

Abstract
Alzheimer’s disease (AD) is a devastating disorder that is clinically characterized by a comprehensive cognitive decline. Accumulation of the amyloid-beta (Aβ) peptide plays a pivotal role in the pathogenesis of AD. In AD, the conversion of Aβ from a physiological soluble monomeric form into insoluble fibrillar conformation is an important event. The most toxic form of Aβ is oligomers, which is the intermediate step during the conversion of monomeric form to fibrillar form. There are at least two types of oligomers: oligomers that are immunologically related to fibrils and those that are not. In transgenic AD animal models, both active and passive anti-Aβ immunotherapies improve cognitive function and clear the parenchymal accumulation of amyloid plaques in the brain. In this report we studied effect of immunotherapy of two sequence-independent non-fibrillar oligomer specific monoclonal antibodies on the cognitive function, amyloid load and tau pathology in 3xTg-AD mice. Anti-oligomeric monoclonal antibodies significantly reduce the amyloid load and improve the cognition. The clearance of amyloid load was significantly correlated with reduced tau hyperphosphorylation and improvement in cognition. These results demonstrate that systemic immunotherapy using oligomer-specific monoclonal antibodies effectively attenuates behavioral and pathological impairments in 3xTg-AD mice. These findings demonstrate the potential of using oligomer specific monoclonal antibodies as a therapeutic approach to prevent and treat Alzheimer’s disease.
Keywords: abeta oligomers, Alzheimer’s disease, amyloid plaques, monoclonal antibodies, tau pathology, vaccination
Accumulation of the amyloid-β (Aβ) peptide plays a pivotal role in the pathogenesis of Alzheimer disease (AD) (Golde et al. 2000; Hardy and Selkoe 2002; and Lee et al. 2007). In animal models of AD, both active and passive anti-Aβ immunotherapies improve cognitive functions and clear the parenchymal accumulation of amyloid plaques in the brain (Schenk et al. 1999; Bard et al. 2000; Janus et al. 2000; Morgan et al. 2000; DeMattos et al. 2001; Dodart et al. 2002; Frazer et al. 2008). Accordingly, immunization against Aβ has offered a promising approach toward the therapeutic management of AD (Schenk 2002; Vasilevko and Cribbs 2006; Brody and Holtzman 2008). Promising pre-clinical findings (Schenk et al. 1999) led to clinical trials with AN1792, a synthetic Aβ42 vaccine, but further development was halted when 6% of immunized patients developed meningoencephalitis. Direct administration of anti-Aβ antibodies is believed to represent a safer alternative that minimizes the risk of a proinflammatory T cell response (Monsonego et al. 2003) and permitting dosage control. Recent pre-clinical studies illustrate that passive immunization with antibodies that target amyloid plaques also increase cerebral amyloid angiopathy (CAA) and CAA-associated microhemorrhage in transgenic mouse models of AD (Pfeifer et al. 2002 and Wilcock et al. 2004, 2006; Racke et al. 2005). The mechanisms proposed for the effects of anti-Aβ immunotherapy on amyloid accumulation remain elusive and generally rely on systemic regimens that yield high titers of anti-Aβ antibodies in the peripheral circulation (Vasilevko and Cribbs 2006 and Levites et al. 2006). High antibody titers bind to substantial amounts of Aβ in the bloodstream, which is believed to shift the overall Aβ equilibrium and create a ‘peripheral sink’ that facilitates an efflux of Aβ from the brain (DeMattos et al. 2001 and Lemere et al. 2003). Furthermore, high doses of antibody in the periphery are required because of the low-level penetration of antibody across the blood–brain barrier to effectively engage the local (i.e., central) mechanisms for clearing the cerebral amyloid (Bard et al. 2000; Levites et al. 2006 and Banks et al. 2002). Interestingly, a single intracerebroventricular injection of anti-Aβ antibodies is able to prevent the Aβ-induced impairment of synaptic plasticity in the hippocampus (Oddo et al. 2003), and also transiently reverse the memory deficit in a transgenic AD mouse model (Oddo et al. 2004). It is also reported that passive immunization reduces oligomers, which directly induce GSK3b(β) activation and tau phosphorylation (Ma et al. 2006).
As antibodies that target plaque amyloid deposits are associated with CAA and microhemorrhage, we examined antibodies that are specific for pre-fibrillar oligomers that do not bind to plaques. In addition to polyclonal oligomeric specific antibody (A11) (Kayed et al. 2003), we isolated a number of different rabbit monoclonal antibodies (Mabs) including 204 and 205 that are specific for pre-fibrillar oligomers (Kayed et al. 2010). To gain further insight into the actions of anti-Aβ antibodies we tested the short time immunotherapeutic effects of anti-oligomeric antibodies in the aged 3xTg-AD mouse model. Our results not only show improvements in the cognitive function but also significantly reduced amyloid deposits and hyperphosphorylated tau.
