

Abstract
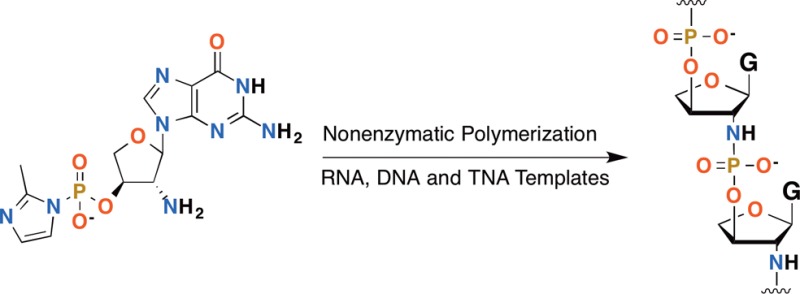
Threose nucleic acid (TNA) is a potential alternative genetic material that may have played a role in the early evolution of life. We have developed a novel synthesis of 2′-amino modified TNA nucleosides (2′-NH2-TNA) based on a cycloaddition reaction between a glycal and an azodicarboxylate, followed by direct nucleosidation of the cycloadduct. Using this route, we synthesized the thymine and guanine 2′-NH2-TNA nucleosides in seven steps with 24% and 12% overall yield, respectively. We then phosphorylated the guanine nucleoside on the 3′-hydroxyl, activated the phosphate as the 2-methylimidazolide, and tested the ability of the activated nucleotide to copy C4 RNA, DNA, and TNA templates by nonenzymatic primer extension. We measured pseudo-first-order rate constants for the first nucleotide addition step of 1.5, 0.97, and 0.57 h–1 on RNA, DNA, and TNA templates, respectively, at pH 7.5 and 4 °C with 150 mM NaCl, 100 mM N-(hydroxylethyl)imidazole catalyst, and 5 mM activated nucleotide. The activated nucleotide hydrolyzed with a rate constant of 0.39 h–1, causing the polymerization reaction to stall before complete template copying could be achieved. These extension rates are more than 1 order of magnitude slower than those for amino-sugar ribonucleotides under the same conditions, and copying of the TNA template, which best represented a true self-copying reaction, was the slowest of all. The poor kinetics of 2′-NH2-TNA template copying could give insight into why TNA was ultimately not used as a genetic material by biological systems.
Introduction
The early development of life must have proceeded through simple cells capable of Darwinian evolution to the complex life forms that exist today. To better understand this process, efforts are underway to synthesize model protocells composed of a self-replicating compartment and self-replicating genetic material.1 Simple processes of growth and division have been developed for fatty acid vesicles.2 However, despite recent advances in autocatalytic RNA synthesis3,4 and ribozyme-catalyzed RNA-dependent RNA polymerization,5 no self-replicating nucleic acid system capable of open-ended evolution has yet been developed.
As an alternative to ribozyme-mediated replication, activated nucleotides can be used to copy sequences without enzymatic catalysis. Building on the pioneering work of Leslie Orgel and colleagues,6−8 we and others have been developing nonenzymatic, template-directed primer extension using amino-sugar nucleotides as models for genetic replication, since these nucleotides show enhanced rates of polymerization due to the greater nucleophilicity of the amine.9−15 In particular, we have studied the polymerization of the acyclic 2′-amino-2′-deoxyglycerol nucleotides,12 2′-amino-2′,3′-dideoxyribose nucleotides,13,15 and 3′-amino-2′,3′-dideoxy-ribose nucleotides.14 These systems are attractive as models of genetic self-replication, since they have the potential to copy all four nucleobases on the order of minutes14 and they are compatible with fatty acid vesicles.15
(l)-α-Threose nucleic acid (TNA) (Figure 1B), based on the four carbon sugar threose, has also generated considerable interest since Eschenmoser and colleagues discovered that it is able to form stable Watson–Crick duplexes with itself, DNA, and RNA.16−18 Since TNA could have been synthesized by the same potentially prebiotic processes that have been proposed for RNA,19,20 it may have played a role in the early evolution of life.21 In support of this hypothesis, TNA has been used as a template for the nonenzymatic polymerization of activated ribonucleotides,22 and in vitro selection techniques have been used to isolate functional TNA sequences23,24 based on the enzymatic copying of DNA to TNA and vice versa.25−29
Figure 1.

Chemical structures of (A) RNA (DNA), (B) threose nucleic acid (TNA), and (C) 2′-amino-2′-deoxythreose nucleic acid (2′-NH2-TNA).
Since TNA has a shortened five atom repeating backbone unit, the interphosphate distance in a TNA–TNA duplex is only 5.9 Å.30 As a result, it has a shallow and wide minor groove, similar to A-form DNA and RNA,30 and binds more tightly to RNA than DNA.31 TNA residues adopt a unique 4′-exo sugar pucker with the 2′- and 3′-hydroxyl groups axial, which maximizes the interphosphate distance.31,32 Since the backbone unit has only four fully rotatable bonds, as opposed to five in RNA, the entropic cost of trapping a single-stranded TNA oligonucleotide into a duplex is less than with RNA or DNA.33,34 As a result, we hypothesized that single-stranded TNA might form a structure preorganized for favorable nucleotide binding and primer extension. Template preorganization has previously been shown to enhance the rate of nonenzymatic polymerization for altritol (ANA), hexitol (HNA), and locked (LNA) nucleic acids.13,35,36
Given the desirable properties of TNA and its potential importance in the origin of life, we became interested in testing the activated nucleotides of 2′-amino-2′-deoxythreose nucleic acid (2′-NH2-TNA) (Figure 1C) in nonenzymatic primer extension experiments. The Eschenmoser group has synthesized the adenine and thymine residues of the 2′- and 3′-amino-modified versions of TNA and shown that they have similar base-pairing properties to regular TNA, although the 2′-amino derivative formed significantly weaker duplexes.33,34 They have also shown that the 2′-amino modification enhances the rate of nonenzymatic ligation of two TNA oligonucleotides on a TNA template.37 Similar to 2′-aminodideoxyribonucleosides,13 the threose ring would hold the activated phosphate and nucleophilic amine of a monomer apart from each other to minimize cyclization, a known issue with amino-sugar nucleotides.12,14
In the route developed by Eschenmoser et al., the thymine 2′-NH2-TNA nucleotide (8t) was synthesized from the thymine TNA nucleoside, and the adenine 2′-NH2-TNA nucleoside was synthesized by trans-nucleosidation of 8t.34 We first sought to develop a direct route for the synthesis of the guanine 2′-NH2-TNA nucleoside (8g). We based our synthesis on a [4 + 2] cycloaddition reaction between an azodicarboxylate and glycal that was previously developed to synthesize trans 2-amino, 3-hydroxyl carbohydates38−40 due to its high efficiency and stereo- and regioselectivities. We then synthesized the 3′-phosphor-2-methylimidazolide derivative (2-MeImpntG) and tested it in nonenzymatic primer extension experiments on C4 RNA, DNA, and TNA templates. This work provides a novel synthetic approach to amino-sugar nucleosides and a better understanding of the potential role threose-based nucleic acids could play as the genetic material in a model protocell.
