

Abstract
The synthesis of novel carbohydrate-based polymers allows the structure to be tailored at the monomer level for a specific property and expands the range of available structures beyond those found in nature. Using a controlled anionic polymerization, a new type of carbohydrate polymer is synthesized in which glucose-derived monomers are joined by an α-1,2 amide linkage to give enantiopure poly-amido-saccharides (PASs). To investigate the effect of adding ionizable carboxylic acid groups, such as those found in natural polysaccharides containing glucuronic acid, the oxidation of the primary alcohol at the C6-position of the repeat unit to a carboxylic acid is reported. TEMPO-mediated oxidation provides control over the degree of oxidation in excellent yield. Based on circular dichroism, the oxidized polymers possess an ordered helical secondary structure in aqueous solution. Finally, oxidized PASs stabilize lysozyme toward dehydration and freezing stresses better than a current, widely used protein stabilizing agent, trehalose.
Polysaccharides, as one of the three major classes of natural biopolymers, play many varied and essential roles that are highly dependent on their molecular composition and structure.1 Natural polysaccharides containing ionizable groups,2 such as the carboxylic acid containing hyaluronic acid,3 aliginic acid,4 and oxidized forms of cellulose,5 are widely investigated and used in a range of biomedical6 and nonbiomedical applications. In the context of biomedical use, however, these materials suffer from one or more of the following limitations, including the need for extensive purfication, variable branching and dispersity, trace contamination by biological toxins, and batch-to-batch variation.7 Notably, the materials used are isolated as polymers, and therefore the ability to control the repeat unit structure at the monomer level and to access structures not found in nature is not possible or significantly limited.
The efficient polymerization of saccharides has been a longtime goal of chemists,8 but carbohydrate polymers remain challenging synthetic targets due to their stereochemical complexity and the many similar functional groups that must often be protected.9 Several strategies have been explored to synthesize polysaccharides and polysaccharide mimics, for example: tethering a sugar to a linear or dendritic polymer to create glycopolymers10 or glycodendrimers,11 opening of the carbohydrate ring via polycondensation or cationic ring-opening polymerization12 to give linear and hyperbranched polymers, stepwise oligosaccharide synthesis,13 and enzymatic synthesis.14 With each strategy possessing strengths and weaknesses, new approaches are in demand.
Herein, we report the synthesis of enantiopure carboxylated poly-amido-saccharides (PASs) composed of glucose(glc)-based monomers. PASs are a new type of carbohydrate polymer in which the ether linkage is replaced with an α-1,2 amide linkage.15 Specifically, we report the (1) synthesis of α-N-1,2-d-glc PASs using a pentafluorophenol initiator; (2) TEMPO mediated oxidation of the polymer to form the oxidized PAS (oxPAS); (3) stability of the helical PAS structure in the presence of acidic and basic conditions; and (4) use of PASs as stabilizing agents against protein denaturation.
The glucose-derived monomer (1, Scheme 1) was synthesized by the stereoselective cycloaddition of tri-O-benzyl-d-glucal and chlorosufonyl isocyanate (CSI) followed by in situ reduction to remove the sulfonyl group, as previously reported.15 Anionic ROP was selected for the polymerization strategy, as it can provide polymers of low dispersity and controlled length.16 Polymerization of the monomer was performed for several degrees of polymerization (DP) from 20 to 50. Briefly, monomer and x mol % BocGly-OPFP, an activated ester initiator, were combined in freshly distilled THF and cooled to 0 °C, and 2.5x mol % LiHMDS was added to begin the reaction. Polymerization was completed within 1 h, as indicated by the disappearance of the monomer by TLC. The reaction was quenched with saturated ammonium chloride, and the polymer was isolated as a white solid from pentane. The benzyl protecting groups were removed by sodium metal in liquid ammonia, followed by purification by dialysis of the now water-soluble polymer.
Scheme 1. Polymer Synthesis.