Material and methods
All animal procedures were performed in accordance with animal protocols approved by the Institutional Animal Care and Use Committee at the University of California, Irvine (UCI). The 3xTg-AD have been described previously (Oddo et al. 2003). Briefly, these mice harbor a knock-in mutation of presenilin 1 (PS1M146V), the Swedish double mutation of amyloid precursor protein (APPKM670/671NL), and a frontotemporal dementia mutation in tau (tauP301L) on a 129/C57BL/6 background. These mice were a kind gift from Dr. Frank Laferla (University of California Irvine, USA). We used 32 female 3xTg-AD mice for our experiment. 3xTg-AD mice are widely used strain that exhibit both intracellular and extracellular Aβ deposits, tau pathology and cognitive deficits.
Vaccination
The detailed purification of these antibodies has already being reported previously (Kayed et al. 2010). These rabbit monoclonal antibodies were produced under a contract with Epitomics, Incorporated, Burlingame, CA. New Zealand white rabbits were immunized subcutaneously with Aβ40-colloidal gold oligomer mimics at a 0.25 mg Aβ dose bit and boosted seven times with the same amount at 3 weeks intervals. The titer and specificity of the immune response was determined by ELISA and dot blot analysis as previously described using serum collected after the third and sixth boosts (Kayed et al. 2003) The mice were distributed equally into four groups with eight mice in each group. Oligomer specific antibodies were injected (intraperitoneal 300 μg/150 μL). One group received the polyclonal oligomer-specific antibody, A11 (Kayed et al. 2003), whereas the other two groups received monoclonal oligomer-specific antibodies 204 and 205 (Kayed 2010). The control group was vaccinated with pre-immune rabbit IgGs (Jackson Immuno-Research Lab, West Grove, PA, USA). The antibodies were administered weekly intraperitoneally for 5 weeks in 13-month-old 3xTg-AD mice. After 5 weeks, mice went through a battery of behavioral test and were killed at 15 months.
Morris water maze
The Morris water maze test was used to assess memory related to the hippocampal function as previously described (Billings et al. 2005). After 5 weeks of antibody administration morris water maze test was performed. For each trial, the mouse was placed into the tank at one of four designated start points in a pseudorandom order. Mice were given four trials a day until they reach the given criterion (< 20 s). The 3xTg-AD mice were trained for 6 days to meet the criterion. After 24 h of retention of spatial memory was assessed. In the probe trial consisted of a 60 s free swim in the pool without the platform. The parameters measured during the probe trial included: (i) initial latency to cross the platform location, (ii) number of platform location crosses, and (iii) time spent in the quadrant opposite to the one containing the platform during training.
Passive inhibitory avoidance
Amygdala dependent tasks were evaluated using the passive inhibitory avoidance (Decker et al. 1990). This was measured using a Gemini Avoidance System instrument (San Diego Instruments, San Diego, CA, USA). During the trial period, the mice were placed inside the lighted compartment and latency was recorded (time to enter in the dark compartment). On complete entry into the dark compartment, the animal received a mildly aversive foot shock (0.15 mA, 1 s). After 24 h the mouse was again placed back in the lighted compartment and the step-through latency before re-entering the dark side was measured for a period up to 180 s. The retention trial was interrupted if the animal took more than 180 s to cross into the dark compartment.
Tissue collection and immunohistochemistry
After mice were anesthetized with pentobarbital (150 mg/kg, IP), blood was collected by cardiac puncture, and mice were perfused transcardially with cold phosphate-buffered saline (PBS). Brain tissues were fixed overnight with 4% paraformaldehyde in PBS, pH 7.4 at 4°C and stored in PBS/0.02% sodium azide (NaN3) at 4°C until use. Fixed brain tissues were sectioned (40 μm) with a vibratome. Coronal sections were collected in PBS (containing 0.02% sodium azide) and stored at 4°C prior to staining. To stain for Aβ plaques, sections were immersed in 70% formic acid for 5 min. Endogenous peroxidase in tissue was blocked by treating with 3% H2O2 in PBS for 10 min at 25°C. Nonspecific background staining was blocked by 1 h incubation in 2% bovine serum albumin, 0.3% Triton X-100 (TX) at 25°C. Tissues were incubated with primary antibodies (6E10, AT8, and CD45) overnight at 4°C, rinsed three times with PBS, 0.1% TX, followed by biotinylated secondary antibodies (anti-rabbit, anti-mouse, and anti-rat), detection with an ABC peroxidase kit, and visualization with a 3,3′-diaminobenzidine substrate kit (Vector, Burlingame, CA, USA). CD45 (1 : 3000; Serotec, Raleigh, NC, USA) staining was done as described previously (Gordon et al. 2002). Control experiments with primary or secondary antibody omitted resulted in no staining.
Image quantification
Image quantification was done as described in our previous publication (Rasool et al. 2012). Briefly immunostaining was observed under a Zeiss Axiovert- 200 inverted microscope (Carl Zeiss, Thornwood, NY, USA) and images were acquired with a Zeiss Axiocam high-resolution digital color camera (1300 × 1030 pixel) using Axiovision 4.1 or 4.6 software. The same software (Carl Zeiss) was used to analyze the digital images. Percent of immunopositive area (% Field Area) (immunopositive area/total image area × 100) was determined for all the markers studied by averaging images of the hippocampus and subiculum area from 4 to 5 sections per animal. Digital images were obtained using the same settings and the segmentation parameters constant within a range per given marker and experiment. The mean value of the % Field Area for each marker in each animal was averaged per genotype group with the number of animals per group indicated in Figure legends.
