

Abstract
The 99 amino acid C-terminal fragment of amyloid precursor protein (C99), consisting of a single transmembrane (TM) helix, is known to form homodimers. Homodimers can be processed by γ-secretase to produce amyloid-β (Aβ) protein, which is implicated in Alzheimer’s disease (AD). While knowledge of the structure of C99 homodimers is of great importance, experimental NMR studies and simulations have produced varying structural models, including right-handed and left-handed coiled-coils. In order to investigate the structure of this critical protein complex, simulations of the C9915–55 homodimer in POPC membrane bilayer and DPC surfactant micelle environments were performed using a multiscale approach that blends atomistic and coarse-grained models. The C9915–55 homodimer adopts a dominant right-handed coiled-coil topology consisting of three characteristic structural states in a bilayer, only one of which is dominant in the micelle. Our structural study, which provides a self-consistent framework for understanding a number of experiments, shows that the energy landscape of the C99 homodimer supports a variety of slowly interconverting structural states. The relative importance of any given state can be modulated through environmental selection realized by altering the membrane or micelle characteristics.
Introduction
The amyloid-β (Aβ) peptide is believed to play a key pathogenic role in Alzheimer’s disease (AD). In vivo, Aβ is characterized by a distribution of isoforms, mostly varying in length from 38 to 43 residues. The most prominent isoform, Aβ40, typically occurs in a 10:1 ratio to the more amyloidogenic isoform, Aβ42. Knowledge of the origin of the Aβ isoform distribution, and its dependence on sequence, environment, and cofactors, is critical to our understanding of the etiology of AD.
Aβ is the product of cleavage of APP-C99 (C99), the 99 amino acid C-terminal fragment of the amyloid precursor protein (APP), by γ-secretase. C99 consists of a single transmembrane (TM) helix flanked by less structured extra- and intracellular domains. Processive cleavage is initiated in the C-terminal TM helical region and proceeds toward the N-terminus.1−3 Cleavage of C99 has been correlated with a number of factors, including peptide sequence4 and stability of the TM helix.5 While the degree of homodimerization of C996−10 has also been discussed as a potentially important factor in C99 processing, the cleavage of C99 dimers has not yet been definitively demonstrated. Environmental influences such as membrane composition,11 membrane curvature,12 and the presence of cholesterol may also play critical roles.13
It has been openly debated whether a quantitative description of C99 homodimerization structure is essential to a complete understanding of the mechanism of cleavage of C99 by γ-secretase and the genesis of the Aβ isoform distribution. Multhaup and co-workers first recognized that modifications in sequence that reduced homodimer affinity impacted cleavage of C99 by γ-secretase.6 Subsequently, studies of homodimer formation in WT and mutant C99 congeners have provided support for the view that C99 homodimerization is critical to C99 processing by γ-secretase and Aβ formation.14 However, it has also been argued that C99 homodimerization is weak and may be largely irrelevant in vivo, suggesting that γ-secretase acts on C99 monomer only as substrate in the production of Aβ.15
Additionally, while Tycko and co-workers suggest that the structure of the TM region of C99 depends on membrane composition,11 recent work by Sanders and co-workers supports the view that at least the backbone structure of C99 is largely independent of membrane lipid composition.16 In contrast, Smith and co-workers suggest that the Aβ product distribution following cleavage of C99 by γ-secretase may depend on the specific structure assumed by the C99 homodimer, which may depend on sequence and membrane composition.14
Although there is debate over the relevance of C99 homodimer in the processing of C99 to form Aβ, there is little doubt that C99 homodimer is an essential species in the overall ensemble of C99 structures. There are two contrasting proposals for the structure of C99 homodimers. The earliest proposed structures for the homodimer of the TM region of C99 (Figure 1) were right-handed coiled-coils stabilized by favorable interactions at the interpeptide interface facilitated by the GxxxG motif.5,6,15,17−20 This motif promotes a right-handed crossing in the α-helices by providing a good surface for packing and permitting close helix proximity.21 In contrast, one recent NMR structure suggests that the structure of GS- C9915–55 (C9915–55 plus two non-native amino acids at the N-terminus) consists of a left-handed coiled-coil structure stabilized by interpeptide contacts facilitated by a heptad-repeat motif involving G38 and A42.22 A more recent NMR structure23 of C9923–55 homodimer finds a right-handed coiled-coil stabilized by interpeptide contacts in the C-terminal region.
Figure 1.
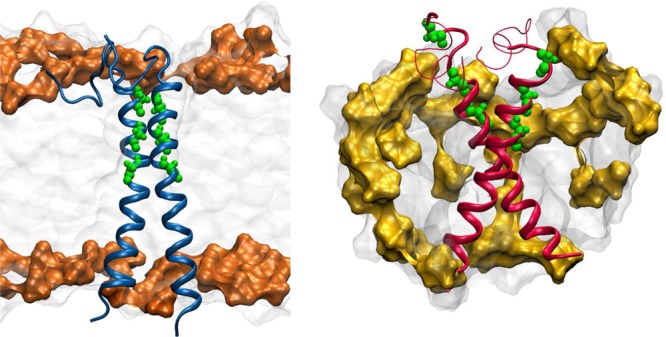
Depiction of C9915–55 dimer in a POPC lipid bilayer (left) and in a DPC micelle (right). The phosphocholine group is shaded orange (POPC) or yellow (DPC). G29, G33 and G37 residues are shown in green.
These contrasting results suggest that a number of fundamental questions related to the structure and processing of C99 in the production of Aβ, including the structure of C99 monomer and homodimer, the sensitivity of monomer and homodimer structure to sequence and membrane, and the relevance of homodimer formation to C99 processing, remain open and require further scrutiny. Here we address a number of critical questions regarding the nature of the C99 homodimer strucure and its dependence on membrane or micelle characteristics.
What is the structural ensemble of the C99 homodimer in a micelle environment? How well do structures of C99 homodimer in a micelle represent the structural ensemble in a lipid bilayer?