Results
Synthesis of 2′-NH2-TNA Nucleosides
We sought to develop an efficient synthesis that could yield multiple 2′-NH2-TNA nucleosides from a common intermediate with high enantiomeric purity. To this end, we decided to base our synthesis on a [4 + 2] cycloaddition reaction between a glycal and an azodicarboxylate, previously developed for the synthesis of 2-amino carbohydrates.38−40 Although this reaction had not been used to synthesize nucleosides or four carbon sugars, the general scheme was compatible with our needs.
We synthesized the glycal 4 from (R)-β-hydroxy-γ-butyrolactone (1), which can be obtained in >99% e.e. As shown in Scheme 1, we first protected the hydroxyl group with a tert-butyldiphenylsilyl (TBDPS) group, since it is bulky and nonelectron withdrawing. Next, we reduced the lactone to lactol 3 with diisobutylaluminum hydride (DIBAL-H) and converted the 1-hydroxy group to a good leaving group by mesylation, after which thermal elimination afforded glycal 4. These three steps were performed at greater than 5 g scale with a 67% overall yield. For the cycloaddition reaction, we mixed the glycal with bis(2,2,2-trichloroethyl) azodicaroboxylate (BTCEAD) in cyclohexane with constant 350 nm irradiation to allow conversion of the trans azodicarboxylate to the more reactive cis isomer.41 Previous reports have used dichloromethane as a cosolvent;39 however, we obtained higher yields in neat cyclohexane.
Scheme 1. Synthesis of the Nucleosidation Precursor by [4 + 2] Cycloaddition.

Reagents and conditions: (a) TBDPSCl, imidazole, DMF, 20 °C, 23 h, 95%; (b) DIBAL-H, THF, −78 °C, 1 h, 95%; (c) MsCl, Et3N, CH2Cl2, −40 °C, 1 h; (d) reflux, 10 h, 74%; (e) BTCEAD, irradiate 350 nm, cyclohexane, 20 °C, 16 h, 83%.
Conveniently, the cycloaddition product 5 proved to be an effective electrophile for one-pot nucleosidation catalyzed by trimethylsilyl trifluoromethanesulfonate (TMSOTf)42 (Scheme 2). Both the thymine and guanine nucleobases were glycosylated with high selectivity for the desired α anomer, likely indicating a mechanism involving direct displacement at C1. For guanine, we used the O6-diphenylcarbamoyl protected nucleobase for high N9 regioselectivity.43 For the thymine hydrazide 6t, the reported reduction conditions39 with zinc powder in acetic acid with one equivalent of acetone were effective in producing the protected nucleoside 7t. However, with the guanine hydrazide 6g these conditions resulted in product degradation and reduction of the N2-acetyl group to an ethyl group.
Scheme 2. Synthesis of 2′-Amino-2′-deoxythreose Nucleosides from Cycloadduct 5.

Reagents and conditions: (a) N2-acetyl-O6-(diphenylcarbamoyl)guanine, BSA, CH3CN, 80 °C, 10 min, cooled to 0 °C and added 5 and then TMSOTf, 0 °C, 15 min, 20 °C, 1.5 h, 79%; (b) indium powder, aluminum foil, 2:1 EtOH–sat. aq. NH4Cl, reflux, 3 h, 45%; (c) TBAF, THF, 20 °C, 3 h; (d) NH4OH, 35 °C, 22 h, 62%; (e) 5, thymine, BSA, CH3CN, 80 °C, 10 min, cooled to 0 °C and added TMSOTf, 0 °C, 30 min, 20 °C, 45 min, 91%; (f) zinc powder, acetic acid, 20 °C, 1 h; (g) acetone, zinc powder, 5 h, 71%; (h) TBAF, THF, 20 °C, 3 h, 68%.
We therefore screened various alternative reduction strategies and ultimately found that refluxing a mixture of indium powder and aluminum foil in ethanol–saturated aqueous ammonium chloride44−46 gave the highest yield of protected nucleoside 7g. Finally, we fully deprotected the intermediates to the free amino nucleosides 8g and 8t. Overall, the yields of 8g and 8t from lactone 1 were 12% and 24%, respectively, in seven steps. The structure of each was solved by single-crystal X-ray diffraction (Figure 2). The pseudorotation phase angle47 for nucleoside 8g and the two molecules of 8t in the crystal asymmetric unit ranged from 0° to 23°, indicating a C3′-endo sugar pucker characteristic of residues in A-form DNA and RNA.
Figure 2.

X-ray crystal structures of the 2′-amino-2′-deoxythreose nucleosides with a (A) guanine (8g) and (B) thymine (8t) nucleobase. Both furanose rings have a C3′-endo sugar pucker.
Synthesis of 2-MeImpntG
After synthesizing the nucleosides, we turned our attention to the synthesis of the activated nucleotide 2-MeImpntG (Scheme S1, Supporting Information and Figure 3A) for use in primer extension reactions. Previously, we have synthesized activated amino-sugar nucleotides using phosphorus(V) oxychloride without protecting the nucleobase.13,14 However, since the 3′-hydroxyl group of nucleoside 8g is a secondary hydroxyl and is further sterically hindered by its proximity to the nucleobase, this reaction was sluggish and not selective. Since the 3′-hydroxyl of TNA nucleosides has proven reactive toward phosphitylation reagents,34 we accessed 2-MeImpntG through phosphitylation-oxidation of the 3′-hydroxyl of protected 8g followed by activation according to the standard procedure developed by Orgel and colleagues.48 We measured the rate of hydrolysis of 2-MeImpntG by 31P NMR spectroscopy under the primer extension reaction conditions and observed a pseudo-first-order rate constant of 0.39 h–1, corresponding to a half-life of 1.8 h (Figure 3B).
Figure 3.

(A) Structure of the activated 2′-NH2-TNA nucleotide 2-MeImpntG. (B) Plot of the hydrolysis of 2-MeImpntG measured by 31P NMR with 100 mM HEPES pH 7.5, 150 mM NaCl, 100 mM HEI, 5 mM potassium phosphate reference, and 10% D2O at 4 °C. The natural logarithm of the fraction of activated nucleotide remaining was fit to a line; the slope yielded a pseudo-first-order rate constant of 0.39 h–1.