Enzyme-linked immunosorbent assay for soluble and insoluble Aβ
We used the same procedure for detecting soluble and insoluble form of Aβ (Rasool et al. 2012). Briefly soluble and insoluble Aβ fractions were isolated from whole brain homogenates using a four step extraction protocol (Kawarabayashi et al. 2001). Frozen hemibrains were sequentially extracted. At each step, sonication in an appropriate buffer was followed by centrifugation at 100 000 g for 1 h at 4°C. The supernatant was then removed, and the pellet was sonicated in the next solution used in the sequential extraction process. For four-step extraction, sonication of the frozen brain (150 mg/mL wet weight) began in Tris-buffered saline (20 mM Tris and 137 mM NaCl, pH 7.6), which contained protease inhibitors (Protease inhibitor cocktail from Sigma St. Louis USA). The next three sequential extraction steps used 1% Triton X-100 in Tris-buffered saline with protease inhibitors, 2% sodium dodecyl sulfate in ddH2O with the same protease inhibitors, and 70% formic acid in ddH2O. To measure the soluble and insoluble form of Aβ, insoluble fractions were diluted to 1 : 20 in a neutralization buffer (1 mol/L Tris base, 0.5 mol/L NaH2PO4) before loading on MaxiSorp immunoplates (Nunc, Rochester, NY, USA), whereas soluble fractions were loaded directly. The immunoplates were coated with Mab 20.1 antibody (kind gift from Dr. David Cribbs, University of California, Irvine) at a concentration of 25 μg/mL in coating buffer (0.1 M/L Na2CO3, pH 9.6) and blocked with 3% bovine serum albumin. Standard solutions for both Aβ1-40 and Aβ1-42 were made in the antigen capture buffer (20 mmol/L NaH2PO4, 2 mmol/EDTA,0.4M NaCl, 0.05% 3-[(3- cholamidopropyl) dimethylammoniopropanesulfonate, and 1% bovine serum albumin, pH 7.0) and loaded onto ELISA plates in duplicate. Samples were then loaded (also in duplicate) and incubated overnight at 4°C. Plates were then washed and probed with either horseradish peroxidase conjugated anti-Aβ1-40(C49) or anti-Aβ1-42(D32) overnight at 4°C. The chromogen was 3,3 – 5,5 tetramethylbenzidine and the reaction was stopped by 30% phosphoric acid. The plates were read at 450 nm using a plate reader (Molecular Dynamics, Sunnyvale, CA, USA). Readings were then normalized to protein concentrations of the samples.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software, San Diego, CA, USA). For comparison between antigen vaccinated groups and control vaccinated group, one-way ANOVA followed by Dunnet post-test was performed. Probability values less than 0.05 were accepted as statistically significant.
Results
Vaccination improves cognitive performance
We first sought to determine whether aged mice harboring an extensive plaque and tangle burden would show improved learning and memory following passive immunization. At the end of the treatment period, we evaluated the behavioral phenotype of the 3xTg-AD mice using two different behavioral paradigms, the Morris water maze and the passive inhibitory avoidance Fig. 1a, shows the time line of the passive immunization trial. 13-month-old 3xTg-AD mice were immunized with A11, 204, and 205 antibody at 1 week intervals for 5 weeks. We evaluated the reference memory after 5 weeks of vaccination in a Morris water maze test. All of the mice vaccinated with anti-oligomer antibodies demonstrated a significant improvement (p < 0.001) in the acquisition and retention of the Morris Water Maze test as compared to control IgG vaccinated mice (Fig. 1b). At 24 h probe trials, mice vaccinated with A11, 204, and 205 showed significant improvement (***p < 0.001) in retention memory as evident by their improved latency to cross the platform location and the number of platform crosses during the 24 h tests as compared with controls (Fig. 1c). Contextual learning and memory was evaluated using the passive avoidance task. In retention trials, the step-through latency of antibody-vaccinated mice significantly increased (**p < 0.01) as compared to that of the control rabbit IgG-vaccinated group (Fig. 1d). These results indicate that vaccination with oligomer specific antibodies showed significant improvement in behavior as compared with control vaccinated mice.
Fig. 1.

Vaccination with antibodies improves spatial cognitive deficits in 3xTg-Alzheimer’s disease (AD) mice. Schematic representation of the passive immunization paradigm. The arrows represent the antibody treatment and the arrowheads represents the behavior testing (a). After 5 weeks of passive vaccination with oligomeric antibodies, 3xTg-AD mice were evaluated using a spatial reference paradigm and compared with control treated mice vaccinated with colloidal gold and adjuvant. (b) Mice were trained on the spatial reference version of the Morris water maze task after treatment. All the vaccinated mice showed significant improvement (*p < 0.05) as compared with control (b). (c) In probe trial during 24 h testing, all vaccinated 3xTg-AD mice also display significantly more number of crosses around to platform than rabbit IgG control vaccinated mice (ANOVA, ***p < 0.001). Vaccination prevents contextual fear memory deficits in a mainly amygdala-dependent task. (d) Mice were tested for retention of memory for fear-associated environments 24 h after training. Mice were taken out after 180 s if they did not cross over. At 24 h all the oligomeric antibody vaccinated mice showed significant improvement (***p < 0.001).