To answer these questions, we carried out multiscale simulations of C9915–55 dimer in POPC bilayers and DPC micelles building on our previous successful predictions of the monomer structures.12 We used coarse-grained (CG) simulations with the MARTINI force field24−26 of a broad sampling of the peptide dimer and lipid/surfactant ensemble. Such coarse-grained models provide an accurate model of TM helical proteins.24,28 Our model is benchmarked against the well-studied glycophorin A (GpA) dimer29−31 and used to develop novel predictions for the C9915–55 dimer.
Following the identification of the predominant structural states through CG simulations, representative conformations in all-atom molecular dynamics simulations were used to refine the atomistic structure and characterize the detailed peptide–peptide, peptide–lipid, and peptide–solvent interactions. Representative structures are shown in Figure 1. The major finding of our work is that the structural ensemble in a bilayer is heterogeneous, consisting of multiple states, whereas in a micelle only one state is predominantly selected. Globally the curvature of the bilayer and micelle is different, which has a profound influence on the conformational heterogeneity. More generally, our study demonstrates that the chemical environment imposes a selection on the nature of the APP dimer ensemble. The environmental selection of the structure is yet another variable, besides sequence, which can affect the plasticity of APP, and hence the product distribution upon cleavage by secretases.
Methods
CG Model Simulations
Initial conditions for the CG parametrization were taken from the experimentally derived structures determined by NMR in dodecylphosphocholine (DPC) micellar environment (PDB 2LOH)22 using the Martinize.py script and the MARTINI 2.2 force field for proteins.24 To build the POPC CG systems, two spatially separated C9915–55 monomers were overlapped with the pre-equilibrated lipid systems taken from the Marrink Web site (http://md.chem.rug.nl/cgmartini/). All lipid and water residues within 1.5 Å of the CG peptide were removed. For the DPC CG systems the same dimeric 2LOH structure was used. A dimer was embeded in a pre-equilibrated DPC CG micelle box also taken from the Marrink Web site. The CG bilayer system consisted of a C9915–55 dimer, 256 POPC lipids, 3863 water particles, and 6 Cl– ions to neutralize the system. The CG micelle system contained C9915–55 dimers, 54 DPC lipids, 5597 water particles, and 6 Cl– ions. Additional CG simulations were performed using 70 and 108 DPC surfactant molecules (see Supporting Information).
For the CG simulations a total of 50 replicas with 1.5 μs of MD were performed on each system in order to see convergence in the computed distribution of homodimer structures (see Figures S1 and S2). Nonbonded interactions were truncated using shift functions (between 0.9 and 1.2 nm for Lennard-Jones interactions and between 0 and 1.2 nm for electrostatics).25 The temperature of the systems was set to 303 K using the Berendsen weak coupling method32 with a coupling time of 0.1 ps. An integration time step of 30 fs was used in all simulations. The pressure was set to 1 bar using a semi-isotropic coupling for POPC and isotropic coupling for DPC using the Berendsen algorithm.
All-Atom Model Simulations
A CG structure from each of the different states in the POPC bilayer and DPC micelle (Gly-in, Gly-side, and Gly-out) was randomly selected and reconstructed into an all-atom representation using PULCHRA.33 The structure with its orientation were resolvated in POPC lipids or DPC surfactant using the CHARMM-GUI Membrane and Micelle Builder34−36 and modeled using the CHARMM36 all-atom lipid and protein force field and TIP3P water model.37−39 Parameters for dodecyl phosphocholine lipids for CHARMM36 were taken from Abel.40 The bilayer systems consisted of the C9915–55 dimers reconstructed from CG models, 128 POPC lipids, TIP3P water molecules extending 15 Å on each side of the bilayer, and 6 Cl– ions to neutralize the systems. The micelle systems contained C9915–55 monomer, 53 DPC molecules, TIP3P waters to solvate a box extending 20 Å from the DPC surfactant and protein, and 6 Cl– ions to neutralize the system.
For simulations in the POPC bilayer a total of 100 ns of MD were performed on each system (following minimization and a short NVT and NPT equilibration with protein backbone fixed). The pressure was set to 1 bar using a semi-isotropic coupling scheme with lateral and perpendicular pressures treated separately with coupling time 0.1 ps using the Parrinello–Rahman barostat methodology. The temperature of the system was set to 303 K and regulated using the Nosé–Hoover weak coupling algorithm.41
The nonbonded interactions were truncated using shift functions (between 0.9 and 1.2 nm for Lennard-Jones interactions and between 0 and 1.2 nm for electrostatics). Long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method42 with a Fourier grid spacing of 0.12 nm. The linear constraint solver (Lincs) method43 was used to constrain all bond lengths, with a 2 fs integration step. All-atom simulations in a DPC micelle were carried out under the same conditions using an isotropic coupling scheme to control the pressure.
The simulations were carried out using GROMACS (v4.5.1) and the analyses were performed using the GROMACS package, the DSSP program, and tailored scripts using python and MD Analysis libraries.44−48 The HELANAL program was used to calculate the kink angle along the TM helix between residues K28 and V50.49 Images were generated using VMD.50 All simulations were performed on the Boston University SCC supercomputers.
Results
(C9915–55)2 Forms Predominantly Right-Handed Coiled-Coil in DPC Micelle and POPC Bilayer
C9915–55 homodimer was simulated using CG molecular dynamics in a POPC bilayer and DPC micelle (see Figure 2). Multiple independent dynamical trajectories were initiated from the experimentally determined left-handed coiled-coil structure in a DPC micelle.22 All simulated replicas were observed to undergo conversion to a distribution that strongly favors right-handed helical packing (see Figure 2).
Figure 2.