The primary alcohol at the C6 position of the deprotected polymer repeat unit was transformed to a carboxylic acid by TEMPO-mediated oxidation,5 with sodium hypochlorite as the reoxidant. For the oxidation reaction, the polymer, 2.4 equiv of NaOCl per repeat unit of polymer, and catalytic amounts of NaBr and TEMPO were dissolved in deionized water and the pH was monitored by a pH-meter with automatic titration with 0.5 M NaOH to maintain a pH of 10.5. The reaction pH remained stable at 10.5 without requiring additional NaOH after 5 min and was maintained for 1 h to ensure complete conversion. Excess oxidant was quenched with ethanol, and the polymer was purified by dialysis and isolated as the sodium salt following lyophilization. By reducing the amount of NaOCl to 1.2 equiv, a polymer in which only 50% of the primary alcohols, on average, were oxidized was synthesized.
The molecular weight and dispersity of the polymers were measured using gel permeation chromatography (Table 1). The protected polymers were analyzed with THF as the eluent against polystyrene standards. For the deprotected and oxidized polymers, aqueous buffer (pH 7.5) was employed as the eluent against polydextran standards. Molecular weights were in agreement with the calculated values, and the dispersities (Đ) were low (1.1–1.3). These results indicate that the polymerization is controlled over a range of molecular weights and that the deprotection and oxidation steps do not degrade the polymer backbone.
Table 1. Polymer Molecular Weights and Dispersities.
| DP | MW (calcd) | BnPAS MWa | (Đ) | PAS MWb | (Đ) | OxPAS MWb | (Đ) |
|---|---|---|---|---|---|---|---|
| 20 | 9.2k | 6.4k | 1.1 | 3.9k | 1.1 | 3.9k | 1.2 |
| 30 | 14k | 13.0k | 1.2 | 5.2k | 1.2 | 5.5k | 1.2 |
| 50 | 23k | 20.0k | 1.1 | 8.7k | 1.2 | 8.9k | 1.3 |
In THF versus polystyrene standards.
In aqueous buffer versus dextran standards.
IR spectra were measured for polymers 4 and 5 (DP = 20) which are 100% and 50% oxidized, respectively. When compared to the unoxidized polymer, a new peak is observed at 1607 cm–1 in the oxidized samples, which is attributed to the C=O stretch of the carboxylate (see Supporting Information (SI) Figure S7). In the half-oxidized polymer, this peak has approximately half the intensity of the fully oxidized polymer. By 1H NMR, the peak corresponding to the C6 protons at 3.8 ppm disappears following oxidation, while the C5 proton shifts downfield, consistent with an oxidized structure.
The oxidized PASs 4 and 5 exhibited a circular dichroism spectrum similar to that of the unoxidized PASs with a maximum at 191 nm and a minimum at 219 nm, indicating a helical structure. The degree of oxidation affected the spectrum, as the fully oxidized polymers showed less helical character than their unoxidized and half-oxidized counterparts (Figure 1). The spectrum is similar to that seen in other β-polypeptides that possess a regular helical secondary structure.17
Figure 1.
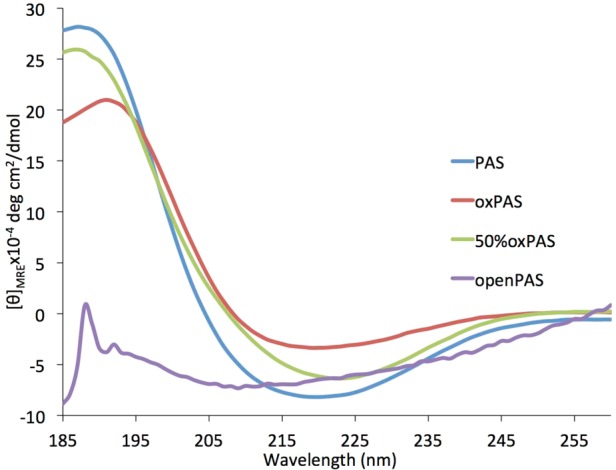
CD spectrum in 7 mM phosphate buffer of unoxidized PAS, 4, 5, and 8 (DP = 20).