Vaccination with oligomer antibodies reduces Aβ plaque accumulation in 3xTg-AD mice
We next determined the effect of passive immunization on plaque burden in the aged mice. Total amyloid deposits in the 3xTg-AD mice after 5 weeks of vaccination were analyzed by immunohistochemistry using 6E10. We compared the effectiveness of the immunization on amyloid deposition by quantifying the amount of anti-Aβ (6E10) immunoreactive material. Fig. 2a, shows representative photomicrographs from the hippocampus of control and vaccinated animals. Image analysis of sections from multiple animals demonstrated that Aβ deposits in A11, 204, and 205 vaccinated mice were decreased significantly as compared with control vaccinated mice. Fig. 2b, shows that all the immunized groups displayed a significant reduction in Aβ plaque load in hippocampus as compared to the respective controls (***p < 0.001, **p < 0.01). These results indicate that administration of oligomer specific antibodies lower the amount of plaque deposits, even in mice with extensive plaque and tangle neuropathology.
Fig. 2.

Passive vaccination decreases total amyloid deposits. (a) Representative photomicrographs of sections from brains (hippocampus) of 3xTg-Alzheimer’s disease mice after 5 weeks of vaccination (Control antibody, A11antibody, 204 antibody, and 205 antibody vaccinated). Immunostaining was done with 6E10 (which reacts with the human amyloid β peptide). Scale bar: 100 μm. Image analysis (% Field area) of Aβ (6E10 antibody) immunoreactivity in hippocampus. Mean of each animal is the average of 4 sections (except untreated control which is 1 section per animal) assessing 4–8 images per section (most to all of the area of the section was analyzed). Bars represent group mean ± SEM of n mice per group: Control antibody vaccinated n = 8, A11 antibody vaccinated n = 8, 204 antibody vaccinated n = 8 and 205 antibody vaccinate n = 8, ***p < 0.001, **p < 0.01 by ANOVA (b).
Vaccination with oligomeric antibodies reduces both soluble and insoluble Aβ40 and Aβ42 levels
3xTg-AD mice increasingly accumulate Aβ as they age with fibrillar amyloid plaques starting to develop from 12 months of age (Oddo et al. 2006a,b). It has been previously reported that the initial cognitive deficits in these mice are caused by intraneuronal Aβ levels and that cognitive deficits increase with increasing brain pathology. To evaluate whether vaccination with oligomer-specific antibodies not only reduce plaque load but also affect cerebral total Aβ levels, brain homogenates were processed using four step extraction which distinguishes between soluble and insoluble Aβ levels. Subsequently, Aβ40 and 42 levels were quantified using a high-sensitivity sandwich ELISA as described in Materials and Methods. Consistent with a decrease in amyloid plaques immunostaining, we observed a significant reduction in brain soluble and insoluble Aβ40 and Aβ42 levels (Fig. 3) in all three of the oligomer-specific antibody vaccinated groups.
Fig. 3.

Vaccination with oligomeric antibodies strongly reduces levels of soluble and insoluble Aβ40 and Aβ42 in brain. Following 5 weeks of passive vaccination, levels of soluble and insoluble Aβ40 (a, b) & Aβ42 (c, d) on homogenates of the whole brain hemisphere of 3xTg-Alzheimer’s disease mice were determined using ELISA and reported in pg of Aβ/mg of protein in case of soluble and pg of Aβ/mg of wet tissue weight (insoluble). Oligomer antibody treatment was associated with significantly lower levels of both soluble and insoluble levels of Aβ40 and Aβ42. Error bars represent standard errors of the mean (n = 8). ***p < 0.001 for soluble and insoluble level of Aβ40 and ***p < 0.001 for soluble Aβ42 and ***p < 0.001 for insoluble levels of Aβ42 in A11 antibody vaccinated mice and 204 antibody vaccinated mice, *p < 0.05 for 205 antibody vaccinated mice by one-way ANOVA test.
Vaccination with antibodies reduces tau pathology
The formation of tau tangles is believed to be dependent on Aβ deposition (Vossel et al. 2010). Therefore, we assessed whether administration of anti-oligomer antibodies could reduce the tau pathology. The hyperphosphorylated micro-tubule-associated protein tau is the major component of the paired helical filament of Alzheimer’s disease, we also examined tau pathology using AT8, an antibody that selectively recognizes tau that is phosphorylated at serine 202 and threonine 205. As hyperphosphorylation of tau is a critical step in the pathway leading to neurofibrillary pathology, we determined whether the Aβ-mediated clearance of tau was dependent on its phosphorylation state. At 14 months of age, homozygous 3xTgAD mice show extensive tau hyperphosphorylation in CA1 pyramidal neurons and were immunopositive for tau antibodies. To investigate the effect of oligomer specific antibodies on hyperphosphorylated, we stained the brain section with AT8 antibody. Fig. 4a, shows the representative photomicrographs from the hippocampus of control and vaccinated mice stained with AT8 antibody. After 5 weeks of vaccination we observed statistically significant reduction in hyperphosphorylated tau aggregates as compared with control IgG vaccinated mice (Fig. 4b, **p < 0.01). We also investigated the effect of antibodies on total tau using HT7Ab. Immunoreactivity with HT7 antibody displays a reduction in antibody vaccinated mice (Fig. 5). Moreover, we cannot exclude the possibility of the reduction in HT7 staining in the 3xTg-AD-treated mice is also due to the decrease in Aβ, as it has been previously showed the lowering Aβ reduces HT7-positive, non-phosphorylated tau (Oddo et al. 2004).