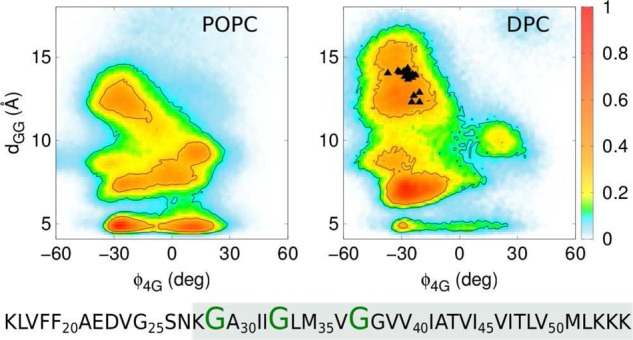
Simulated distributions for a CG model of the C9915–55 homodimer in POPC membrane (left panel) and DPC micelle (right panel) projected onto the order parameters ϕ4G and dGG. ϕ4G is the dihedral angle formed by G29A-G37A-G37B-G29B, where A and B label the two C9915–55 monomers, and dGG is the interhelical distance between G33A-G33B. The colored scale on the right defines the relative population. The system sequence is shown below the panels where G29, G33, and G37 are highlighted and the TM helical domain is shaded. The black triangles depict the values of ϕ4G and dGG obtained from the experimentally derived NMR structure23 of C9923–55.
The structure of the homodimer is conveniently characterized in terms of an interhelical distance, dGG and a dihedral angle, ϕ4G that differentiates the handedness of the coiled-coil. The ϕ4G order parameter is positive for left-handed structures and negative for right-handed structures. Structures stabilized by interpeptide interactions facilitated by the GxxxG repeat region are characterized by small values of the dGG parameter.
The first experimentally derived solution phase NMR structure of the C9915–55 homodimer in a micelle is characterized by a left-handed coiled-coil geometry with ϕ4G = +31° and dGG = 20 Å, forming an X-like homodimer configuration with minimal interpeptide contact in the G38xxxA42 region.22 In contrast, a more recent experimentally derived NMR structure of the shorter C9923–55 homodimer in a a micelle is characterized by a right-handed coiled-coil geometry with ϕ4G ≈ −25° and dGG ≈ 14 Å.23
The simulated homodimer distribution in POPC bilayer and DPC micelle environments are dominated by right-handed coiled-coil conformations. However, the broad distribution of homodimer conformations as a function of dGG (see Figure 2) reflects the existence of distinct conformational states, implying considerable heterogeneity in the structural ensemble.
(C9915–55)2 Ensemble in POPC Micelle and DPC Bilayer Is Characterized by Multiple Conformational States
Structural ensembles of two C9915–55 monomers in a POPC bilayer, derived from 50 independent CG replica simulations (see above), are presented in Figures 2 and 4. Interestingly, the C9915–55 homodimer assembled spontaneously on the time scale of a few hundred nanoseconds. This suggests that the sampling achieved with the CG model effectively represents the equilibrium homodimer structural ensemble in this single-component POPC lipid bilayer.
Figure 4.

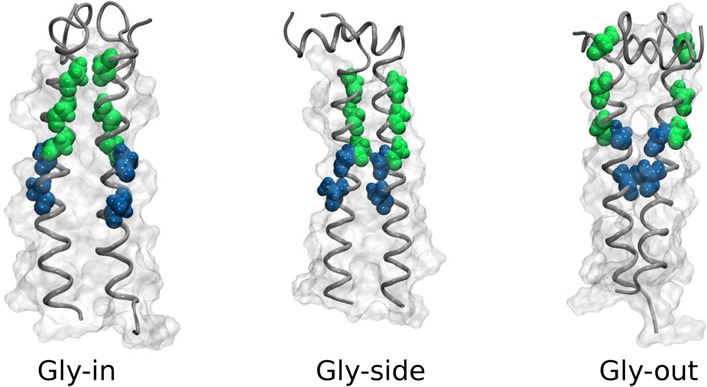
Most representative structure is a Gly-in state for the POPC bilayer (top left) and Gly-out state for the DPC micelle (top right). Cα atoms of the key glycines are shown in green. Simulated distributions for a CG model of the C9915–55 homodimer in POPC membrane (bottom left) and DPC micelle (bottom right) projected onto the Crick angles characterize the relative orientation of peptides within a homodimer. The black triangles depict the values of ψCrick obtained from the experimentally derived NMR structure23 of C9923–55. The atomic coordinates of the most representative structures (Gly-in, Gly-side and Gly-out) have been deposited as Supporting Information.
Although there are three characteristic states in the POPC bilayer, a significant shift in population between substates is observed in the DPC micelle environment. In particular, while the homodimer in a POPC bilayer is predominantly found in the Gly-in substate, in the DPC micelle the homodimer is dominanted by Gly-side and Gly-out conformations. This result suggest that global membrane characteristics influence structural heterogeneity.
Analysis of the dimer ensemble (see Figures 2 and 4) clearly shows the existence of multiple conformational state populations. ψCrick identifies the location of a residue relative to the axis between the two helices (Figure 3). Smaller values (between 0 and 60°) identify residues closer to the dimer interface, while larger angles (close to 180°) denote residues on opposite sides of the interface. For the competing structural states, we find (1) Gly-in configurations with close interpeptide contacts facilitated by exposure of backbone carbonyls in the GxxxG repeat region (small dGG, small ψCrick), (2) Gly-out configurations characterized by glycine repeats facing the outside of the homodimer interface (large dGG, large ψCrick), and (3) Gly-side configurations characterized by “out-of-phase” values of ψCrick (intermediate dGG, small/large or large/small ψCrick). Similar observations have been made in past computational studies of C99 homodimerization employing simplified models.20 Our results are consistent with those general observations, while providing a more detailed analysis of the homodimer ensemble and its dependence on environment.
Figure 3.
