software, San Jose, CA). The Student t test was used to compare 2 groups of normally distributed data. The Mann Whitney rank-sum test was used to compare data that were not normally distributed. A P value < 0.05 in a two-tailed test was considered as statistically significant.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The authors thank Victoria Gabayan for her invaluable work with the mouse breeding. Flow cytometry was performed in the UCLA Jonsson Comprehensive Cancer Center (JCCC) and Center for AIDS Research Flow Cytometry Core Facility that is supported by National Institutes of Health awards CA-16042 and AI-28697, and by the JCCC, the UCLA AIDS Institute, and the David Geffen School of Medicine at UCLA. The UCLA Vector Core is supported by JCCC/P30 CA016042 and CURE/P30 DK041301.
Sources of funding—This research was supported by the NIH grant R01 DK 065029 to T.G, R01 DK 090554 & R01 DK 090554 to S.R and the ASH scholar award to L.K.
Footnotes
DATABASE ACCESSION NUMBERS:
Microarray gene-expression profiling data from mouse bone marrow samples after hemorrhage are available in the GEO repository under the accession number GSE52740. The human FAM132B mRNA sequence is available under Genbank # KF984314.
AUTHOR CONTRIBUTIONS:
L.K. designed and performed the experiments, analyzed the data and wrote the paper. G.J. assisted with experiments, S.R. provided HbbTh3/+ mouse samples for preliminary experiments, and E.V.V. prepared a key reagent. E.N. and T.G. conceived the project and wrote the paper.
Disclosures—Drs. Nemeth and Ganz are shareholders and scientific advisors of Intrinsic LifeSciences and Merganser Biotech, and consultants for Xenon Pharmaceuticals. Dr Rivella receives laboratory support from Isis, Bayer and Merganser and is a shareholder of Merganser Biotech. The rest of the authors have nothing to disclose.
Reference List
- 1.Finch C. Regulators of iron balance in humans. Blood. 1994;84:1697–1702. [PubMed] [Google Scholar]
- 2.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823:1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicolas G, et al. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia, hypoxia, and inflammation. J Clin Invest. 2002;110:1037–1044. doi: 10.1172/JCI15686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pak M, Lopez MA, Gabayan V, Ganz T, Rivera S. Suppression of hepcidin during anemia requires erythropoietic activity. Blood. 2006;108:3730–3735. doi: 10.1182/blood-2006-06-028787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vokurka M, Krijt J, Sulc K, Necas E. Hepcidin mRNA levels in mouse liver respond to inhibition of erythropoiesis. Physiol Res. 2006;55:667–674. doi: 10.33549/physiolres.930841. [DOI] [PubMed] [Google Scholar]
- 6.Ashby DR, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95:505–508. doi: 10.3324/haematol.2009.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ramos P, et al. Iron metabolism and ineffective erythropoiesis in beta-thalassemia mouse models. Ann N Y Acad Sci. 2010;1202:24–30. doi: 10.1111/j.1749-6632.2010.05596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tanno T, et al. Identification of TWSG1 as a second novel erythroid regulator of hepcidin expression in murine and human cells. Blood. 2009;114:181–186. doi: 10.1182/blood-2008-12-195503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tanno T, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 10.Casanovas G, et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444–447. doi: 10.3324/haematol.2012.069807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Theurl I, et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. 2010;148:449–455. doi: 10.1111/j.1365-2141.2009.07961.x. [DOI] [PubMed] [Google Scholar]
- 12.Waalen J, von LK, Lee P, Xu X, Friedman JS. Erythropoietin, GDF15, IL6, hepcidin and testosterone levels in a large cohort of elderly individuals with anaemia of known and unknown cause. Eur J Haematol. 2011;87:107–116. doi: 10.1111/j.1600-0609.2011.01631.x. [DOI] [PubMed] [Google Scholar]
- 13.Meynard D, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41:478–481. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 14.Andriopoulos B, Jr, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–487. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kautz L, et al. Iron regulates phosphorylation of Smad1/5/8 and gene expression of Bmp6, Smad7, Id1, and Atoh8 in the mouse liver. Blood. 2008;112:1503–1509. doi: 10.1182/blood-2008-03-143354. [DOI] [PubMed] [Google Scholar]
- 16.Seldin MM, Peterson JM, Byerly MS, Wei Z, Wong GW. Myonectin (CTRP15), a novel myokine that links skeletal muscle to systemic lipid homeostasis. J Biol Chem. 2012;287:11968–11980. doi: 10.1074/jbc.M111.336834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Socolovsky M. Molecular insights into stress erythropoiesis. Curr Opin Hematol. 2007;14:215–224. doi: 10.1097/MOH.0b013e3280de2bf1. [DOI] [PubMed] [Google Scholar]