Fig. 4.

Passive vaccination with oligomer antibodies led to a reduction in total tau levels: (a) Representative photomicrographs of sections from brains (hippocampus) of 3xTg-Alzheimer’s disease mice after 5 weeks of vaccination (Control antibody, A11antibody, 204 antibody, and 205 antibody vaccinated). Immunostaining was done with HT7 (which recognizes the total tau). Immunohistochemical analysis of oligomer antibody vaccinated mice showed a prominent reduction of total tau in the hippocampus (scale bar = 100 μm) as compared with control antibody vaccinated mice (b). The reduction was statistically significant **p < 0.01.
Fig. 5.

Antibodies against oligomeric Aβ decrease hyperphosphorylated tau using AT8 antibody. (a) Representative photomicrographs of sections from brains (hippocampus) of 3xTg-Alzheimer’s disease mice after 5 weeks of vaccination (Control antibody, A11antibody, 204 antibody, and 205 antibody vaccinated). Brain sections were subjected to immunostaining using anti-phosphorylated tau (Ser202) (AT8 antibody) Immunohistochemical analysis of oligomer antibody vaccinated mice showed a prominent reduction of hyperphosphorylated tau in the hippocampus (scale bar = 100 μm) as compared with control antibody vaccinated mice. The reduction was statistically significant **p < 0.01 (b).
Vaccination reduces microglial activation
One of the main characteristics accompanying the accumulation of Aβ plaques in both human AD brains and in transgenic mouse models of AD is an enhanced neuroinflammatory response characterized by activation of microglia. It has been previously reported that immunization with Aβ reduces the microglial activation (Wilcock et al. 2003). We assessed the effect of vaccination by anti-oligomeric antibodies on microglial reactivity as measured by CD45, which is a protein-tyrosine phosphatase that is normally moderately expressed on microglia around amyloid deposits in aged APP transgenic mice and is a widely used marker for microglial activation. Fig. 6a, shows representative photomicrographs from the cortex including white matter of control and vaccinated animals. Image analysis of sections from multiple animals demonstrated that CD45 immunore-activity in A11, 204, and 205 vaccinated mice were decreased significantly as compared with control vaccinated mice. After 5 weeks of vaccination, we observed a statistically significant decrease in % age of CD45 immunostaining relative to the respective control vaccinated mice (Fig. 6b, ***p < 0.001).
Fig. 6.
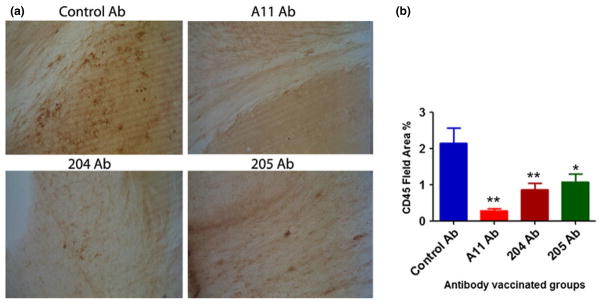
Vaccination with oligomeric antibodies decreases activated microglia. Brain sections were subjected to immunostaining using microglial marker (CD45 antibody). Representative photomicrographs of CD45 (brown) staining in the cortex and white matter of brain sections from 3xTg-Alzheimer’s disease mice (a). Mean of each animal is the average of two sections in which most to all the area of study was analyzed (4–8 images per section) (b) Bars represent mean ± SD of n = 8 mice per group. **p < 0.01 for A11 antibody and 204 antibody vaccinated mice, *p < 0.05 for 205 antibody vaccinated mice.