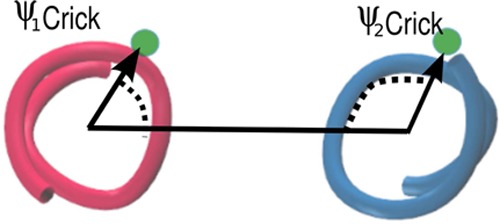
Definition of ψCrick corresponding to the angle between (1) the vector connecting the axis points of the two helices and (2) the vector connecting the Cα of a given residue to its corresponding α-helical axis point.51−53 We define ψCrick to be the average of the G33 and G37 dimer Crick angles in order to characterize the G33xxxG37 interface.
ll- or λ-like Structures Prominent in POPC Bilayer Are Replaced by X- or Y-like Structures in DPC Micelle
Figure 5 shows the distance between residues AK28 and BK28 (KK28) or AK54 and BK54 (KK53) where A and B indicate different monomers. The two distances represent the separation between the interfacial residues of the TM helices, and is a good measure to characterize the global topology of the dimer structure. Parallel or ll-like structures are characterized by both smaller KK28 and KK54 distances (5 Å), λ-like structures show smaller KK28 distances and larger KK54 distances, X-like structures show large KK28 and KK54 distances (15 Å), and Y-like structures have small KK54 and large KK28 distances. Simulations in POPC adopt predominantly ll-or λ-like conformations consistent with our previous computational predictions of C9923–55 homodimer in a membrane environment.17 We predicted a λ-like right-handed helical dimer structure in agreement with solid state NMR studies5 with a predominantly Gly-in orientation between the helices. In contrast, the DPC micelle simulations show larger populations of Y- or X-like structures with a predominantly Gly-out orientation between the helices (Figure 4).
Figure 5.

Distance between AK28 and BK28 (KK28) and AK54 and BK54 (KK54) in POPC bilayer (left) and DPC micelle (right) colored by the most populated ψ4G: red, green, and blue correspond to Gly-in, Gly-side, and Gly-out conformations for data of CG simulations. The spot size corresponds to the number of structures for that particular KK28 and KK54 conformation. The black triangles are the values of KK28 and KK54 obtained from the experimentally derived NMR structure23 of C9923–55. On the lower section the most representative λ-like (far left) and ll-like (center left) conformations for the simulations in POPC bilayer are shown in blue. X-like (center right) and Y-like (far right) conformations for the DPC micelle environment are shown in red. The K28 and K55 residues are indicated with orange spheres.
Results of additional CG simulations performed using 70 and 108 DPC surfactant molecules are shown in Figure 6. Qualitatively, we see that the X-like strucures are dominant for all micelle sizes. As the number of surfactant molecules is increased, there is a broadening of the distribution of X-like states. These results suggest that the micelle environment suppresses the sampling of Gly-in conformations and favors X- and Y-like structures, largely independent of the size of the micelle.
Figure 6.
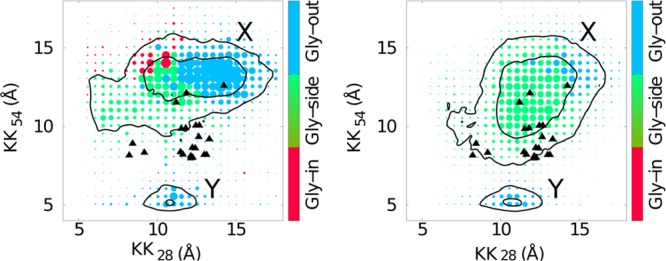
Left: distance between AK28 and BK28 (KK28) and AK54 and BK54 (KK54) colored by the most populated ψ4G: red, green, and blue correspond to Gly-in, Gly-side, and Gly-out conformations for data of CG simulations in 70 DPC surfactant molecules. The right panel corresponds to KK28 and KK54 for the C9915–55 system in 108 DPC surfactant molecules. The spot size corresponds to the number of structures for that particular KK28 and KK54 conformation.
Helicity of C9915–55 Is Unchanged by Dimerization in Bilayer but Diminished by Dimerization in Micelle
We performed all-atom simulations using CHARMM36 in POPC bilayer and DPC micelle environments, starting from representative CG structures from the Gly-in, Gly-side and Gly-out homodimer conformational states.
Experimental data suggest that the average stability of the TM helix is similar for monomeric peptide and peptide associated as a homodimer.54 Average helicity of each peptide in the micelle and bilayer is shown in Figure 7. We also show results for the simulated helicity of C9915–55 monomer in DPC micelle and POPC lipid for comparison, along with experimentally derived helicity values for monomeric C99 in a micelle.54 The average helicity in the TM domain of the simulated peptide is in good agreement with the experimentally measured helicity, while helicity in the juxtamembrane (JM)12 domain is somewhat larger in the simulated structures. This could result from differing size of the hydrophobic core in the micelle versus the bilayer, as the higher water accessibility in the micelle is expected to impact the stability of the helix structure, as well as the differing head groups and interfacial environments of the DPC (zwitterionic, simulation) and LMPG (anionic, experiment) micelles.
Figure 7.
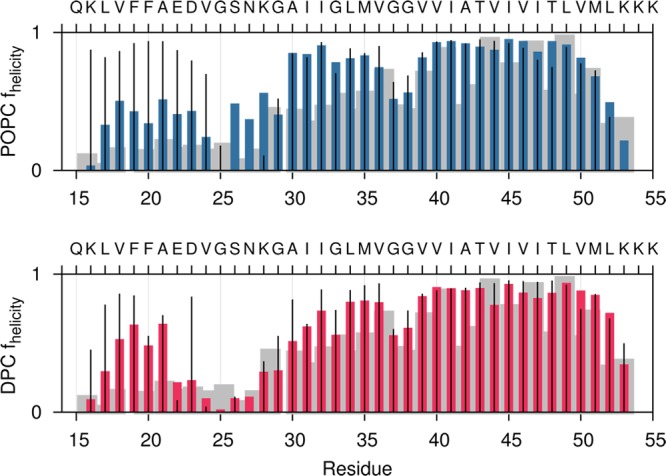
Average helicity over the three different states (Gly-in, Gly-side, and Gly-out) calculated from all-atom simulations of C9915–55 homodimer in a POPC bilayer (above) and DPC micelle (below). Thin black lines show results for helicity of C9915–55 monomer in the corresponding micelle or bilayer. The gray shadow shows experimentally determined helicity based on Cα NMR chemical shift measurements for monomeric C991–55 in an LMPG (lysomyristoylphosphatidylglycerol) micelle.54
Structural fluctuations in the kink angle are enhanced and less symmetric in the C9915–55 homodimer in a DPC micelle relative to the homodimer in a POPC bilayer, while fluctuations in homodimeric C9915–55 are smaller in magnitude than those observed for the C9915–55 monomer (see Figure 8). In particular, in the VGSN region we found more substantial fluctuations in the helicity that can be related to the surface curvature of the micelle environment.12
Figure 8.