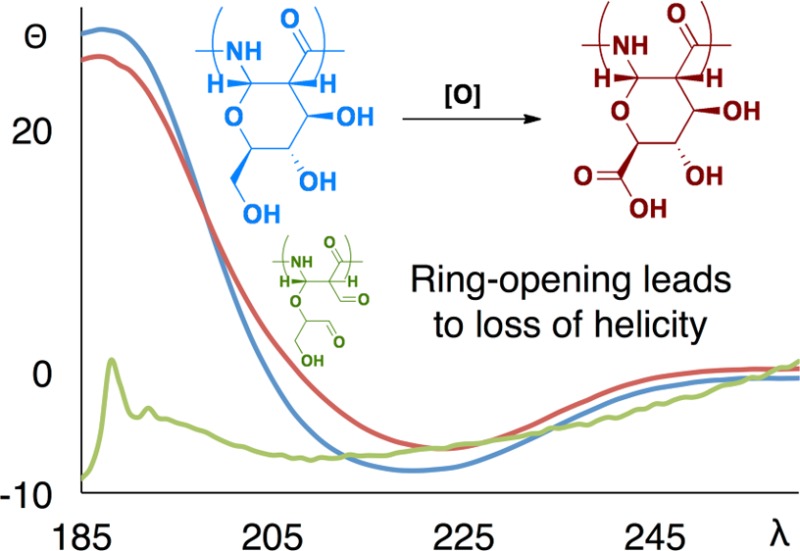
We hypothesized that the conformationally restricted pyranose ring present in the polymer backbone contributed to the helical secondary structure. To test this hypothesis, we opened the pyranose ring by oxidizing the vicinal diols of C3 and C4 to the aldehyde using sodium periodate.21 The resulting aldehyde in the position β to an amide tautomerized to a conjugated enol (Scheme 2), as seen by 1HNMR (see SI Figure S6). Following dialysis and lyophilization, the CD spectrum of this polymer was measured in aqueous buffer. As shown in Figure 1, the helical character was no longer present in the polymer (openPAS, 8). The secondary structure of the oxidized PASs did not significantly change over a wide pH range, from 2 to 12, after 2 or 48 h with only a slight decrease in helical character seen at extremely low pH (Figure 2). These results show that the helical structure adopted by these polymers is robust.
Scheme 2. Sodium Periodate Oxidation and Tautomerization.
Figure 2.
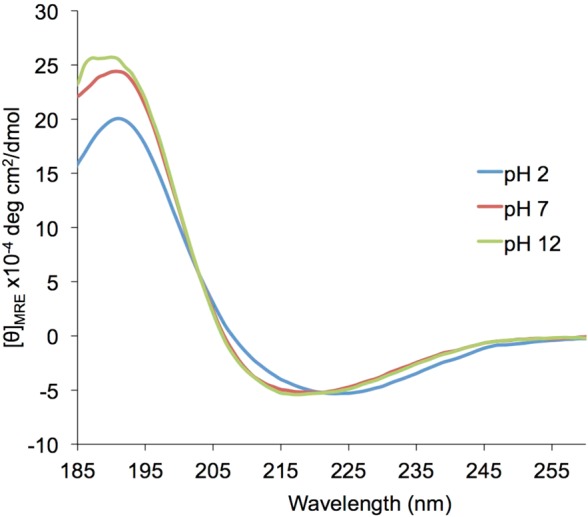
CD spectrum of oxPAS 4 (DP = 20) in 7 mM phosphate buffer at pH values of 2, 7, and 12.