Discussion
Passive immunization has been shown to improve cognition by reducing amyloid plaque levels (Chen et al. 2000; Morgan et al. 2000). Passive immunotherapy in APP transgenic mice has been shown to be applicable to human clinical trials (Bard et al. 2000; DeMattos et al. 2001; Dodart et al. 2002) and so are other treatments that reduce Aβ burden (Blennow et al. 2006). Diffuse Aβ plaques in mouse models such as Tg2576, PDAPP and 3xTg-AD were efficiently reduced by most N-terminal anti-Aβ antibodies (Bard et al. 2000; Levites et al. 2006; Oddo et al. 2006b). It has also been reported that thioflavin-S positive plaques are cleared by certain anti-Aβ antibodies of the IgG2a isotype (Bussiere et al. 2004). In transgenic models with highly insoluble dense-cored plaques, like tg-ArcSwe mice (Philipson et al. 2009), the effects of anti-Aβ immunotherapy were subtle and purely preventive (Levites et al. 2006; Tucker et al. 2008) with only diffuse non-fibrillar Aβ deposits being cleared (Levites et al. 2006). In such studies, induced transgene driven intracerebral injections of Aβ antibodies lacked efficiency as only nascent Aβ production was blocked (Tucker et al. 2008). Aβ protofibril levels were lowered while levels of insoluble Aβ were unaffected as detected using protofibril-selective antibody mAb158 (Lord A et al. 2009). In contrast, our data indicate that the formation of amyloid deposits can be reversed with passive immunization using pre-fibrillar oligomer-specific antibodies. These antibodies are specific for oligomers that are immunologically distinct from fibrils and do not bind plaques (Kayed et al. 2010). We observed a significant reduction in both soluble and insoluble levels of Aβ40 and 42. This reduction may be affected by the opsonization of antibody-bound oligomeric Aβ42 or 40 by microglia or infiltrated macrophages. It has been previously reported that intracranial injection of anti-Aβ in 19-month-old Tg2576 mice showed a dramatic reduction in cerebral amyloid by 24 h after a single dose, which was maintained up to 7 days post-injection (Wilcock et al. 2003)
Current evidence suggests that hyperphosphorylation and aggregation of tau protein is an important pathway driving neurodegeneration in AD. Tau is a microtubule-binding protein involved in regulation of neuronal microtubule assembly and stabilization (Caceres and Kosik 1990; Avila et al. 1994; Esmaeli-Azad et al. 1994; Harada et al. 1994). Aggregates of Aβ are candidate triggers of tau hyperphosphorylation, aggregation and the subsequent degeneration of affected neurons (Greenberg et al. 1992; Iqbal et al. 1994; Sigurdsson et al. 1997; Lovestone and Reynolds 1997; Spillantini and Goedert 1998; Lee and Trojanowski 1999). In 3xTg-AD mice, only the early, but not the late forms of phosphorylated human tau were removed by a single injection of anti-Aβ antibody into the brain (Oddo et al. 2004).
Clearance of tau is critically dependent on its phosphorylation state. At late-stages, hyperphosphorylated aggregates appear to be unaffected by anti-Aβ antibody treatment. Notably, the injection of the anti-oligomeric Aβ antibody A11, leads to the reduction of tau pathology and Aβ pathology in 3x Tg-AD mice and strongly suggest that Aβ oligomers may represent a link between Aβ and tau pathology (Oddo et al. 2006a,b). It has been previously reported that passive immunization reduces Aβ oligomeric levels, which has been shown to directly induce GSK3β activation and tau phosphorylation (Ma et al. 2006). Anti-Aβ active immunization has been shown to decrease tau hyperphosphorylation in two other mouse models that sequentially develop amyloid plaques and neuronal aggregates of hyperphosphorylated native murine tau, thus adding further evidence for its potential beneficial effect on tau pathology (Wilcock et al. 2009). These data are consistent with our results showing that passive immunization with anti-oligomer antibodies efficiently clears hyperphosphorylated tau. It has been shown that Aβ fibrils accelerate the formation of abnormally phosphorylated neurofibrillary tangles in a tau transgenic mouse (Gotz et al. 2001), and that Aβ could induce tau phosphorylation and toxicity in cultured septal cholinergic neurons (Zheng et al. 2002). More recently, it has been shown that Aβ oligomers cause abnormal tau phosphorylation and morphology changes of spines by missorting of endogenous tau into dendrites (Zempel et al. 2010). Naturally occurring Aβ dimers isolated from AD brains are sufficient to induce AD-type tau phosphorylation and, consequently, neuritic dystrophy (Jin et al. 2011). For synaptic plasticity, it has been found that the absence of tau protein inhibits the impairment of LTP and neurotoxicity caused by Aβ (Shipton et al. 2011) and targeting tau by immunotherapy prevents cognitive decline in a novel tangle mouse model (Asuni et al. 2007; Boutajangout et al. 2010).
CD45 is a protein-tyrosine phosphatase that is elevated with microglial activation. Loss of microglia activation after intracranial antibody administration (Wilcock et al. 2003) and active immunization (Wilcock et al. 2001) has been previously reported. In our study, we also observed a significant reduction in CD45 levels as compared with control IgG vaccinated mice. This suggests that the reduced microglial activation could possibly be attributable to the clearance of most amyloid plaques. It is also conceivable that the microglia could be undergoing apoptosis attributable to the robust activation, as described previously by Liu et al. (2001), when microglia are over-activated by lipopolysaccharide. An alternative explanation could be tolerance of the microglia to antibody-opsonized Aβ.
Our findings demonstrated that conformation-dependent antibodies efficiently clear amyloid plaque, tau pathology and improve cognition, which highlight that these oligomer specific antibodies have a therapeutic potential to prevent and treat Alzheimer’s disease. Because these antibodies do not target plaques and other fibrillar amyloid deposits, they may have a more favorable safety profile than antibodies that increase CAA and the risk of hemorrhage.
Acknowledgments
This study was supported by a University of California BioStar Discovery grant and the Cure Alzheimer’s Fund. We thank undergraduate students Nazia Farah and Chatman Yu for their help in cognitive function tests of mice.
Abbreviations used
- Aβ
amyloid-beta
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- CAA
cerebral amyloid angiopathy
- PBS
phosphate-buffered saline
Footnotes
There is no conflict of interest.