Primer Extension of 2-MeImpntG on RNA, DNA, and TNA Templates
Once we had synthesized the activated 2′-NH2-TNA nucleotide 2-MeImpntG, we tested its reactivity in nonenzymatic template-directed primer extension experiments. To allow for a direct comparison to previous results from other amino-modified nucleic acid systems,13,14 we used the same reaction conditions: 5 mM monomer, pH 7.5, 150 mM NaCl, and 100 mM N-(hydroxyethyl)imidazole (HEI). The HEI additive is an imidazole derivative that catalyzes both aminonucleotide polymerization and degradation,13,14 likely by exchanging with 2-methylimidazole to form a highly reactive zwitterionic intermediate. We used a DNA primer with a 3′-amino dideoxyribose terminal nucleotide and 3′-C4A2-5′ RNA, DNA, and TNA templates (Figure 4A).
Figure 4.

Nonenzymatic primer extension of 2-MeImpntG on C4 RNA, DNA, and TNA templates. (A) Schematic diagram of the primer extension reaction. A 5′-TAMRA-labeled DNA primer with a 3′-amino terminus was hybridized to each template and incubated with 5 mM 2-MeImpntG monomer at pH 7.5 and 4 °C with 150 mM NaCl and 100 mM HEI as catalyst. (B) Plot of the amount of primer remaining as a fraction of the total lane integration against time. The plot was fit to a single exponential decay to measure pseudo-first-order rate constants of 1.5, 0.97, and 0.57 h–1 and plateau values of 8%, 9%, and 9% for the RNA (•), DNA (■), and TNA (▲) templates, respectively. (C) PAGE analysis of the products of the primer extension reactions.
Activated nucleotide 2-MeImpntG was an effective monomer for nonenzymatic primer extension, with only 28% of the primer remaining after 1 h and 42% converted to full-length product after 6 h on the RNA template (Figure 4). To compare the reaction rates on the different templates, we first fit the fraction of primer remaining to a first-order exponential decay and obtained pseudo-first-order rate constants of 1.5, 0.97, and 0.57 h–1 for the RNA, DNA, and TNA templates, respectively (Figure 4B). As expected, the RNA template, which binds more tightly to TNA,17 was a more effective template than DNA. However, the TNA template, which we expected to form a preorganized single strand for favorable primer extension, was the least effective template. The reactions reached a plateau with 8–9% primer remaining, possibly indicating a population of primer that was chemically modified or not properly bound to the template. In the absence of HEI catalyst, the rate of polymerization was 19 times slower on average, and hydrolysis was 87 times slower (Figure S1, Supporting Information).
Discussion
We have developed a novel synthesis of 2′-NH2-TNA nucleosides 8g and 8t in 12% and 24% overall yield, respectively, from lactone 1 in seven steps (Schemes 1 and 2). For comparison, Eschenmoser and colleagues synthesized 8t in nine steps from l-ascorbic acid in 10% overall yield.17,18,34 Both nucleosides crystallized with a C3′-endo sugar pucker characteristic of residues in A-form DNA and RNA (Figure 2). However, TNA residues prefer a C4′-exo sugar pucker in the context of a duplex.30−32 The C3′-endo and C3′-exo sugar puckers have also been reported in crystallized N6-benzoyladenine and thymine TNA nucleosides,17 respectively, suggesting that the C4′-exo sugar pucker is preferred only in the context of a duplex. Since both the C3′-endo and C4′-exo sugar puckers hold the 2′- and 3′-substituents in an extended axial conformation required for the duplex, the C4′-exo sugar pucker is likely required for the optimal orientation of the nucleobase and phosphodiester backbone. The 2′-amino group may also affect the preferred conformation.
The cycloaddition–nucleosidation–reduction scheme we have developed could potentially be applied to other 2′-amino nucleosides. Stereoselectivity of the cycloaddition reaction has been maintained with 3-deoxy glycals by using more distal bulky protecting groups.40 Therefore, it could potentially be used to access the 2′-amino-2′,3′-dideoxyribose nucleoside series, which performs well in nonenzymatic polymerization experiments but is currently difficult to synthesize.13 The more common 2′-hydroxyl nucleosides could potentially be accessed by using phenanthrenequinone instead of azodicarboxylates. It has previously been shown that phenanthrenequinone can undergo similar stereoselective [4 + 2] cycloaddition chemistry with glycals to form new C1–O and C2–O bonds,49 although it is not known if the cycloadduct formed is a good substrate for nucleosidation.
The monomer 2-MeImpntG was able to extend a primer on C4 RNA, DNA, and TNA templates (Figure 4). This finding demonstrates the first nonenzymatic polymerization of a threose-based nucleotide, a reaction that is critical for its potential use as a self-replicating genetic material. The pseudo-first-order rate constants for the first addition to the primer ranged from 0.57 to 1.5 h–1 (Figure 4B). For comparison, in the 2′-amino and 3′-amino-2′,3′-dideoxyribose systems, the activated guanine nucleotides completely copy a C4 RNA template in under 5 min with the same conditions, indicating a rate constant of at least 42 h–1. It is possible that the constrained sugar of 2-MeImpntG limits its ability to adopt the geometry necessary for the phosphoryl transfer when bound to the template. We also found that the addition steps did not progress evenly, with less +2 product than +1, +3, and +4 present at intermediate time points (Figure 4C). A similar trend was observed for the 3′-amino-2′,3′-dideoxyribose system14 and may be due to a change in helical geometry as the amino nucleotides extend from the DNA primer. Alternatively, the monomers could form short oligomers that subsequently add to the primer. The monomer hydrolyzed surprisingly quickly with a half-life of 1.8 h (Figure 3B), making it only slightly more stable than its 3′-amino-2′,3′-dideoxyribose analogue, which cyclizes with a half-life of 1.2 h.14
Interestingly, the ranking of the polymerization rates on the different templates was RNA > DNA > TNA. We expected polymerization to be faster on the A-form RNA template than the B-form DNA template, since TNA prefers an A-form geometry due to its shortened backbone.31 However, we similarly expected polymerization to be fastest on the TNA template. TNA is also a worse template than RNA for the polymerization of activated ribonucleotides.22 The restrained geometry of TNA either makes it less able to bind tightly to nucleotides or for them to adopt the geometry necessary for phosphoryl transfer once bound. These alternate hypotheses could be tested by measuring the rate of polymerization at a range of monomer concentrations on the different templates, combined with direct physical measurements of monomer binding to templates.
