

Abstract
Purpose
To identify the spectrum and frequency of five candidate genes in Chinese patients with congenital ectopia lentis (EL).
Methods
Forty consecutive and unrelated congenital probands with EL were collected and underwent ocular, skeletal, and cardiovascular examinations. Sanger sequencing was used to analyze all of the coding and adjacent regions of five candidate genes: FBN1, ADAMTS10, ADAMTSL4, TGFBR2, and CBS. Mutation analysis was performed to evaluate the pathogenic variants and to identify the cause of congenital EL.
Results
The FBN1 gene screen revealed 25 pathogenic variants in 34 of the 40 families with congenital EL, including three novel (c.1955G>T, c.2222delA, and c.4381T>C) and 22 known mutations. The ADAMTSL10 gene screen revealed a compound heterozygous variant (c.1586G>A and c.2485T>A) in a family with Weill-Marchesani syndrome (WMS). In the remaining five probands, no pathogenic variant was detected in any of the five screened genes.
Conclusions
In this study, we identified three novel and 22 known mutations in FBN1 in 34 of 40 EL families. The results expand the mutation spectrum of the FBN1 gene and suggest that FBN1 mutations may be the major cause of congenital EL in Chinese patients.
Introduction
Congenital ectopia lentis (EL) is the most common form of lenticular dislocation and is a result of partial zonular dysplasia. EL can manifest mildly or severely, with symptoms ranging from refractive errors to amblyopia, complicated glaucoma, and retinal detachment, and can seriously impair visual quality. EL is the second most prevalent reason for lens surgery after cataracts, particularly in juveniles [1]. According to a nationwide survey in Denmark, the estimated prevalence rate is 6.4/100,000 [2]. Congenital EL can be broadly divided into two groups: isolated forms and syndromic forms. Isolated EL mainly includes simple dominant EL (ECTOL1; OMIM# 129600) and EL et pupillae (ECTOL2; OMIM# 225200). Syndromic EL mainly includes Marfan syndrome (MFS; OMIM# 154700), homocystinuria (HCU; OMIM# 236200), Weill-Marchesani syndrome (WMS; OMIM# 277600), and sulfite oxidase deficiency (SOD; OMIM# 272300) [3].
The fibrillin-1 (FBN1; OMIM# 134797) gene is well-known to be linked with MFS [4], and FBN1 mutations could be present in up to 60% of patients with congenital EL [5]. Furthermore, the transforming growth factor beta receptor II (TGFBR2; OMIM# 190182) gene has been identified as a pathogenic gene in Loeys-Dietz-syndrome type 2B (OMIM# 610380), known as MFS type 2 (MFS2; OMIM# 154705), which causes serious cardiovascular problems but only mild ocular tissue problems [6]. Mutations in the ADAMTSL4 gene (OMIM# 610113) were identified in 2009 in patients with autosomal recessive inherited EL et pupillae [7], which was defined as an ADAMTSL4-related eye disorder, and is the second most common cause of congenital EL in the Caucasian population [8,9]. Simple dominant EL is also due to FBN1 gene mutations but does not involve other organs [10]. In addition, WMS is either linked to FBN1 and inherited in an autosomal-dominant manner or to ADAMTS10 (OMIM# 608990) in an autosomal-recessive manner [11,12]. HCU is linked to the cystathionine-beta-synthase gene (CBS; OMIM# 613381) in autosomal recessive inheritance [13]. Molecular genetic studies have shown that congenital EL has a common pathological basis and is associated with defects in microfibril assembly [14,15]. The ocular zonule consists of microfibrils, with fibrillin-1 as the chief molecular component. