

Abstract
Thymidylate synthase (TSase) catalyzes the intracellular de novo formation of thymidylate (a DNA building block) in most living organisms, making it a common target for chemotherapeutic and antibiotic drugs. Two mechanisms have been proposed for the rate-limiting hydride transfer step in TSase catalysis: a stepwise mechanism in which the hydride transfer precedes the cleavage of the covalent bond between the enzymatic cysteine and the product and a mechanism where both happen concertedly. Striking similarities between the enzyme-bound enolate intermediates formed in the initial and final step of the reaction supported the first mechanism, while QM/MM calculations favored the concerted mechanism. Here, we experimentally test these two possibilities using secondary kinetic isotope effect (KIE), mutagenesis study, and primary KIEs. The findings support the concerted mechanism and demonstrate the critical role of an active site arginine in substrate binding, activation of enzymatic nucleophile, and the hydride transfer studied here. The elucidation of this reduction/substitution sheds light on the critical catalytic step in TSase and may aid future drug or biomimetic catalyst design.
Thymidylate synthase (TSase) catalyzes the last committed step in the de novo biosynthesis of 2′-deoxythymidine-5′-monophosphate (dTMP) from 2′-deoxyuridine-5′-monophosphate (dUMP). Since the product of this reaction is a nucleotide required for DNA biosynthesis, the activity of TSase is crucial for cell proliferation, making TSase a common target for many chemotherapeutic and antibiotic drugs.1 Because of its tremendous importance in health and disease, the detailed mechanistic features of TSase have been a subject of intensive investigations for many decades.2 In a TSase-catalyzed reaction, the cofactor methylene tetrahydrofolate (CH2H4folate) donates a methylene group and a hydride at different stages of the reaction (Scheme 1). Among the series of bond cleavages and formations catalyzed by TSase, the hydride transfer from C6 of tetrahydrofolate (H4folate) to exocyclic methylene intermediate (compound D in Scheme 1) is of particular interest. The hydride transfer is essentially irreversible2 and rate-determining on both first-order (kcat) and second-order rate constants (kcat/Km) for the overall reaction.3,4
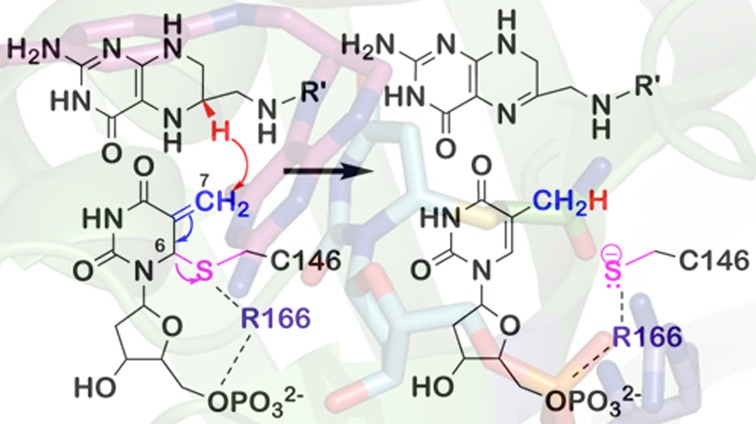
Scheme 1. Proposed Chemical Mechanism for TSase.

(Top panel) Proposed mechanism for TSase (adapted from ref (2)). The reaction is initiated by Michael addition of the enzymatic cysteine (C146) at C6 of dUMP (step 2) that leads to the formation of a covalent TSase-dUMP enolate intermediate (B), followed by Mannich condensation (step 3) to form a covalent ternary complex of TSase-dUMP-CH2H4folate (C). Following the Hoffman elimination (step 4), the methylene group forms an enzyme-bound exocyclic methylene intermediate (D). Finally, a hydride is transferred from C6 of H4folate to C7 of compound D and C146 departs from the product dTMP (step 5). (Bottom panels) Proposed mechanisms for step 5. (A). A stepwise mechanism that proceeds through the formation of a covalent TSase-dTMP enolate intermediate (compound E, similar to the compound B in the top panel).8,9 (B) A concerted mechanism where the hydride transfer takes place concomitantly with the C–S bond cleavage.5,7
Recently, we tested two possible mechanisms for the hydride transfer in the step 5 of Scheme 1 using E. coli TSase as a model system.5 We used hybrid quantum mechanics/molecular mechanics (QM/MM) simulations in the theoretical framework of the ensemble-averaged variational transition-state theory with multidimensional tunneling (EAVTST/MT) combined with Grote–Hynes theory.5−7 We compared the energy landscapes and reaction barriers for mechanisms A and B in Scheme 1. For the stepwise mechanism (panel A), the supporting evidence offered by Santi and co-workers8,9 comes from the close similarities between the enolate intermediates in steps 2 and 5A (compounds B and E, Scheme 1), where the formation of the former enolate was supported by several experimental findings including a TSase-catalyzed H/D exchange reaction of 5D-2′-deoxyuridine-5′-monophosphate (dUMP) and debromination of 5-bromo-dUMP that require Michael addition but no CH2H4folate cofactor8,10 and many covalent binary complexes of TSase with dUMP analogues.2 In contrast to that logical expectation,2,8,9 the QM/MM calculations predicted that the hydride transfer and C6–S cleavage (steps 5A and 6) occur concertedly.5−7 The calculations also identified a highly conserved arginine (R166) that at the ground state serves as part of the nucleotide phosphoester binding pocket but at the transition state (TS) activates the C6–S bond cleavage and concomitantly facilitates the hydride transfer. In the calculations, the positively charged R166 approaches the thioether at the TS for the hydride transfer and promotes the C6–S bond cleavage by polarizing it (Supplementary Figure S3).7 This makes this particular residue an inextricable part of the hydride transfer reaction coordinate in the concerted mechanism (panel B, Scheme 1). Several other studies also recognized the catalytic importance of R166 and identified it as one of the five “most essential” residues for TSase catalysis.10,11 Apart from structural contributions,11,12 the postulated roles of R166 in the chemical steps included to help initiate the catalysis by activating the cysteine through electrostatic interactions10 and to facilitate the reversal of the C146-nucleotide covalent adduct.13
In order to distinguish between the two mechanisms (bottom panels of Scheme 1) and to examine the role of R166 (if any) in the rate-limiting hydride transfer step, we applied two types of experimental investigations: secondary (2°) kinetic isotope effect (KIE) measurements on C6 of dUMP and mutation of R166 followed by an examination of primary (1°) KIEs on the hydride transfer. While both 2° and 1° KIEs are the ratio of rates of two isotopologues, they report on different chemical events. A 2° KIE is observed when an isotopically labeled atom’s bond is not cleaved or formed, but its bond order and vibrational states change as the system moves form ground state to TS. A hydrogen 2° KIE can probe the structure of the TS as it reflects a change in the carbon hybridization between ground state and TS. A 2° KIE can be normal (greater than unity) for a change of hybridization from sp3 to sp2 or inverse (less than unity) for sp2 to sp3 rehybridization of the pertinent atom.14 In enzyme kinetics, due to kinetic complexity, a 2° KIE measured on the second order rate constant (kcat/KM) will be experimentally detectable only if the rehybridization process occurs at or before the first irreversible step.14,15 Thus, in the stepwise mechanism (Scheme 1A), no 2° KIE on kcat/KM for the conversion of compound D to dTMP is expected, because the irreversible hydride transfer precedes the C6–S cleavage. For the concerted hydride transfer mechanism (Scheme 1B), on the other hand, the sp3 to sp2 rehybridization of C6 from compound D to dTMP is concomitant with the irreversible hydride transfer, thus predicting a normal 2° KIE for the rate-limiting hydride transfer step. Since all of the steps that precede the hydride transfer are reversible and only the hydride transfer is irreversible (Scheme 1), the observed 2° KIE on kcat/KM follows the equation9
| 1 |
where D is the exocyclic methylene intermediate (Scheme 1), and EIE is the equilibrium isotope effect on all steps preceding the hydride transfer. A maximal value of 0.835 was estimated for 2° H/T equilibrium isotope effect (EIE(dUMP→D)) using QM calculations of vibrational frequencies as described in the Supporting Information. With 6T-labeled dUMP, an inverse 2° H/T (KIE(dUMP→dTMP)obs) of 0.922 ± 0.004 was measured on kcat/KM, which, using eq 1 and the maximal EIE(dUMP→D), yields a 2° KIE(D→dTMP) of 1.104 ± 0.004. Any significant normal 2° KIE on kcat/KM at C6 of dUMP is consistent only with the cleavage of the C6–S bond being concerted with the irreversible hydride transfer, and therefore, the assessed normal 2° KIE(D→dTMP) of 1.104 ± 0.004 on kcat/KM strongly supports the concerted mechanism (Scheme 1B). Bruce and Santi9 measured an EIE of 0.808 for the conversion of free dUMP to an inhibitory covalent ternary complex of TSase-5F-dUMP-CH2H4folate (equivalent to compound C). This value is even more inverse than the EIE calculated here, which would lead to even larger normal 2° KIE on the hydride transfer step.
While the normal 2° KIE supported the concerted mechanism, to assess the role of R166, we substituted R166 with lysine (R166K), which is the only active variant of R166.11 Steady-state kinetic measurements indicate that this mutation causes about a 90-fold decrease in turnover rate (kcat) and increases Km for dUMP and CH2H4folate more than 300-fold and 18-fold, respectively (Supplementary Table S1). The inflated KM’s indicate a crucial role of R166 in ligand binding and especially in dUMP binding, which is consistent with crystal structures where that arginine is part of the dUMP’s phosphoester binding pocket. Although there are four arginines anchoring the phosphate of dUMP (Figure 1), mutations in R166 are the most detrimental and lead to an inactive mutant (except lysine).11 In the crystal structure of R166Q (PDB ID 1FWM),12 the active site structure is disoriented though the phosphate binding site is largely intact, indicating the importance of the R166 side chain in maintaining the active site architecture. While the structural roles of R166 are evident from crystal structures, probing its role in the chemical step(s) by steady-state kinetic studies is difficult.
Figure 1.
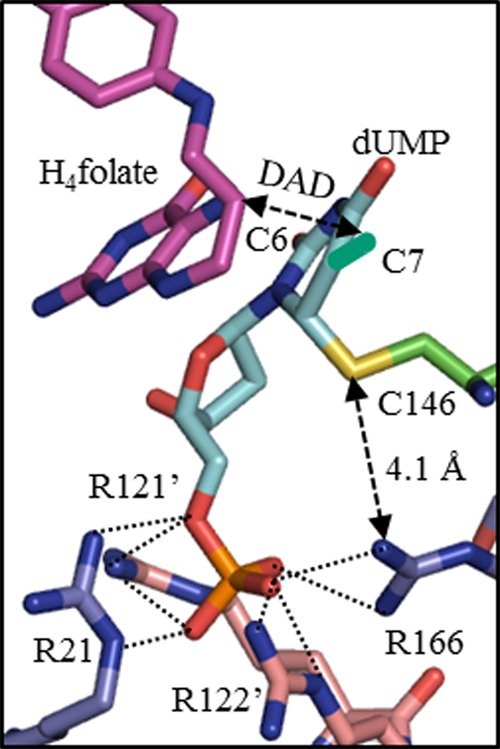
Crystal structure of E. coli TSase-dUMP-H4folate complex (PDB ID 1KZI) showing the phosphate-binding site. All of the arginines presented are within hydrogen bond lengths from the phosphoester of dUMP (2.7–2.9 Å). Methylene C7 (turquoise) was built on C5 of dUMP to mimic the exocyclic methylene intermediate (compound D in Scheme 1). C6 of H4folate and C7 of methylene intermediate are the hydride donor and acceptor, respectively.
To test the effect of the R166K mutation on the hydride transfer step, we measured the temperature dependence of intrinsic KIEs for that step and compared it to the same measurements with the wild-type enzyme.4 This tool probes the effect of the mutation on the nature of hydride transfer per se, despite kinetic complexity caused by other steps along the complex reaction catalyzed by TSase.16 In the framework of Marcus-like models, which have been extensively discussed in several reviews by us16−18 and others,19−23 an increase in temperature dependence of intrinsic KIEs results from a broader distribution of H-donor and H-acceptor distances (DADs) at the tunneling ready state (TRS, i.e. the QM delocalized TS). Furthermore, regardless of a specific theoretical model used to analyze the data, a mutation of a residue that is part of the reaction coordinate for a certain chemical conversion alters the distribution of DADs at the TRS, which will be probed as a change in the temperature dependence of the intrinsic KIEs. Alternatively, if a residue is not part of that reaction coordinate for the specific step under study, its mutation may have no effect on the kinetic probe. If R166 activates the C–S bond cleavage, and that cleavage is concerted with the hydride transfer step (Scheme 1B), the mutation would alter the TS and TRS for the hydride transfer, which would result in significant temperature-dependent intrinsic KIEs.7 On the other hand, in the stepwise mechanism (Scheme 1A), since R166 is not predicted to be a component of the hydride transfer coordinate, no significant alteration in the temperature dependence of the intrinsic KIEs on this step is anticipated for the mutant enzyme (R166K). In the past, most mutations to TSase residues had little or no effect on the temperature dependence of the intrinsic KIEs for the hydride transfer.18,24−26
To assess the intrinsic KIEs, we measured competitive primary (1°) KIEs of hydrogen to tritium (H/T) and deuterium to tritium (D/T) in 5–35 °C for the R166K mutant and extracted the intrinsic KIEs from the observed H/T and D/T KIEs using the Swain–Schaad relationships14 as described before for the wild-type TSase4 and in the enclosed Supporting Information. Both observed and intrinsic H/T KIEs are presented in Figure 2. As apparent from Figure 2 and Supplementary Table S3, while the intrinsic KIEs of wild-type TSase are temperature-independent, the intrinsic KIEs for R166K TSase display a very steep temperature dependence. Regardless of a model used to analyze the data, this finding indicates that R166 is part of the TS for the hydride transfer and the coupled C6–S bond cleavage. In the context of Marcus-like models,16−23 the observed steep temperature dependence of intrinsic KIEs for R166K results from a broader distribution of DADs at the TRS, and the larger values of the intrinsic KIEs are due to the longer average DAD at the TRS for the hydride transfer compared to the wild-type. Even though there is no direct contact between the residue R166 and either the hydride donor (C6 of H4folate) or the hydride acceptor (C7 of methylene intermediate) (Figure 1), R166 appears to modulate both the distribution of and average DAD at the TRS. As R166 is close to C146 (Figure 1), the current finding supports the prediction of the QM/MM calculations,5,7 suggesting a concerted C6–S cleavage and hydride transfer (Scheme 1B). In the stepwise mechanism (Scheme 1A), the charge stabilization effect by R166 occurs after the rate-limiting hydride transfer, excluding its stabilization by R166 from being a component of the hydride transfer coordinate.
Figure 2.