- 18.Porpiglia E, Hidalgo D, Koulnis M, Tzafriri AR, Socolovsky M. Stat5 Signaling Specifies Basal versus Stress Erythropoietic Responses through Distinct Binary and Graded Dynamic Modalities. PLoS Biol. 2012;10:e1001383. doi: 10.1371/journal.pbio.1001383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang T, et al. A mouse knockout library for secreted and transmembrane proteins. Nat Biotechnol. 2010;28:749–755. doi: 10.1038/nbt.1644. [DOI] [PubMed] [Google Scholar]
- 20.Gardenghi S, et al. Hepcidin as a therapeutic tool to limit iron overload and improve anemia in beta-thalassemic mice. J Clin Invest. 2010;120:4466–4477. doi: 10.1172/JCI41717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Peyssonnaux C, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117:1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pinto JP, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood. 2008;111:5727–5733. doi: 10.1182/blood-2007-08-106195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Q, Davidoff O, Niss K, Haase VH. Hypoxia-inducible factor regulates hepcidin via erythropoietin-induced erythropoiesis. J Clin Invest. 2012;122:4635–4644. doi: 10.1172/JCI63924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merryweather-Clarke AT, et al. Global gene expression analysis of human erythroid progenitors. Blood. 2011;117:e96–e108. doi: 10.1182/blood-2010-07-290825. [DOI] [PubMed] [Google Scholar]
- 25.Diaz V, et al. Liver iron modulates hepcidin expression during chronically elevated erythropoiesis in mice. Hepatology. 2013;58:2122–2132. doi: 10.1002/hep.26550. [DOI] [PubMed] [Google Scholar]
- 26.Pippard MJ, Warner GT, Callender ST, Weatherall DJ. Iron absorption and loading in [beta]-thalassemia intermedia. The Lancet. 1979;314:819–821. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 27.Papanikolaou G, et al. Hepcidin in iron overload disorders. Blood. 2005;105:4103–4105. doi: 10.1182/blood-2004-12-4844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Centis F, et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with b-thalassemia major. Blood. 2000;96:3624–3629. [PubMed] [Google Scholar]
- 29.Kattamis A, et al. The effects of erythropoetic activity and iron burden on hepcidin expression in patients with thalassemia major. Haematologica. 2006;91:809–812. [PubMed] [Google Scholar]
- 30.Origa R, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92:583–588. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 31.Nai A, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119:5021–5029. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guo S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123:1531–1541. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schmidt PJ, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121:1200–1208. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goodnough LT, Nemeth E, Ganz T. Detection, evaluation, and management of iron-restricted erythropoiesis. Blood. 2010;116:4754–4761. doi: 10.1182/blood-2010-05-286260. [DOI] [PubMed] [Google Scholar]
- 35.Fleming RE, et al. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. P N A S. 2002;99:10653–10658. doi: 10.1073/pnas.162360699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang FW, Pinkus JL, Pinkus GS, Fleming MD, Andrews NC. A mouse model of juvenile hemochromatosis. J Clin Invest. 2005;115:2187–2191. doi: 10.1172/JCI25049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramos E, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120:3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pfaffl MW, Horgan GW, Dempfle L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002;30:e36. doi: 10.1093/nar/30.9.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Handel B, et al. The first trimester human placenta is a site for terminal maturation of primitive erythroid cells. Blood. 2010;116:3321–3330. doi: 10.1182/blood-2010-04-279489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Latour C, et al. Testosterone perturbs systemic iron balance through activation of epidermal growth factor receptor signaling in the liver and repression of hepcidin. Hepatology. 2014;59:683–694. doi: 10.1002/hep.26648. [DOI] [PubMed] [Google Scholar]
- 41.Hod EA, Francis RO, Spitalnik SL, Winters A, Cooke KS, Sasu BJ. Validation and Preclinical Correlation of a New Sandwich ELISA for Measuring Murine Hepcidin. Blood (ASH Annual Meeting Abstracts) 2012;120:2100. [Google Scholar]
- 42.Goodnough JB, Ramos E, Nemeth E, Ganz T. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56:291–299. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.