Taken together, our results show that 2′-NH2-TNA could potentially be used as a self-replicating genetic material, but its slow rate of polymerization compared to monomer hydrolysis, especially on the TNA template, makes other systems more attractive. Furthermore, the limited reactivity of the 3′-hydroxyl group could make the prebiotic phosphorylation of TNA nucleosides difficult. If these results hold for TNA monomers, TNA would be inferior to RNA as a genetic polymer in the origin of life, which may be why it was ultimately not used despite the likely coproduction of threose- and ribose-based nucleosides through prebiotic chemistry.19,20
Materials and Methods
General
All reagents and solvents were purchased from Sigma–Aldrich, Alfa Aesar, or Toronto Research Chemicals, except for deuterated solvents, which were purchased from Cambridge Isotope Laboratories. Flash chromatography was performed on a Biotage SP1 instrument with HP-Sil columns. NMR spectroscopy was performed on a 400 MHz Varian spectrometer (Oxford AS-400) operating at 25 °C, unless otherwise specified. Spectra were referenced to the solvent peak according to published values,50 except for 31P spectra, which were referenced to orthophosphate or phosphoric acid (0 ppm). High-resolution mass spectrometry was carried out on a Waters Q-TOF micro LC-MS or an Agilent 6520 QTOF LC-MS. Low resolution electrospray ionization mass spectrometry (ESI-MS) was performed on a Bruker Esquire 6000 with direct injection. Preparative high-performance liquid chromatography (HPLC) purification was performed on a Varian ProStar instrument with a Dynamax Microsorb C18 column (250 mm × 21.4 mm). Data analysis was performed using Prism software (GraphPad).
Primer Extension Reaction
The conditions for the primer extension reaction were as follows: 200 mM HEPES pH 7.5, 150 mM NaCl, 100 mM HEI pH 7.5, 5 mM 2-MeImpntG, 0.3 μM primer, 1.5 μM template, 4 °C. The primer sequence was 5′-TAMRA-GCG TAG ACT GAC TGG-3′-NH2. The sequence of the RNA, DNA, and TNA templates was 5′-AAC CCCCCA G(U/T)C AG(U/T) C(U/T)A CGC-3′, with the C4 template region in bold. The TNA template was composed of DNA, except for the underlined region. The primer was provided by Dr. Sergei Gryaznov (Geron Corp.), the TNA template was provided by Dr. John Chaput (Arizona State University), and the RNA and DNA templates were purchased from IDT DNA.
The total reaction volume was 30 μL. At each time point, a 5 μL aliquot was taken, mixed with 10 μL of kill buffer (100 mM EDTA, 4.8 M urea, 1X TBE), and precipitated by ethanol. Time points were taken at 1, 2, 3, 4.5, 6, and 24 h. Samples were analyzed by 20% urea–TBE denaturing polyacrylamide gel electrophoresis (PAGE) and imaged on a Typhoon Scanner 9400 (GE Healthcare) and integrated using ImageQuant TL software (GE Healthcare).
Kinetics of 2-MeImpntG Hydrolysis
The kinetics of 2-MeImpntG hydrolysis were measured under the same conditions as the primer extension reaction, except with 3 mM 2-MeImpntG, 100 mM HEPES pH 7.5, 5 mM potassium phosphate reference, and 10% D2O. Time points were taken over the course of 6 h. At each time point, a 31P NMR spectrum was measured, the phosphate signal was set to a chemical shift of 0 ppm and an integration of 1, and the integrations of the activated (−12.02 ppm) and hydrolyzed (1.15 ppm) nucleotide were then measured and normalized to the initial integration.
(R)-4-((tert-Butyldiphenylsilyl)oxy)dihydrofuran-2(3H)-one (2)
A mixture of lactone 1 (10 g, 94.0 mmol) and imidazole (16.7 g, 245 mmol) in DMF (36 mL) under argon was cooled on ice, and then, TBDPSCl (32 mL, 119 mmol) was added slowly. The mixture was warmed to 20 °C, stirred for 23 h, and then, poured into ddH2O (400 mL). The product was extracted with CH2Cl2 (3 × 250 mL), and the combined organic fractions were washed with brine (200 mL), dried over MgSO4, filtered, and concentrated to 100 mL. The concentrate was filtered through a bed of silica with CH2Cl2 and then triturated with hexanes to afford protected lactone 2 (28.3 g, 95%) as a white solid: Rf = 0.69, CH2Cl2; 1H NMR (400 MHz, CDCl3) δ 7.62 (d, J = 6.9 Hz, 4H, H-Ph), 7.46 (dd, J = 7.2 Hz, 2H, H-Ph), 7.40 (dd, J = 7.1 Hz, H-Ph), 4.56 (m, 1H, H–C3), 4.21 (dd, 1H, J = 9.8, 2.5 Hz, H–C4), 4.16 (dd, 1H, J = 9.8, 4.6 Hz, H–C4), 2.50 (d, 2H, J = 3.9 Hz, H–C2), 1.06 (s, 9H, H-t-Bu); 13C NMR (100 MHz, CDCl3) δ 175.9, 135.8, 133.0, 130.4, 128.2, 75.9, 69.2, 38.1, 26.9, 19.2; HRMS (m/z): [M + H+]+ calcd. for C20H24O3Si, 341.1568, obsd. 341.1563.
(4R)-4-((tert-Butyldiphenylsilyl)oxy)tetrahydrofuran-2-ol (3)
Protected lactone 2 (10.02 g, 29.4 mmol) was dissolved in anhydrous THF (100 mL) under argon and cooled to −78 °C with an acetone–dry ice bath. A 1 M solution of DIBAL-H in toluene (38 mL, 38 mmol) was added dropwise over 1 h. The reaction was quenched with methanol (3 mL) and then warmed to room temperature. Saturated sodium potassium tartrate (150 mL) was added, and the mixture was poured into 400 mL of EtOAc and stirred for 2 h. The organic layer was washed with water (150 mL), washed with brine (150 mL), dried over MgSO4, and filtered, and the crude was purified by flash chromatography (5–20% EtOAc in hexanes) to afford protected lactol 3 (9.58 g, 95%) as a clear oil: Rf = 0.28, 1:4 EtOAc–hexanes; 1H NMR (400 MHz, DMSO-d6) anomer 1: δ 7.63–7.54 (m, 4H, H-Ph), 7.48–7.37 (m, 6H, H-Ph), 6.06 (d, J = 5.1 Hz, 1H, H–O–C1), 5.44 (m, 1H, H–C1), 4.49 (m, 1H, H–C3), 3.76 (dd, J = 9.1, 4.9 Hz, 1H, H–C4), 3.55 (dd, J = 9.2, 2.4 Hz, 1H, H–C4), 1.94 (m, 1H, H–C2), 1.77 (ddd, J = 13.3, 6.3, 3.0 Hz, 1H, H–C2), 0.97 (s, 9H, H-t-Bu); anomer 2: δ 7.63–7.54 (m, 4H, H-Ph), 7.48–7.37 (m, 6H, H-Ph), 6.11 (d, J = 5.0, 1H, H–O–C1), 5.20 (m, 1H, H–C1), 4.28 (m, 1H, H–C3), 3.68–3.60 (m, 2H, H–C4), 2.10 (m, 1H, H–C2), 1.67 (m, 1H, H–C2), 0.98 (s, 9H, H-t-Bu); 13C NMR (100 MHz, DMSO-d6) δ 135.2, 135.1, 133.3, 133.2, 129.9, 127.9, 97.6, 97.3, 72.9, 72.6, 72.1, 71.5, 43.3, 42.4, 26.7, 18.7, 18.6; HRMS (m/z): [M + Na+]+ calcd. for C20H26O3Si, 365.1543, obsd. 365.1544.