References
- Asuni AA, Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau conformers in a tangle mouse model reduces brain pathology with associated functional improvements. J Neurosci. 2007;27:9115–9129. doi: 10.1523/JNEUROSCI.2361-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila J, Dominguez J, Diaz-Nido J. Regulation of microtubule dynamics by microtubule-associated protein expression and phosphorylation during neuronal development. Int J Dev Biol. 1994;38:13–25. [PubMed] [Google Scholar]
- Banks WA, Terrell B, Farr SA, Robinson SM, Nonaka N, Morley JE. Passage of amyloid beta protein antibody across the blood-brain barrier in a mouse model of Alzheimer’s disease. Peptides. 2002;23:2223–2226. doi: 10.1016/s0196-9781(02)00261-9. [DOI] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer’s disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368:387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- Boutajangout A, Quartermain D, Sigurdsson EM. Immunotherapy targeting pathological tau prevents cognitive decline in a new tangle mouse model. J Neurosci. 2010;30:16559–16566. doi: 10.1523/JNEUROSCI.4363-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere T, Bard F, Barbour R, et al. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am J Pathol. 2004;165:987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caceres A, Kosik KS. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature. 1990;343:461–463. doi: 10.1038/343461a0. [DOI] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, et al. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer’s disease. Nature. 2000;408:975–979. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Decker MW, Gill TM, McGaugh JL. Concurrent muscarinic and beta-adrenergic blockade in rats impairs place-learning in a water maze and retention of inhibitory avoidance. Brain Res. 1990;513:81–85. doi: 10.1016/0006-8993(90)91091-t. [DOI] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer’s disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Esmaeli-Azad B, McCarty JH, Feinstein SC. Sense and antisense transfection analysis of tau function: tau influences net microtubule assembly, neurite outgrowth and neuritic stability. J Cell Sci. 1994;107(Pt 4):869–879. doi: 10.1242/jcs.107.4.869. [DOI] [PubMed] [Google Scholar]
- Frazer ME, Hughes JE, Mastrangelo MA, Tibbens JL, Federoff HJ, Bowers WJ. Reduced pathology and improved behavioral performance in Alzheimer’s disease mice vaccinated with HSV amplicons expressing amyloid-beta and interleukin-4. Mol Ther. 2008;16:845–853. doi: 10.1038/mt.2008.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golde TE, Eckman CB, Younkin SG. Biochemical detection of Abeta isoforms: implications for pathogenesis, diagnosis, and treatment of Alzheimer’s disease. Biochim Biophys Acta. 2000;1502:172–187. doi: 10.1016/s0925-4439(00)00043-0. [DOI] [PubMed] [Google Scholar]
- Gordon MN, Holcomb LA, Jantzen PT, DiCarlo G, Wilcock D, Boyett KW, Connor K, Melachrino J, O’Callaghan JP, Morgan D. Time course of the development of Alzheimer-like pathology in the doubly transgenic PS1+ APP mouse. Exp Neurol. 2002;173:183–195. doi: 10.1006/exnr.2001.7754. [DOI] [PubMed] [Google Scholar]
- Gotz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated tau PHF proteins display the same biochemical properties as normal tau. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Zaidi T, Bancher C, Grundke-Iqbal I. Alzheimer paired helical filaments. Restoration of the biological activity by dephosphorylation. FEBS Lett. 1994;349:104–108. doi: 10.1016/0014-5793(94)00650-4. [DOI] [PubMed] [Google Scholar]
- Janus C, Pearson J, McLaurin J, et al. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–5824. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid (beta) protein in the Tg2576 transgenic mouse model of Alzheimer’s disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Kayed R, Canto I, Breydo L, et al. Conformation dependent monoclonal antibodies distinguish different replicating strains or conformers of prefibrillar Abeta oligomers. Mol Neurodegener. 2010;5:57. doi: 10.1186/1750-1326-5-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee VM, Trojanowski JQ. Neurodegenerative tauopathies: human disease and transgenic mouse models. Neuron. 1999;24:507–510. doi: 10.1016/s0896-6273(00)81106-x. [DOI] [PubMed] [Google Scholar]
- Lee HG, Zhu X, Castellani RJ, Nunomura A, Perry G, Smith MA. Amyloid-beta in Alzheimer disease: the null versus the alternate hypotheses. J Pharmacol Exp Ther. 2007;321:823–829. doi: 10.1124/jpet.106.114009. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Spooner ET, LaFrancois J, et al. Evidence for peripheral clearance of cerebral Abeta protein following chronic, active Abeta immunization in PSAPP mice. Neurobiol Dis. 2003;14:10–18. doi: 10.1016/s0969-9961(03)00044-5. [DOI] [PubMed] [Google Scholar]