To better understand the origin of the CD spectrum observed with PASs, we compared our results to those obtained with homooligomeric 1,2-substituted cyclohexane β-peptides, which form helical secondary structures in solution. The trans-substituted isomer, synthesized from trans-2-aminocyclohexanecarboxylic acid (trans-ACHC), exhibits a 3–14 helix stabilized by hydrogen bonding.18 In contrast, the homooligomeric cis-2-aminocyclohexanecarboxylic acid (cis-ACHC) β-peptides, which are more closely related in structure to the PASs reported here, display extended conformations rather than compact helices stabilized by hydrogen bonds between residues.19 Additionally, α/β-peptides containing cis-ACHC residues favor the formation of 11/9-helices.20 However, the glc-derived PAS structures differ from the cis-ACHC repeat unit of these previous examples in several important ways that may influence secondary structure formation. Most importantly, the PAS repeat unit has a strong preference for the 4C1 chair conformation with the nitrogen substituent at C1 in an axial position and the carbon at C2 in an equatorial position. In contrast, the extended and helical structures of cis-ACHC-containing oligomers reported in the literature show the nitrogen substituent in an equatorial position and the carbon substituent in an axial position. These differences in conformational preference, combined with the presence of the pyranose oxygen and the hydroxyl and carboxyl substituents at positions C3–C5, make direct extrapolation from the cyclohexyl derivatives challenging. It is unlikely that PASs, due to their cis-geometry of PASs, form the 3–14 helix observed for trans-ACHC oligomers. Additionally, the 4C1 chair conformational preference imposed by the carbohydrate ring may prevent PASs from adopting the extended or helical structures observed in reported cis-ACHC oligomers. Thus, the exact nature of the helical secondary structure and the forces stabilizing its formation are actively under investigation.
Proteins are widely used in biochemical research and as pharmaceutical agents, and their stability and retention of activity are primary concerns for storage and subsequent use.22 Consequently, protein stabilization agents are added to protein samples before lyophilization to lessen the loss of activity upon freeze-drying. Trehalose, an α-linked glucose disaccharide, is one of the most common agents used. It stabilizes proteins toward lyophilization by retaining water molecules near the protein structure and by hydrogen bonding to the protein. In fact, trehalose side chain polymers, prepared by RAFT polymerization, when conjugated to an enzyme impart enhanced stabilization toward environmental stressors.23 We hypothesized that PASs, both in their oxidized and unoxidized forms when mixed with an enzyme, may perform a similar role to trehalose due to the presence of multiple hydrogen bond donors and acceptors on each repeat unit, but have increased potency due to their polymeric structure. Lysozyme was chosen as a model protein to perform the stabilization studies.24 Samples of lysozyme alone, lysozyme with 100-fold excess by mass trehalose, PAS (DP50), or oxPAS (DP50, 7) were subjected to 10 lyophilization cycles, and the lysozyme activity was subsequently measured and compared to the activity before lyophilization, using a well-established assay (Figure 3).23b All three carbohydrates tested showed statistically significantly (p < 0.001) higher lysozyme activity than the untreated control, with the oxPAS performing the best (p < 0.01) as compared to trehalose. To confirm if this stabilizing effect arises partly from electrostatic interactions with lysozyme, which has an isoelectric point of 11.35,25 we performed several experiments. First, we observed a complexation between lysozyme and oxPAS by gel electrophoresis (see SI Figure S9). Second, we performed stabilization studies with two additional anionic polysaccharides (sodium alginate and sodium hyaluronate; 100-fold excess by mass) both of which exhibited a similar stabilizing effect as the oxPAS (p > 0.05).
Figure 3.
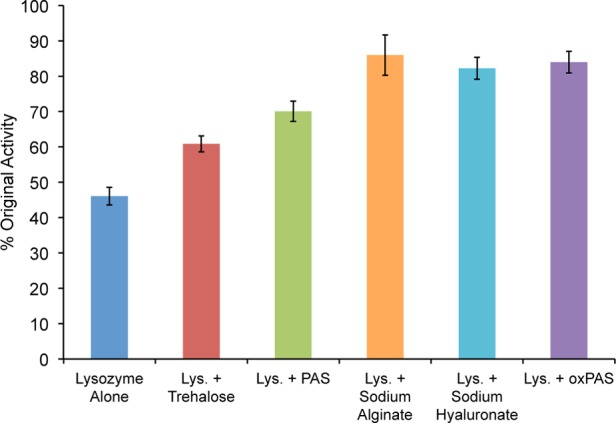
Lysozyme activity following 10 lyophilization cycles versus untreated controls (N = 5 for all samples).