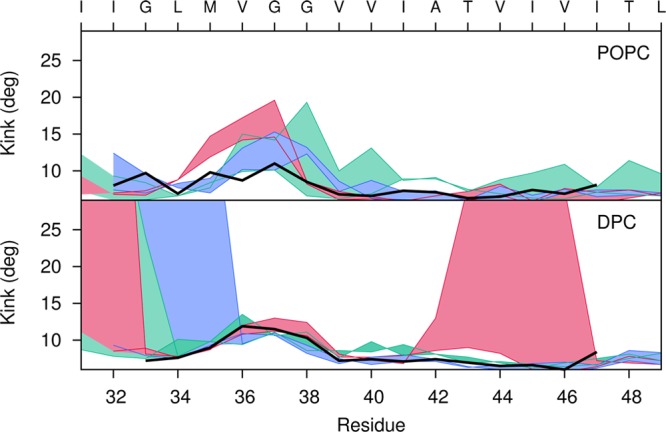
Measurement of the observed kink angle for each peptide of the all-atom simulations of C9915–55 homodimer in a POPC bilayer (top) and DPC micelle (bottom). For the three different systems, Gly-in (dark pink), Gly-out (blue) and Gly-side (green), the filled curve represents the difference ΔA–Bkink = θA – θBkink, where θA and θBkink are the kink angles of peptides A and B, respectively. The hinge angle in the C9915–55 monomer12 is shown in black for POPC bilayer and DPC micelle simulations.
Homodimer structures were analyzed for kink angle. Our results indicate that a structural kink appears near G37/G38 in the C9915–55 monomer in a POPC bilayer and DPC micelle, as has been proposed for the monomer structure based on experimental results for C991–55 in a LPMG micelle.54 A structural kink is also observed near G37/G38 in the C9915–55 homodimer in the DPC micelle environment. However, simulations of C9915–55 in DPC micelles show additional structural kinks in the TM helix. Greater kink angles are induced in an attempt to match the hydrophobic length of the TM helix with the hydrophobic thickness of the small DPC micelle. Gly-in conformations that destabilize the TM-C domain show a large kink at T43, while Gly-out structures that destabilize the TM-N helix show a large kink at G33.
Location of TM Helix γ-Site Shifted in DPC Micelle Relative to POPC Bilayer
Densities of the lipid phase of the POPC bilayer and DPC micelle were computed using all-atom simulations of the C9915–55 homodimer (Figure 9). Superimposed on the density profiles are distributions of key peptide residues. Importantly, although the density profiles of the lipophilic phase of the bilayer and surfactant micelle are similar, the solvent distributions in the two environments are dramatically different. We observe a substantially deeper penetration of water in the micelle simulations. In addition, the relative positions of key residues, including the γ-cleavage site, are significantly shifted relative to the center of the lipophilic phase in the DPC micelle when compared to the POPC bilayer.
Figure 9.
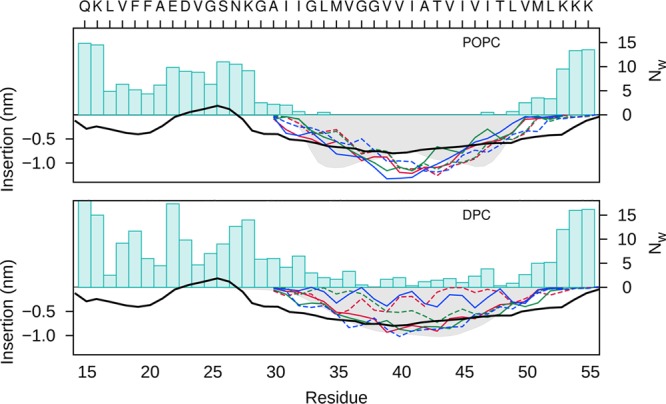
Density distribution of the lipid phases (shadow) for the all-atom simulations of the POPC bilayer (above) and DPC micelle (below) . Superimposed are the distributions of Cα positions of key residues along the z-axis for dimer A (solid lines) and dimer B (dashed lines) of C9915–55 for Gly-in (red), Gly-out (blue), and Gly-side (green) conformations. The number of waters molecules (Nw) within 4 Å of each amino acid of the dimers are indicated by blue bars. The EPR power saturation data derived from experimental measurements13 is shown for comparison (black dashed line).
A recent study involving H/D exchange experiments on the C99 peptide, complemented by molecular dynamics simulations of C9928–55 in a POPC bilayer, provided insight into the stability of helical regions of C99 including the TM helix.19 Considering the hinge at G37/38 to be a flexible divider in the TM helix, the N-terminal region of the TM domain (TM-N helix) showed enhanced H/D exchange relative to the C-terminal portion (TM-C helix).19 Our simulation results for the C9915–55 homodimer in a DPC micelle, in which a dominant “hinge” and less stable TM-N domain are observed, are consistent with those experimental and computational results.
Discussion
Observed Impact of Environment on Homodimer Structures
Studies have indicated that membrane protein structure in micelles and membranes can be similar for certain systems.55 The GpA homodimer has been extensively studied in micelle and bilayer environments, through both experimental and computational approaches, making it an excellent reference system for understanding environmental effects on the C9915–55 homodimer. Homodimerization of GpA in a micelle has been explored using all-atom molecular dynamics simulation,29 yielding good agreement with known experimental structures. Moreover, simulations employing an all-atom model of the GpA homodimer in DPC surfactant micelle and DMPC/DHPC lipid bicelles56 found the general topology of the homodimer to be similar in both environments.