(R)-tert-Butyl((2,3-dihydrofuran-3-yl)oxy)diphenylsilane (4)
Protected lactol 3 (8.94 g, 27.6 mmol) and Et3N (14.6 mL, 104.7 mmol) were dissolved in anhydrous CH2Cl2 (200 mL) under argon and then cooled to −40 °C with an acetonitrile–dry ice bath. MsCl (2.7 mL, 34.9 mmol) was added slowly, and the reaction was stirred at −40 °C for 1 h, warmed to room temperature, and refluxed for 10 h. The reaction mixture was concentrated to 100 mL and then purified by flash chromatography (1:1 CH2Cl2–hexanes) to afford protected glycal 4 (6.59 g, 74%) as a clear oil: Rf = 0.57, 1:1 CH2Cl2–hexanes; 1H NMR (400 MHz, CDCl3) δ 7.68–7.64 (m, 4H, H-Ph), 7.45–7.41 (m, 2H, H-Ph), 7.40–7.36 (m, 4H, H-Ph), 6.49 (apt. d, J = 2.6 Hz, 1H, H–C1), 5.01 (dddd, J = 7.2, 2.6, 2.6, 0.9 Hz, 1H, H–C3), 4.96 (dd, J = 2.6, 2.6 Hz, 1H, H–C2), 4.23 (dd, J = 10.5, 2.6 Hz, 1H, H–C4), 4.04 (dd, J = 10.5, 7.20 Hz, 1H, H–C4), 1.04 (s, 9H, H-t-Bu); 13C NMR (100 MHz, DMSO-d6) δ 150.5, 135.5, 135.4, 133.8, 133.6, 130.2, 130.1, 128.3, 128.2, 103.7, 77.1, 74.8, 27.0, 19.0; ESI-MS (m/z): [M + Na+]+ calcd. for C20H24O2Si, 347.1, obsd. 347.1.
(4aR,7R,7aR)-2,2,2-Trichloroethyl-7-((tert-butyl-diphenylsilyl)oxy)-3-(2,2,2-trichloroethoxy)-4a,6,7,7a-tetrahydro-1H-furo[3,2-e][1,3,4]oxadiazine-1-carboxylate (5)
Protected glycal 4 (1.00 g, 9.25 mmol) and bis(2,2,2-trichloroethyl)azodicarboxylate (1.65 g, 13.0 mmol) were dissolved in 18 mL of anhydrous cyclohexane under argon in a sealed 40 mL glass vial. The solution was irradiated at 350 nm while stirred at room temperature (rt) for 16 h. Volatiles were removed in vacuo, and the crude residue was purified by flash chromatography (0–10% EtOAc in hexanes) to afford cycloadduct 5 (1.83 g, 83%) as a colorless foam: Rf = 0.52, 3:7 ether–hexanes; 1H NMR (400 MHz, CDCl3) δ 7.6–7.3 (m, 10H, H-Ph), 5.69 (d, J = 4.6 Hz, 1H, H–C1), 5.20 (dd, J = 4.6, 6.6 Hz, 1H, H–C2), 4.70 (br. s, 4H, H2C), 4.43 (apt. q, J = 6.4 Hz, 1H, H–C3), 3.90 (dd, J = 9.3, 7.0 Hz, 1H, H–C4), 3.80 (dd, J = 9.4, 5.9 Hz, 1H, H–C4), 1.07 (s, 9H, H3C); 13C NMR (100 MHz, CDCl3) δ 135.8, 135.8, 135.3, 134.9, 132.2, 130.4, 130.3, 129.8, 128.1, 128.1, 127.9, 99.3, 95.2, 94.2, 74.2, 26.9, 26.7, 19.2; HRMS (m/z): [M + H+]+ calcd. for C26H28Cl6N2O6Si, 702.9921, obsd. 702.9895.
Bis(2,2,2-trichloroethyl) 1-((3R,4R)-2-(2-Acetamido-6-((diphenylcarbamoyl)-oxy)-9H-purin-9-yl)-4-((tert-butyldiphenylsilyl)oxy)tetrahydrofuran-3-yl)hydrazi-ne-1,2-dicarboxylate (6g)
N2-Acetyl-O6-(diphenylcarbamoyl)-guanine (2.15 g, 5.54 mmol) was suspend in anhydrous acetonitrile (40 mL) under argon. Bis(trimethylsilyl)acetamide (2.8 mL, 11.5 mmol) was added, and the suspension was stirred at 80 °C for 10 min to dissolve the silylated guanine. The solution was cooled to 0 °C, and then, cycloadduct 5 (3.26 g, 4.62 mmol) was added in acetonitrile (30 mL) followed by the dropwise addition of TMSOTf (1.08 mL, 5.98 mmol). The reaction was stirred at 0 °C for 15 min and then at rt for 1.5 h. Volatiles were removed in vacuo, and the residue was dissolved in EtOAc (100 mL), washed with 5% NaHCO3 (2 × 100 mL), washed with brine (100 mL), dried over MgSO4, filtered, and concentrated in vacuo to a crude. The crude was purified by flash chromatography (20–60% EtOAc in hexanes) to afford protected hydrazide 6g as a white foam (4.01 g, 79%). Rf = 0.72, 6% MeOH in CH2Cl2; 1H NMR (400 MHz, DMSO-d6, 40 °C) Note: broad multiplets were observed due to stable rotamers δ 8.47 (br d, 1H, H–C8), 7.77–7.32 (m, 20H, H-Ph), 6.20 (s, 1H, H–C1′), 5.65 (br s, 1H, H–C3′), 5.00–4.30 (br m, 6H, H–C4′ and H-Troc), 3.87 (br m, 1H, H–C2′), 2.29 (br s, 3H, H–Ac), 1.04 (br s, 9H, H-tBu); 13C NMR (100 MHz, DMSO-d6, 60 °C) δ 169.9, 155.2, 153.8, 152.1, 149.7, 143.9, 143.5, 141.4, 135.5, 135.3, 135.0, 132.4, 132.1, 130.0, 129.9, 129.6, 129.5, 129.3, 128.8, 127.7, 127.3, 126.8, 126.4, 120.3, 95.1, 94.5, 74.9, 59.4, 54.5, 26.5, 26.3, 18.4; HRMS (m/z): [M + H+]+ calcd. for C46H44Cl6N8O9Si, 1091.1204, obsd. 1091.1254.