In conclusion, the first synthesis of carboxylated glucuronic poly-amido-saccharides is reported. The polymers are prepared using a controlled anionic ring-opening polymerization followed by a selective oxidation of the C6 alcohol of the repeat unit to a carboxylic acid. The helical structure formed by these polymers, as indicated by CD, is unaffected by strong acid or base conditions, but is lost upon cleavage of the pyranose ring structure in the polymer backbone. In addition, PASs and oxPASs show promise as new protein stabilizing agents, with the oxPASs being significantly more effective than trehalose at stabilizing lysozyme to repeat freeze-drying. These acidic, water-soluble, carbohydrate polymers provide a unique opportunity to study structurally defined polysaccharide mimics and to identify potential applications for this new class of biopolymers.
Acknowledgments
The authors thank Boston University and NIHT32EB006359 for financial support. E.L.D. acknowledges receipt of an NIH/NIGMS Postdoctoral Fellowship (F32GM097781).
Supporting Information Available
NMR, IR, CD, gel electrophoresis, and details of synthesis and lyophilization protocol. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Stern R.; Jedrzejas M. J. Chem. Rev. 2008, 108, 5061–5085. [DOI] [PubMed] [Google Scholar]
- a Courtois-Samberg J.; Courtois B.; Heyraud A.; Colin-Morel P.; Rinaudo-Duhem M.. WO9318174, 1993.; b Elboutachfaiti R.; Delattre C.; Petit E.; Michaud P. Carbohydr. Polym. 2011, 84, 1–13. [Google Scholar]
- Burdick J. A.; Prestwich G. D. Adv. Mater. 2011, 23, H41–H56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka T.; Tsuru K.; Hayakawa S.; Osaka A. Biomaterials 2003, 24, 2889–2894. [DOI] [PubMed] [Google Scholar]
- a de Nooy A. E. J.; Besemer A. C.; van Bekkum H. Carbohydr. Res. 1995, 269, 89–98. [Google Scholar]; b Kato Y.; Matsuo R.; Isogai A. Carbohydr. Polym. 2003, 51, 69–75. [Google Scholar]
- Rinaudo M. Polym. Int. 2008, 57, 397–430. [Google Scholar]
- a Dumitriu S.Polysaccharides: Structural Diversity and Functional Versatility, 2nd ed.; Marcel Dekker, Inc.: New York, 2005. [Google Scholar]; b Lichtenthaler F.Ulman’s Encyclopedia of Industrial Chemistry; Wiley-VCH Verlag: New York, 2000. [Google Scholar]
- Pacsu E.; Mora P. T. J. Am. Chem. Soc. 1950, 72, 1045–1045. [Google Scholar]
- a Seeberger P. H.; Haase W.-C. Chem. Rev. 2000, 100, 4349–4394. [DOI] [PubMed] [Google Scholar]; b Gruner S. A. W.; Locardi E.; Lohof E.; Kessler H. Chem. Rev. 2002, 102, 491–514. [DOI] [PubMed] [Google Scholar]
- a Nishimura S.-I.; Matsuoka K.; Furuike T.; Ishii S.; Kurita K. Macromolecules 1991, 24, 4236–4241. [Google Scholar]; b Fraser C.; Grubbs R. H. Macromolecules 1995, 28, 7248–7255. [Google Scholar]; c Klein J.; Hüttermann C. F.; Skeries B. J. Macromol. Sci. A 2003, 40, 21–35. [Google Scholar]
- a Roy R.; Zanini D.; Meunier S. J.; Romanowska A. J. Chem. Soc., Chem. Commun. 1993, 1869. [Google Scholar]; b Turnbull W. B.; Stoddart J. F. Rev. Mol. Biotechnol. 2002, 90, 231–255. [DOI] [PubMed] [Google Scholar]