In contrast, other studies have noted a dependence of protein structure and association on the differing structural environments of micelles and bilayers, as well as the particular detergent composition of micelles or lipid composition of membrane bilayers. There is substantial evidence that for a particular membrane system, a careful choice of detergent must be made to create a micellular environment in which the protein conformational ensemble is similar to that in a membrane bilayer.57 It has been observed that dimerization of GpA can be modulated by detergents,58,59 with variations in alkyl chain length and headgroup nature (ionic, zwitterionic, and nonionic) potentially influencing helix stability and helix dimerization. As a result, variations in detergent may impact helix dimerization while having little impact on helix stability. Finally, it is known that TM helical structure and stability can show a strong dependence on lipid composition in membrane bilayers.60
As in the case of C99, dimerization of the GpA TM domain has been proposed to be a consequence of favorable intermolecular interactions facilitated by GxxxG motif repeats. We have simulated the sequence of GpA62–101 using the same multiscale simulation approach employed in our study of C99. The GpA62–101 sequence includes a TM helical domain and N-terminal juxtamembrane domain, as is the case in C9915–55. Good order parameters for the homodimer structures in GpA (C99) are (1) dihedral angle ϕGT formed by G18A-T26A-T26B-G18B, where A and B label the two GpA62–101 monomers, (2) interhelical distance dGG between G22A and G22B, and (3) the Crick angle ψCrick of the GxxxG motif. The ϕGT order parameter is positive for left-handed structures and negative for right-handed structures. Structures stabilized by interpeptide interactions facilitated by the GxxxG repeat region are characterized by small values of the dGG parameter and small values of ψGT. Our results are summarized in Figure 10. Experimentally derived NMR structures are found to agree well with the simulation predictions in terms of (ϕGT , dGG). Differences of 15° are observed in the comparison of ψCrick angles between experimentally derived and computationally predicted structures. The observed differences may result from inherent limitations in the spatial resolution of both the coarse-grained model employed in our study and experimental data.
Figure 10.

Left: simulated distributions for a CG model of the GpA62–101 homodimer in a POPC bilayer projected onto the order parameters ϕGT and dGG (top), and onto the Crick angles ψCrick (bottom). The panels on the right corresponds to ϕGT and dGG (top) and Crick angles (ψCrick) (bottom) for the GpA62–101 system in 56 DPC surfactant molecules. The colored scale defines the relative population. Triangles represent NMR structures derived from GpA62–101 in DPC micelles (1AFO, black), GpA70–98 in DPC micelles (2KPE, green), and GpA61–98 in DMPC/DHPC bicelles (2KPF, red).
Our simulations results suggest similarities between between the DPC micelle and POPC bilayer simulations of GpA62–101 and C9915–55 homodimers, as well as sequence specific effects differentiating the two dimer ensembles. (1) The strucural distribution of C9915–55 dimer is signficantly more diverse than in the case of the GpA62–101 homodimer. (2) In both GpA and C99 homodimers, the structural ensembles are found to be significantly more diverse in the bilayer environment, relative to the micelle, consisting of multiple distinct conformational substates. In the DPC micelle, only one of the substates tends to be represented as it is selectively stabilized by the micelle geometry and surface curvature. It is useful to note that while GpA explores Gly-side and Gly-out homodimer conformations in a bilayer environment, the distribution is substantially more focused and dominated by right-handed Gly-in structures than in the case of C9915–55 homodimer. The results of this study, showing a clear dependence of C9915–55 homodimer structure on micelle and bilayer environment, are consistent with this understanding.
Handedness of Coiled-Coils in the Homodimer Ensemble
There remains an outstanding question regarding the helicity of the C99 homodimer. The earliest predicted structures for the C9923–55 fragment, containing the TM helical domain, proposed a right-handed coiled-coil structure consistent with the Gly-in topology described in this work.5,6,15,17−20 A recent NMR structure of the C9923–55 homodimer in a DPC micelle environment is also a right-handed coiled-coil, although of the Gly-out topology.23 An earlier NMR structure for the C9915–55 homodimer in a DPC micelle environment led to the proposal of a left-handed coiled-coil of the Gly-out topology.22 However, it was noted the homodimer ensemble may well contain right-handed and left-handed coiled-coil structures.
It is expected that both the length and sequence of the C99 fragment are critical to the ultimate homodimer structure. While dimers formed from the WT TM domain alone are almost certainly right-handed coiled-coils in most environments, it is possible that introducing mutations within the TM domain or the addition of the JM domain could lead to a shift in the relative population of one of the various dimer states (Gly-in, Gly-side, or Gly-out) or a change in the handedness of the homodimer. It has been observed that small changes in sequence can strongly impact binding affinities for TM homodimers. Our study has focused on the C9915–55, while the work of the Arseniev laboratory22 is based on GS-C9915–55, in which two non-native amino acids (GS) have been added to the N-terminus. It is possible that the addition of these residues, not included in our computational study, could impact the structure of the JM domain and also the handedness of the resulting homdimer relative to C9915–55. In this context, it is important to note that our study focuses on C9915–55 rather than full-length C99. Therefore, our results must be considered to provide insight into, but not fully represent, the properties of the full-length peptide.
Additional analysis shows good agreement between chemical shifts computed from our simulated homodimer structures and experimentally derived chemical shifts22 (see Figures S3 and S4). However, the experimentally derived NOE data provide unambiguous support for a left-handed coiled-coil structure of GS- C9915–55 homodimer in DPC micelle.22 This suggests that the chemical shifts are largely determined by the secondary structure of the peptide and local environment and are weak reporters on the nature of the tertiary coiled-coil geometry.
The findings of this paper provide a complete and self-consistent framework for organizing the existing experimental and computational results. For structures of the TM domain represented by the C9923–55 peptide, computational and experimental results suggest that in most membrane and micelle environments the structure is a right-handed coiled coil. The particular homodimer topology will depend on the membrane thickness or micelle size and curvature, with thinner environments (DMPC bilayer) and those with substantial surface curvature (DPC micelle) selecting Gly-out topologies, with thicker environments (POPC bilayer) selecting Gly-in structures.
Evidence from this study as well as the available NMR structures suggest that the handedness of the coiled-coil structure adopted by C9915–55 is ultimately primarily determined by (1) a preference of the TM domain of the WT protein to form a right-handed coilded coil, (2) characteristics of the membrane or micelle thickness and surface curvature, and (3) the secondary structure and relative position of the JM domain. In our study of the WT C9915–55 in POPC bilyaer and DPC micelle, we have largely assumed that the JM domain is helical, leading to a preference for a right-handed coiled-coil structure. However, fluctuations in the structure of the JM domain and its orientation relative to the TM domain may lead to a relative stabilization of left-handed coiled-coil strutures.