2-Acetamido-9-((3R,4R)-3-amino-4-((tert-butyl-diphenylsilyl)oxy)tetrahydro-furan-2-yl)-9H-purin-6-yl diphenylcarbamate (7g)
Protected hydrazide 6g (4.54 g, 4.15 mmol), indium powder (1.90 g, 16.6 mmol), and aluminum foil (1.29 g, 47.9 mmol) were suspended in EtOH (67 mL) and sat. aq. NH4Cl (33 mL) and refluxed under argon with vigorous stirring for 3 h. The hot reaction mixture was then filtered through a short bed of Celite, diluted with water (200 mL), adjusted to pH 11 with 1 M NaOH, and then, extracted with CH2Cl2 (3 × 100 mL). The combined organic phase was dried over MgSO4, filtered, and concentrated in vacuo to a crude. The crude was purified by flash chromatography (0–4% MeOH in CH2Cl2) to afford protected aminonucleoside 7g as a white foam (1.35 g, 45%). Rf = 0.29, 6% MeOH in CH2Cl2; 1H NMR (400 MHz, DMSO-d6) δ 8.56 (s, 1H, H–C8), 7.57–7.27 (m, 20H, H-Ph), 5.80 (d, J = 2.7 Hz, 1H, H–C1′), 4.17 (dd, J = 7.4, 3.3 Hz, 1H, H–C3′), 4.11 (dd, J = 9.1, 3.1 Hz, 1H, H–C4′), 4.04 (dd, J = 9.4, 5.1 Hz, 1H, H–C4′), 4.01 (m, 1H, H–C2′), 2.22 (s, 3H, H–Ac), 0.88 (s, 9H, H-tBu); 13C NMR (100 MHz, DMSO-d6) δ 169.1, 155.1, 154.2, 152.1, 150.1, 143.5, 141.7, 135.3, 135.2, 132.6, 132.6, 130.1, 130.0, 128.0, 127.9, 120.4, 91.3, 78.4, 74.7, 63.6, 26.6, 24.8, 18.5; HRMS (m/z): [M + H+]+ calcd. for C40H41N7O5Si, 728.3011, obsd. 728.2943.
9-(2′-Amino-2′-deoxy-α-l-threofuranosyl)guanine (8g)
Protected aminonucleoside 7g (234 mg, 321 mmol) was dissolved in THF (15 mL) under argon. A 1 M solution of TBAF in THF (0.48 mL, 480 mmol) was added, and the reaction was stirred at rt for 3 h. Volatiles were removed in vacuo, and the residue was transferred to a pressure vessel in MeOH (5 mL), 28% NH4OH (15 mL) was added, and the sealed flask was heated at 35 °C for 22 h. Volatiles were removed in vacuo, and the residue was purified by flash chromatography (2–20% MeOH in CH2Cl2) to afford aminonucleoside 8g (50 mg, 62%) as a white solid. Rf = 0.04, 3:7 methanol–dichloromethane; 1H NMR (400 MHz, DMSO-d6) δ 7.88 (s, 1H, H–C8), 5.54 (d, J = 3.4 Hz, 1H, H–C1′), 4.06 (dd, J = 8.8, 4.8 Hz, 1H, H–C4′), 4.00 (m, 1H, H–C3′), 3.89 (dd, J = 8.8, 3.2 Hz, 1H, H–C4′), 3.55 (apt. t, J = 3.1 Hz, 1H, H–C2′); 13C NMR (100 MHz, DMSO-d6) δ 157.7, 154.5, 152.0, 137.0, 117.4, 90.5, 77.4, 75.0, 65.0; HRMS (m/z): [M + H+]+ calcd. for C9H12N6O3, 253.1044, obsd. 253.1045. Cambridge Crystallographic Data Centre deposition number 967793.
Bis(2,2,2-trichloroethyl)-1-((3R,4R)-4-((tert-butyl-diphenylsilyl)oxy)-2-(5-methyl-2,4-dioxo-3,4-dihydro-pyrimidin-1(2H)-yl)tetrahydrofuran-3-yl)hydrazine-1,2-dicarboxylate (6t)
Thymine (1.25 g, 9.91 mmol) and cycloadduct 5 were suspended in anhydrous acetonitrile (76 mL) under argon. Bis(trimethylsilyl)acetamide (7.25 mL, 29.7 mmol) was added, and the suspension was stirred at 80 °C for 10 min to dissolve the silylated thymine. The solution was cooled to 0 °C, and then, TMSOTf (1.24 mL, 6.86 mmol) was added dropwise. The reaction was stirred at 0 °C for 30 min and then at rt for 45 min. Volatiles were removed in vacuo, and the residue was dissolved in CH2Cl2 (400 mL) and washed with 5% NaHCO3 (300 mL). The aqueous phase was extracted further with CH2Cl2 (2 × 100 mL), dried over MgSO4, filtered, and concentrated in vacuo to a crude. The crude was purified by flash chromatography (0–2% MeOH in CH2Cl2) to afford protected hydrazide 6t as a white foam (3.71 g, 91%). Rf = 0.40, 5% MeOH in CH2Cl2; 1H NMR (400 MHz, DMSO-d6, 50 °C) δ 11.30 (s, 1H, H–N3), 10.41 (br s, 1H, H–N-Troc), 7.63–7.35 (m, 11H, H-Ph and H–C6), 6.07 (br s, 1H, H–C1′), 5.00–4.65 (m, 6H, H2–CCCl3 and H2–C4′), 4.07 (app d, J = 9.3 Hz, H–C3′), 3.76 (br s, 1H, H–C2′), 1.73 (s, 3H, H2–CC5), 1.04 (s, 9H, H-tBu); 13C NMR (100 MHz, DMSO-d6) δ 163.8, 155.0, 153.0, 150.2, 135.2, 132.4, 130.2, 128.0, 95.4, 95.0, 83.4, 75.2, 73.9, 70.8, 26.6, 18.6, 12.2; HRMS (m/z): [M + H+]+ calcd. for C31H34Cl6N4O8Si, 829.0350, obsd. 829.0341.