- Levites Y, Smithson LA, Price RW, et al. Insights into the mechanisms of action of anti-Abeta antibodies in Alzheimer’s disease mouse models. FASEB J. 2006;20:2576–2578. doi: 10.1096/fj.06-6463fje. [DOI] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- Lord A, Gumucio A, Englund H, et al. An amyloid-beta protofibril-selective antibody prevents amyloid formation in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2009;36:425–434. doi: 10.1016/j.nbd.2009.08.007. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Reynolds CH. The phosphorylation of tau: a critical stage in neurodevelopment and neurodegenerative processes. Neuroscience. 1997;78:309–324. doi: 10.1016/s0306-4522(96)00577-5. [DOI] [PubMed] [Google Scholar]
- Ma QL, Lim GP, Harris-White ME, Yang F, Ambegaokar SS, Ubeda OJ, Glabe CG, Teter B, Frautschy SA, Cole GM. Antibodies against beta-amyloid reduce Abeta oligomers, glycogen synthase kinase-3beta activation and tau phosphorylation in vivo and in vitro. J Neurosci Res. 2006;83:374–384. doi: 10.1002/jnr.20734. [DOI] [PubMed] [Google Scholar]
- Monsonego A, Imitola J, Zota V, Oida T, Weiner HL. Microglia-mediated nitric oxide cytotoxicity of T cells following amyloid beta-peptide presentation to Th1 cells. J Immunol. 2003;171:2216–2224. doi: 10.4049/jimmunol.171.5.2216. [DOI] [PubMed] [Google Scholar]
- Morgan D, Diamond DM, Gottschall PE, et al. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43:321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Tran L, Lambert MP, Glabe CG, Klein WL, LaFerla FM. Temporal profile of amyloid-beta (Abeta) oligomerization in an in vivo model of Alzheimer disease. A link between Abeta and tau pathology. J Biol Chem. 2006a;281:1599–1604. doi: 10.1074/jbc.M507892200. [DOI] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble Abeta and tau, but not soluble Abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006b;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Philipson O, Hammarström P, Nilsson KP, et al. A highly insoluble state of Abeta similar to that of Alzheimer’s disease brain is found in Arctic APP transgenic mice. Neurobiol Aging. 2009;30:1393–1405. doi: 10.1016/j.neurobiolaging.2007.11.022. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, et al. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasool S, Albay R, 3rd, Martinez-Coria H, et al. Vaccination with a non-human random sequence amyloid oligomer mimic results in improved cognitive function and reduced plaque deposition and micro hemorrhage in Tg2576 mice. Mol Neurodegener. 2012;7:37. doi: 10.1186/1750-1326-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D. Amyloid-beta immunotherapy for Alzheimer’s disease: the end of the beginning. Nat Rev Neurosci. 2002;3:824–828. doi: 10.1038/nrn938. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Shipton OA, Leitz JR, Dworzak J, et al. Tau protein is required for amyloid {beta}-induced impairment of hippocampal long-term potentiation. J Neurosci. 2011;31:1688–1692. doi: 10.1523/JNEUROSCI.2610-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Lee JM, Dong XW, Hejna MJ, Lorens SA. Bilateral injections of amyloid-beta 25–35 into the amygdala of young Fischer rats: behavioral, neurochemical, and time dependent histopathological effects. Neurobiol Aging. 1997;18:591–608. doi: 10.1016/s0197-4580(97)00154-1. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–433. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- Tucker SM, Borchelt DR, Troncoso JC. Limited clearance of pre-existing amyloid plaques after intracerebral injection of Abeta antibodies in two mouse models of Alzheimer disease. J Neuropathol Exp Neurol. 2008;67:30–40. doi: 10.1097/nen.0b013e31815f38d2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilevko V, Cribbs DH. Novel approaches for immunotherapeutic intervention in Alzheimer’s disease. Neurochem Int. 2006;49:113–126. doi: 10.1016/j.neuint.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L. Tau reduction prevents Abeta-induced defects in axonal transport. Science. 2010;330:198. doi: 10.1126/science.1194653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Gordon MN, Ugen KE, et al. Number of Abeta inoculations in APP+PS1 transgenic mice influences antibody titers, microglial activation, and congophilic plaque levels. DNA Cell Biol. 2001;20:731–736. doi: 10.1089/10445490152717596. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Alamed J, Gottschall PE, Grimm J, Rosenthal A, Pons J, Ronan V, Symmonds K, Gordon MN, Morgan D. Deglycosylated anti-amyloid-beta antibodies eliminate cognitive deficits and reduce parenchymal amyloid with minimal vascular consequences in aged amyloid precursor protein transgenic mice. J Neurosci. 2006;26:5340–5346. doi: 10.1523/JNEUROSCI.0695-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Gharkholonarehe N, Van Nostrand WE, Davis J, Vitek MP, Colton CA. Amyloid reduction by amyloid-beta vaccination also reduces mouse tau pathology and protects from neuron loss in two mouse models of Alzheimer’s disease. J Neurosci. 2009;29:7957–7965. doi: 10.1523/JNEUROSCI.1339-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E, Mandelkow EM. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010;30:11938–11950. doi: 10.1523/JNEUROSCI.2357-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng WH, Bastianetto S, Mennicken F, Ma W, Kar S. Amyloid beta peptide induces tau phosphorylation and loss of cholinergic neurons in rat primary septal cultures. Neuroscience. 2002;115:201–211. doi: 10.1016/s0306-4522(02)00404-9. [DOI] [PubMed] [Google Scholar]