Overall, the homodimer ensemble must be considered to consist of left- and right-handed coiled coils, representing Gly-in, Gly-side, and Gly-out topologies. The handedness and topology of the dominant homodimer structure will be determined by the protein sequence and the bilayer or micelle.
Conclusions
We find the C9915–55 homodimer structural ensemble in POPC micelles and DPC bilayers consists of multiple conformational states that are structurally distinct and largely characterized by the relative orientation of the peptide helices. A dynamic “hinge” near G37/G38 is observed to divide the TM helix, with structural fluctuations being greater in the micelle than in the POPC bilayer environment. Dimerization results in little change in helix stability in the POPC bilayer, but a measurable decrease in helix stability is observed in the DPC micelle. Although the dimer ensemble in either environment is characterized by multiple conformational states, the dominant structures observed in our simulations in both the DPC micelle and POPC bilayer are consistently right-handed coiled-coil structures, supporting the conclusions of earlier experimental5 and computational studies.17,19
The relative importance of particular states is modulated by the C9915–55 homodimer environment. The Gly-in substate (stabilized by interpeptide contacts facilitated by the GxxxG motif repeats) is predominant in a POPC bilayer environment, while the Gly-out conformation (stabilized by interpeptide contacts consistent with a heptad repeat motif including G38 and A42) is dominant in a DPC surfactant micelle environment. Our results suggest the DPC micelle environment suppresses interactions mediated by GxxxG repeats in the TM region, leading to an X-shaped structure that best satisfies the boundaries of the surfactant/solvent interface. In this way, the environment “selects” a predominant substate through membrane thickness, interfacial curvature, and peptide-lipid interactions.
Past computational studies of homodimers of the TM domain of C99 have noted similar heterogeneity in the homodimer ensemble.20 Moreover, it has been proposed that the TM domain of C99 may be “processed to the γ sites depending on its dimerzation state and on the orientation of the TM helices in the dimers”.14 Our findings support these prior studies suggesting a role for membrane in modulating the formation of specific C99 homodimer structures for processing by secretases, as well as our interpretation of structures derived in diverse micelle environments.
Acknowledgments
The authors gratefully acknowledge the support of the National Science Foundation (CHE-1114676 and CHE-1361946) and the National Institutes of Health (RO1 GM076688). J.E.S. and L.D. thank the Schlumberger Foundation “Faculty for the Future Program” and CONACYT for the generous support of our research. We thank Chuck Sanders for his valuable comments and Dr. Eduard Bocharov for kindly sharing experimental chemical shift data presented in this work. We are also thankful for the resources of the Center for Computational Science at Boston University.
Supporting Information Available
Additional data describing the convergence of coarse-grained simulations and the comparison between computed and experimental chemical shifts are presented; atomic coordinates of the most representative structures (Gly-in, Gly-side, and Gly-out) in PDB format. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Qi-Takahara Y.; Morishima-Kawashima M.; Tanimura Y.; Dolios G.; Hirotani N.; Horikoshi Y.; Kametani F.; Maeda M.; Saido T. C.; Wang R.; Ihara Y. J. Neurosci. 2005, 25, 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; Tan J.; Mao G.; Cui M.-Z.; Xu X. J. Neurochem. 2007, 100, 1234–1246. [DOI] [PubMed] [Google Scholar]
- Kienlen-Campard P.; Tasiaux B.; Van Hees J.; Li M.; Huysseune S.; Sato T.; Fei J. Z.; Aimoto S.; Courtoy P. J.; Smith S. O. J. Biol. Chem. 2008, 283, 7733–7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munter L.-M.; Botev A.; Richter L.; Hildebrand P. W.; Althoff V.; Weise C.; Kaden D.; Multhaup G. J. Biol. Chem. 2010, 285, 21636–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T.; Tang T.-c.; Reubins G.; Fei J. Z.; Fujimoto T.; Kienlen-Campard P.; Constantinescu S. N.; Octave J.-N.; Aimoto S.; Smith S. O. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munter L.-M.; Voigt P.; Harmeier A.; Kaden D.; Gottschalk K. E.; Weise C.; Pipkorn R.; Schaefer M.; Langosch D.; Multhaup G. EMBO J. 2007, 26, 1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann S.; Hambsch B.; Hesse L.; Stumm J.; Schmidt C.; Beher D.; Bayer T. A.; Beyreuther K.; Multhaup G. J. Biol. Chem. 2001, 276, 33923–33929. [DOI] [PubMed] [Google Scholar]
- Gorman P.; Kim S.; Guo M.; Melnyk R.; McLaurin J.; Fraser P.; Bowie J.; Chakrabartty A. BMC Neurosci. 2008, 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggert S.; Midthune B.; Cottrell B.; Koo E. H. J. Biol. Chem. 2009, 284, 28943–28952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo J. H.; Park W. J. DNA Cell Biol. 2004, 23, 59–65. [DOI] [PubMed] [Google Scholar]