1-((3R,4R)-3-Amino-4-((tert-butyldiphenylsilyl)oxy)-tetrahydrofuran-2-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (7t)
Protected hydrazide 6t (250 mg, 301 μmol) was dissolved in glacial acetic acid (10 mL) under argon. Zinc powder (950 mg) was added, and the suspension was stirred vigorously for 1 h at rt. Acetone (0.04 mL, 544 μmol) was added, and the reaction was stirred for another 1 h. Finally, more zinc powder (750 mg) was added, and the reaction was stirred for another 4 h. The suspension was filtered, washed with MeOH, concentrated in vacuo, and coevaporated with toluene to obtain a crude foam. The crude was purified by flash chromatography (0 to 2% MeOH in CH2Cl2 with 0.1% Et3N to afford protected nucleoside 7t as a white foam (99 mg, 71%). Rf = 0.35, 6% MeOH in CH2Cl2; 1H NMR (400 MHz, DMSO-d6, 40 °C) δ 7.62–7.42 (m, 11H, H-Ph and H–C6), 5.50 (d, J = 2.1 Hz, H–C1′), 4.10–4.00 (m, 3H, H–C4′ and H–C3′), 3.43 (m, 1H, H–C2′), 1.73 (d, J = 1.2, H3C–C5), 0.98 (s, 9H, H-tBu); 13C NMR (100 MHz, DMSO-d6) δ 163.9, 150.5, 136.4, 135.2, 132.6, 130.0, 128.0, 108.3, 92.6, 78.5, 75.4, 64.4, 26.5, 18.6, 12.3; ESI-MS (m/z): [M + H+]+ calcd. for C25H31N3O4Si, 466.2, obsd. 466.1.
1-(2′-Amino-2′-deoxy-α-l-threofuranosyl)thymine (8t)
Protected nucleoside 7t (203 mg, 450 μmol) was dissolved in anhydrous THF (15 mL) under argon. A 1 M solution of tetrabutylammonium fluoride in THF (0.67 mL, 670 μmol) was added, and the solution was stirred at rt for 3 h. The solution was concentrated in vacuo, and the crude was purified by flash chromatography (2–8% MeOH in CH2Cl2) and then precipitated from ether and trituration with CH2Cl2 to remove excess tetrabutylammonium to afford nucleoside 8t as a white powder (70 mg, 68%). Rf = 0.08, 8% MeOH in CH2Cl2; 1H NMR (400 MHz, DMSO-d6) δ 7.61 (d, J = 1.2 Hz, 1H, H–C6), 5.55 (d, J = 3.1 Hz, 1H, H–C1′), 4.04 (dd, J = 5.0, 9.7 Hz, 1H, H–C4′), 3.93 (m, J = 2.5 Hz, 1-H, H–C4′), 3.92 (m, 1H, H–C3′), 3.23 (dd, J = 2.7 Hz, 1H, H–C2′), 1.76 (d, J = 1.2 Hz, 3H, H3C–C5); 13C NMR (100 MHz, DMSO-d6) δ 163.9, 150.7, 137.1, 108.3, 91.8, 75.9, 74.5, 64.0, 12.3; HRMS (m/z): [M + H+]+ calcd. for C9H13N3O4, 228.0979, obsd. 228.0990. Cambridge Crystallographic Data Centre deposition number 967794.
Acknowledgments
We thank Dr. John Chaput for the TNA template, Dr. Sergei Gryaznov for the primer oligonucleotide, and members of our laboratory for helpful discussions and comments on the manuscript. We also thank Dr. Shao-Liang Zheng for his help with X-ray data collection and structure determination. This research was funded in part by grant CHE-0809413 from the National Sciences Foundation. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Supporting Information Available
Scheme of the synthesis of the activated 2′-NH2-TNA nucleotide 2-MeImpntG, figure of the nonenzymatic primer extension and hydrolysis of 2-MeImpntG in the absence of HEI with associated procedures, thermal ellipsis representations of X-ray crystal structures, experimental procedures and characterization data for the synthesis of 2-MeImpG, experimental procedures for X-ray crystallography, and X-ray crystallography data in cif file format. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
† Ra Pharmaceuticals, One Kendall Square, Suite B14301, Cambridge, Massachusetts 02139, United States.
The authors declare no competing financial interest.