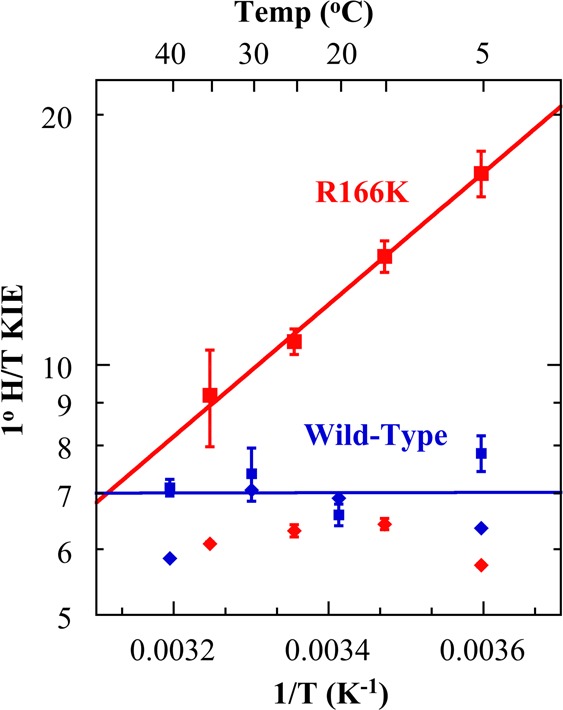
Arrhenius plots of observed (◆) and intrinsic KIEs (■) on the hydride transfers catalyzed by wild-type TSase (blue)4 and R166K (red). The intrinsic KIEs were fitted to the Arrhenius equation (eq S5 in Supporting Information) to obtain an isotope effect on the energy of activation (ΔEa(T-H)) and on the pre-exponential factors (AH /AT). These parameters are presented in Supplementary Table S3.
Alteration in temperature dependence of KIEs for R166K may alternatively arise from changes in protein’s overall conformational landscape or alteration of global dynamic network across the enzyme owing to the mutation. However, numerous X-ray studies indicate that the structural backbone of wild-type TSase and most of its mutants including that of R166 (R166Q)12 are perfectly superimposable, indicating an extremely rigid overall structure of TSase.27 Furthermore, most of the active site mutants as well as a remote one thus far studied displayed only minor, or no effect on temperature dependence of KIEs except R166K studied here.24−26 Therefore, the structural rigidity of TSase together with invariance in temperature dependence of KIEs for most mutants does not support an indirect structural or dynamic effect as a source of altered temperature dependence of KIEs observed for R166K, although such an option may not be explicitly eliminated. The agreement between our experimental observations reported here and the predictions of QM/MM calculations7 makes the interpretation of 1° KIEs as consistent with direct effect of R166 on the concerted cleavage of C6–S bond and the hydride transfer quite appealing.
In summary, there is strong experimental evidence for the formation of substrate enolate (intermediate B in Scheme 1) before the attack on the methylene of CH2H4folate.2,8,9 However, QM/MM calculations predicted that the hydride transfer in step 5 occurs concertedly with the C6–S bond cleavage (step 5B in Scheme 1),5,7 so no equivalent product enolate is formed (intermediate E in Scheme 1). This prediction was not an obvious outcome as, thermodynamically, the enolate intermediate (E) in the stepwise mechanism in Scheme 1A is not significantly different than the one formed after step 2 (compound B), although the kinetics of its formation could be very different. As reported above, we have tested the mechanism for the rate-limiting hydride transfer in TSase employing two experimental probes, 2° KIE on C6 of dUMP and the temperature dependence of intrinsic 1° KIEs on the hydride transfer for R166K mutant. The significant normal 2° KIE (1.104 ± 0.004) on kcat/KM indicates that the irreversible hydride transfer does not occur prior to C6–S cleavage, thus supporting the concerted mechanism as predicted by QM/MM calculations. The dramatic change in the temperature dependence of intrinsic 1° KIEs between the wild-type TSase and R166K suggests a critical role of R166 in the TS of the hydride transfer step. Since R166 is far from the hydride transfer site and situated in close proximity to the C6–S bond (Figure 1), its involvement in facilitating the C6–S bond breakage as predicted in the calculations is most conceivable. The combination of these two experimental probes provides unique support for the calculated concerted mechanism on the rate-limiting step of the TSase reaction and distinguishes the crucial role of R166 in the TS of that concerted chemical conversion.
Acknowledgments
The authors would like to thank Dr. Daniel Roston (University of Wisconsin, Madison) for the assistance with QM calculations. This work was supported by NIH R01 GM065368 and NSF CHE 1149023 to A.K. and the Iowa Center of Biocatalysis and Bioprocessing associated with NIH T32 GM008365 to Z.I. and I.G.