- Lu J.-X.; Yau W.-M.; Tycko R. Biophys. J. 2011, 100, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez L.; Meredith S. C.; Straub J. E.; Thirumalai D. J. Am. Chem. Soc. 2014, 136, 854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett P. J.; Song Y.; Van Horn W. D.; Hustedt E. J.; Schafer J. M.; Hadziselimovic A.; Beel A. J.; Sanders C. R. Science 2012, 336, 1168–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalifa N. B.; Van Hees J.; Tasiaux B.; Huysseune S.; Smith S. O.; Constantinescu S. N.; Octave J.-N.; Kienlen-Campard P. Cell Adhes. Migrat. 2010, 4, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Hustedt E. J.; Brandon S.; Sanders C. R. Biochemistry 2013, 52, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y.; Mittendorf K. F.; Lu Z.; Sanders C. R. J. Am. Chem. Soc. 2014, 136, 4093–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita N.; Straub J. E.; Thirumalai D.; Sugita Y. J. Am. Chem. Soc. 2009, 131, 3438–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pester O.; Barrett P. J.; Hornburg D.; Hornburg P.; Pröbstle R.; Widmaier S.; Kutzner C.; Dürrbaum M.; Kapurniotu A.; Sanders C. R.; Scharnagl C.; Langosch D. J. Am. Chem. Soc. 2013, 135, 1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Barreyro L.; Provasi D.; Djemil I.; Torres-Arancivia C.; Filizola M.; Ubarretxena-Belandia I. J. Mol. Biol. 2011, 408, 879–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKenzie K. R.; Prestegard J. H.; Engelman D. M. Science 1997, 276, 131–133. [DOI] [PubMed] [Google Scholar]
- Nadezhdin K. D.; Bocharova O. V.; Bocharov E. V.; Arseniev A. S. FEBS Lett. 2012, 586, 1687–1692. [DOI] [PubMed] [Google Scholar]
- Chen W.; Gamache E.; Rosenman D. J.; Xie J.; Lopez M. M.; Li Y.-M.; Wang C. Nat. Commun. 2014, 5, 3037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monticelli L.; Kandasamy S. K.; Periole X.; Larson R. G.; Tieleman D. P.; Marrink S.-J. J. Chem. Theor. Comput. 2008, 4, 819–834. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; Risselada H. J.; Yefimov S.; Tieleman D. P.; de Vries A. H. J. Phys. Chem. B 2007, 111, 7812–7824. [DOI] [PubMed] [Google Scholar]
- Marrink S. J.; de Vries A. H.; Tieleman D. P. Biochim. Biophys. Acta—Biomembranes 2009, 1788, 149–168. [DOI] [PubMed] [Google Scholar]
- Bond P. J.; Holyoake J.; Ivetac A.; Khalid S.; Sansom M. S. J. Struct. Biol. 2007, 157, 593–605. [DOI] [PubMed] [Google Scholar]
- Braun R.; Engelman D. M.; Schulten K. Biophys. J. 2004, 87, 754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuthbertson J. M.; Bond P. J.; Sansom M. S. Biochemistry 2006, 45, 14298–14310. [DOI] [PubMed] [Google Scholar]
- Psachoulia E.; Fowler P. W.; Bond P. J.; Sansom M. S. Biochemistry 2008, 47, 10503–10512. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J.; Postma J. P. M.; van Gunsteren W. F.; DiNola A.; Haak J. J. Chem. Phys. 1984, 81, 3684. [Google Scholar]
- Rotkiewicz P.; Skolnick J. J. Comput. Chem. 2008, 29, 1460–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Im W. PLoS One 2007, 2, e880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Kim T.; Iyer V. G.; Im W. J. Comput. Chem. 2008, 29, 1859–1865. [DOI] [PubMed] [Google Scholar]
- Cheng X.; Jo S.; Lee H. S.; Klauda J. B.; Im W. J. Chem. Inf. Model. 2013, 53, 2171–2180. [DOI] [PubMed] [Google Scholar]
- MacKerell A. D.; Bashford D.; Bellott M.; Dunbrack R.; Evanseck J.; Field M. J.; Fischer S.; Gao J.; Guo H.; Ha J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- Klauda J. B.; Venable R. M.; Freites J. A.; O’Connor J. W.; Tobias D. J.; Mondragon-Ramirez C.; Vorobyov I.; MacKerell A. D. Jr.; Pastor R. W. J. Phys. Chem. B 2010, 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; MacKerell A. D. J. Comput. Chem. 2013, 34, 2135–2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abel S.; Dupradeau F.-Y.; Marchi M. J. Chem. Theor. Comput. 2012, 8, 4610–4623. [DOI] [PubMed] [Google Scholar]
- Hoover W. G. Phys. Rev. A 1985, 31, 1695. [DOI] [PubMed] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar]
- Hess B.; Bekker H.; Berendsen H. J.; Fraaije J. G. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. J. Comput. Chem. 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; Van Der Spoel D.; Lindahl E. J. Chem. Theor. Comput. 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- Kabsch W.; Sander C. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- Joosten R. P.; te Beek T. A.; Krieger E.; Hekkelman M. L.; Hooft R. W.; Schneider R.; Sander C.; Vriend G. Nucleic Acids Res. 2011, 39, D411–D419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N.; Denning E. J.; Woolf T. B.; Beckstein O. J. Comput. Chem. 2011, 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal M.; Kumart S.; Velavan R. J. Biomol. Struct. Dyn. 2000, 17, 811–819. [DOI] [PubMed] [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. J. Mol. Graph. 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- Crick F. H. C. Acta Crystallogr. 1953, 6, 689–697. [Google Scholar]
- Strelkov S. V.; Burkhard P. J. Struct. Biol. 2002, 137, 54–64. [DOI] [PubMed] [Google Scholar]
- Lupas A. N.; Gruber M. In Fibrous Proteins: Coiled-Coils, Collagen and Elastomers; Parry D. A. D., Squire J. M., Eds.; Advances in Protein Chemistry70; Academic Press: San Diego, CA, 2005; pp 37–38. [Google Scholar]
- Beel A. J.; Mobley C. K.; Kim H. J.; Tian F.; Hadziselimovic A.; Jap B.; Prestegard J. H.; Sanders C. R. Biochemistry 2008, 47, 9428–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzin C. M.; Teriete P.; Marassi F. M. J. Am. Chem. Soc. 2007, 129, 8078–8079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mineev K.; Bocharov E.; Volynsky P.; Goncharuk M.; Tkach E.; Ermolyuk Y. S.; Schulga A.; Maslennikov V. C. I.; Efremov R.; Arseniev A. Acta Natur. 2011, 3, 90. [PMC free article] [PubMed] [Google Scholar]
- Stangl M.; Veerappan A.; Kroeger A.; Vogel P.; Schneider D. Biophys. J. 2012, 103, 2455–2464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher L. E.; Engelman D. M.; Sturgis J. N. J. Mol. Biol. 1999, 293, 639–651. [DOI] [PubMed] [Google Scholar]
- Fisher L. E.; Engelman D. M.; Sturgis J. N. Biophys. J. 2003, 85, 3097–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White S. H.; Wimley W. C. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 319–365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.