- a García-Martín M. d. G.; Pérez R. R.; Hernández E. B.; Galbis J. A. Macromolecules 2006, 39, 7941–7949. [Google Scholar]; b Imai T.; Satoh T.; Kaga H.; Kaneko N.; Kakuchi T. Macromolecules 2004, 37, 3113–3119. [Google Scholar]
- Stick R. V.; Williams S. J. In Carbohydrates: The Essential Molecules of Life, 2nd ed.; Elsevier: Oxford, 2009; p 203. [Google Scholar]
- Kadokawa J.-i. Chem. Rev. 2011, 111, 4308–4345. [DOI] [PubMed] [Google Scholar]
- a Dane E. L.; Ballok A. E.; O’Toole G. A.; Grinstaff M. W. Chem. Sci. 2013, 5, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dane E. L.; Chin S. L.; Grinstaff M. W. ACS Macro Lett. 2013, 2, 887–890. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Dane E. L.; Grinstaff M. W. J. Am. Chem. Soc. 2012, 134, 16255–16264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sebenda J.; Hauer J.; Biros J. J. Polym. Sci. Polym. Chem. Ed. 1976, 14, 2357–2363. [Google Scholar]; b Sebenda J.; Hauer J. Polym. Bull. 1981, 5, 529–534. [Google Scholar]; c Hashimoto K. Prog. Polym. Sci. 2000, 25, 1411–1462. [Google Scholar]; d Hashimoto K.; Oi T.; Yasuda J.; Hotta K.; Okada M. J. Polym. Sci. A: Polym. Chem. 1997, 35, 1831–1838. [Google Scholar]; e Hashimoto K.; Yasuda J.; Kobayashi M. J. Polym. Sci. A: Polym. Chem. 1999, 37, 909–915. [Google Scholar]; f Cheng J.; Deming T. J. J. Am. Chem. Soc. 2001, 123, 9457–9458. [DOI] [PubMed] [Google Scholar]; g Zhang J.; Kissounko D. A.; Lee S. E.; Gellman S. H.; Stahl S. S. J. Am. Chem. Soc. 2009, 131, 1589–1597. [DOI] [PubMed] [Google Scholar]; h Zhang J.; Markiewicz M. J.; Weisblum B.; Stahl S. S.; Gellman S. H. ACS Macro Lett. 2012, 1, 714–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Seebach D.; Overhand M.; Kuhnle F. N. M.; Martioni B. Helv. Chim. Acta 1996, 79, 913–941. [Google Scholar]; b Mándity I. n. M.; Fulop L.; Vass E. r.; Tóth G. b. K.; Martinek T. s. A.; Fulop F. Org. Lett. 2010, 12, 5584–5587. [DOI] [PubMed] [Google Scholar]; c Cheng R. P.; Gellman S. H.; DeGrado W. F. Chem. Rev. 2001, 101, 3219–3232. [DOI] [PubMed] [Google Scholar]
- Gellman S. H. Acc. Chem. Res. 1998, 31, 173–180. [Google Scholar]
- Choi S. H.; Ivancic M.; Guzei I. A.; Gellman S. H. Eur. J. Org. Chem. 2013, 3464–3469. [Google Scholar]
- Lee M.; Shim J.; Kang P.; Guzei I. A.; Choi S. H. Angew. Chem., Int. Ed. 2013, 52, 12564–12567. [DOI] [PubMed] [Google Scholar]
- Bertoldo M.; Zampano G.; Suffner L.; Liberati E.; Ciardelli F. Polym. Chem. 2013, 4, 653. [Google Scholar]
- a Veronese F. M. Biomaterials 2001, 22, 405–417. [DOI] [PubMed] [Google Scholar]; b Keefe A. J.; Jiang S. Nat. Chem. 2011, 4, 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Cummings C.; Murata H.; Koepsel R.; Russell A. J. Biomaterials 2013, 34, 7437–7443. [DOI] [PubMed] [Google Scholar]; b Mancini R. J.; Lee J.; Maynard H. D. J. Am. Chem. Soc. 2012, 134, 8474–8479. [DOI] [PubMed] [Google Scholar]
- Liao Y. H.; Brown M. B.; Martin G. P. Eur. J. Pharm. Biopharm. 2004, 58, 15–24. [DOI] [PubMed] [Google Scholar]
- Wetter L. R.; Deutsch H. F. J. Biol. Chem. 1951, 192, 237–242. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.