Supplementary Material
References
- Szostak J. W.; Bartel D. P.; Luisi P. L. Nature 2001, 409, 387–390. [DOI] [PubMed] [Google Scholar]
- Zhu T. F.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 5705–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincoln T. A.; Joyce G. F. Science 2009, 323, 1229–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaidya N.; Manapat M. L.; Chen I. A.; Xulvi-Brunet R.; Hayden E. J.; Lehman N. Nature 2012, 491, 72–77. [DOI] [PubMed] [Google Scholar]
- Wochner A.; Attwater J.; Coulson A.; Holliger P. Science 2011, 332, 209–212. [DOI] [PubMed] [Google Scholar]
- Inoue T.; Orgel L. E. Science 1983, 219, 859–862. [DOI] [PubMed] [Google Scholar]
- Lohrmann R.; Orgel L. E. Nature 1976, 261, 342–344. [DOI] [PubMed] [Google Scholar]
- Wu T. F.; Orgel L. E. J. Am. Chem. Soc. 1992, 114, 317–322. [DOI] [PubMed] [Google Scholar]
- Kaiser A.; Richert C. J. Org. Chem. 2013, 78, 793–799. [DOI] [PubMed] [Google Scholar]
- Kaiser A.; Spies S.; Lommel T.; Richert C. Angew. Chem., Int. Ed. 2012, 51, 8299–8303. [DOI] [PubMed] [Google Scholar]
- Röthlingshöfer M.; Kervio E.; Lommel T.; Plutowski U.; Hochgesand A.; Richert C. Angew. Chem., Int. Ed. 2008, 47, 6065–6068. [DOI] [PubMed] [Google Scholar]
- Chen J. J.; Cai X.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 2119–2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrum J. P.; Ricardo A.; Krishnamurthy M.; Blain J. C.; Szostak J. W. J. Am. Chem. Soc. 2009, 131, 14560–14570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Zhang N.; Blain J. C.; Szostak J. W. J. Am. Chem. Soc. 2013, 135, 924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansy S. S.; Schrum J. P.; Krishnamurthy M.; Tobé S.; Treco D. A.; Szostak J. W. Nature 2008, 454, 122–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschenmoser A. Science 1999, 284, 2118–2124. [DOI] [PubMed] [Google Scholar]
- Schoning K.; Scholz P.; Guntha S.; Wu X.; Krishnamurthy R.; Eschenmoser A. Science 2000, 290, 1347–1351. [DOI] [PubMed] [Google Scholar]
- Schoning K.; Scholz P.; Wu X.; Guntha S.; Delgado G.; Krishnamurthy R.; Eschenmoser A. Helv. Chim. Acta 2002, 85, 4111–4153. [Google Scholar]
- Kim H.-J.; Ricardo A.; Illangkoon H. I.; Kim M. J.; Carrigan M. A.; Frye F.; Benner S. A. J. Am. Chem. Soc. 2011, 133, 9457–9468. [DOI] [PubMed] [Google Scholar]
- Islam S.; Aguilar J. A.; Powner M. W.; Nilsson M.; Morris G. A.; Sutherland J. D. Chemistry 2013, 19, 4586–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgel L. E. Science 2000, 290, 1306–1307. [DOI] [PubMed] [Google Scholar]
- Heuberger B. D.; Switzer C. Org. Lett. 2006, 8, 5809–5811. [DOI] [PubMed] [Google Scholar]
- Ichida J.; Zou K.; Horhota A.; Yu B.; McLaughlin L.; Szostak J. W. J. Am. Chem. Soc. 2005, 127, 2802–2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Zhang S.; Chaput J. C. Nat. Chem. 2012, 4, 183–187. [DOI] [PubMed] [Google Scholar]
- Chaput J. C.; Szostak J. W. J. Am. Chem. Soc. 2003, 125, 9274–9275. [DOI] [PubMed] [Google Scholar]
- Chaput J. C.; Ichida J. K.; Szostak J. W. J. Am. Chem. Soc. 2003, 125, 856–857. [DOI] [PubMed] [Google Scholar]
- Ichida J.; Horhota A.; Zou K.; McLaughlin L.; Szostak J. W. Nucleic Acids Res. 2005, 33, 5219–5225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro V. B.; Taylor A. I.; Cozens C.; Abramov M.; Renders M.; Zhang S.; Chaput J. C.; Wengel J.; Peak-Chew S.-Y.; McLaughlin S. H.; Herdewijn P.; Holliger P. Science 2012, 336, 341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H.; Zhang S.; Dunn M. R.; Chaput J. C. J. Am. Chem. Soc. 2013, 135, 3583–3591. [DOI] [PubMed] [Google Scholar]
- Ebert M.-O.; Mang C.; Krishnamurthy R.; Eschenmoser A.; Jaun B. J. Am. Chem. Soc. 2008, 130, 15105–15115. [DOI] [PubMed] [Google Scholar]
- Pallan P. S.; Wilds C. J.; Wawrzak Z.; Krishnamurthy R.; Eschenmoser A.; Egli M. Angew. Chem., Int. Ed. 2003, 42, 5893–5895. [DOI] [PubMed] [Google Scholar]
- Wilds C.; Wawrzak Z.; Krishnamurthy R.; Eschenmoser A.; Egli M. J. Am. Chem. Soc. 2002, 124, 13716–13721. [DOI] [PubMed] [Google Scholar]
- Wu X.; Guntha S.; Ferencic M.; Krishnamurthy R.; Eschenmoser A. Org. Lett. 2002, 4, 1279–1282. [DOI] [PubMed] [Google Scholar]
- Ferencic M.; Reddy G.; Wu X.; Guntha S.; Nandy J.; Krishnamurthy R.; Eschenmoser A. Chem. Biodiversity 2004, 1, 939–979. [DOI] [PubMed] [Google Scholar]
- Kozlov I.; Politis P.; Van Aerschot A.; Busson R.; Herdewijn P.; Orgel L. E. J. Am. Chem. Soc. 1999, 121, 2653–2656. [DOI] [PubMed] [Google Scholar]
- Kozlov I.; Zielinski M.; Allart B.; Kerremans L.; Van Aerschot A.; Busson R.; Herdewijn P.; Orgel L. E. Chem.—Eur. J. 2000, 6, 151–155. [DOI] [PubMed] [Google Scholar]
- Wu X.; Delgado G.; Krishnamurthy R.; Eschenmoser A. Org. Lett. 2002, 4, 1283–1286. [DOI] [PubMed] [Google Scholar]
- Fitzsimmons B. J.; Leblanc Y.; Rokach J. J. Am. Chem. Soc. 1987, 109, 285–286. [Google Scholar]
- Leblanc Y.; Fitzsimmons B. J. Tetrahedron Lett. 1989, 30, 2889–2892. [Google Scholar]
- Leblanc Y.; Fitzsimmons B. J.; Springer J. P.; Rokach J. J. Am. Chem. Soc. 1989, 111, 2995–3000. [Google Scholar]
- Koerner von Gustorf E.; White D. V.; Kim B.; Hess D.; Leitich J. J. Org. Chem. 1970, 35, 1155–1165. [Google Scholar]
- Wright G. E.; Dudycz L. W. J. Med. Chem. 1984, 27, 175–181. [DOI] [PubMed] [Google Scholar]
- Robins M. J.; Zou R. M.; Guo Z. Q.; Wnuk S. J. Org. Chem. 1996, 61, 9207–9212. [Google Scholar]
- Valluri M.; Mineno T.; Hindupur R.; Avery M. Tetrahedron Lett. 2001, 42, 7153–7154. [Google Scholar]
- Mineno T.; Choi S.; Avery M. Synlett 2002, 883–886. [Google Scholar]
- Cicchi S.; Bonanni M.; Cardona F.; Revuelta J.; Goti A. Org. Lett. 2003, 5, 1773–1776. [DOI] [PubMed] [Google Scholar]
- Altona C.; Sundaralingam M. J. Am. Chem. Soc. 1972, 94, 8205–8212. [DOI] [PubMed] [Google Scholar]
- Joyce G. F.; Inoue T.; Orgel L. E. J. Mol. Biol. 1984, 176, 279–306. [DOI] [PubMed] [Google Scholar]
- Lichtenthaler F.; Weimer T.; Immel S. Tetrahedron: Asymmetry 2004, 15, 2703–2709. [Google Scholar]
- Gottlieb H.; Kotlyar V.; Nudelman A. J. Org. Chem. 1997, 62, 7512–7515. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.