Supporting Information Available
Materials and methods for competitive KIE experiments. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Sergeeva O. A.; Khambatta H. G.; Cathers B. E.; Sergeeva M. V. Biochem. Biophys. Res. Commun. 2003, 307, 297. [DOI] [PubMed] [Google Scholar]
- Carreras C. W.; Santi D. V. Annu. Rev. Biochem. 1995, 64, 721. [DOI] [PubMed] [Google Scholar]
- Spencer H. T.; Villafranca J. E.; Appleman J. R. Biochemistry 1997, 36, 4212. [DOI] [PubMed] [Google Scholar]
- Agrawal N.; Hong B.; Mihai C.; Kohen A. Biochemistry 2004, 43, 1998. [DOI] [PubMed] [Google Scholar]
- Kanaan N.; Marti S.; Moliner V.; Kohen A. J. Phys. Chem. A 2009, 113, 2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaan N.; Martí M.; Moliner V.; Kohen A. Biochemistry 2007, 46, 3704. [DOI] [PubMed] [Google Scholar]
- Kanaan N.; Ferrer S.; Marti S.; Garcia-Viloca M.; Kohen A.; Moliner V. J. Am. Chem. Soc. 2011, 133, 6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Santi D. V. Biochemistry 1997, 36, 1869. [DOI] [PubMed] [Google Scholar]
- Bruice T. W., Santi D. V. In Enzyme Mechanism from Isotope Effects; CRC Press: Boca Raton, 1991; p 457. [Google Scholar]
- Finer-Moore J. S.; Santi D. V.; Stroud R. M. Biochemistry 2003, 42, 248. [DOI] [PubMed] [Google Scholar]
- Kawase S.; Cho S. W.; Rozelle J.; Stroud M. R.; Finer-Moore J.; Santi D. V. Protein Eng. 2000, 13, 557. [DOI] [PubMed] [Google Scholar]
- Sotelo-Mundo R. R.; Changchien L.; Maley F.; Montfort W. R. J. Biochem. Mol. Toxicol. 2006, 20, 88. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Ferrer S.; Moliner V.; Kohen A. Biochemistry 2013, 52, 2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook P. F., Cleland W. W. In Enzyme Kinetics and Mechanism; Garland Science: London, New York, 2007; p 253. [Google Scholar]
- Cook P. F.Enzyme Mechanism from Isotope Effects; CRC Press: Boca Raton, 1991. [Google Scholar]
- Klinman J. P.; Kohen A. Annu. Rev. Biochem. 2013, 82, 5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston D.; Islam Z.; Kohen A. Arch. Biochem. Biophys. 2014, 544, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roston D.; Islam Z.; Kohen A. Molecules 2013, 18, 5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammes-Schiffer S. Acc. Chem. Res. 2006, 39, 93. [DOI] [PubMed] [Google Scholar]
- Nagel Z. D.; Klinman J. P. Chem. Rev. 2010, 110, PR41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus R. A. Philos. Trans. R. Soc., B 2006, 361, 1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antoniou D.; Basner J.; Nunez S.; Schwartz S. D. Chem. Rev. 2006, 106, 3170. [DOI] [PubMed] [Google Scholar]
- Sutcliffe M. J.; Masgrau L.; Roujeinikova A.; Johannissen L. O.; Hothi P.; Basran J.; Ranaghan K. E.; Mulholland A. J.; Leys D.; Scrutton N. S. Philos. Trans. R. Soc., B 2006, 361, 1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Abeysinghe T.; Finer-Moore J. S.; Stroud R. M.; Kohen A. J. Am. Chem. Soc. 2012, 134, 17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B.; Maley F.; Kohen A. Biochemistry 2007, 46, 14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B.; Haddad M.; Maley F.; Jensen J. H.; Kohen A. J. Am. Chem. Soc. 2006, 128, 5636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud R. M.; Finer-Moore J. S. Biochemistry 2003, 42, 239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.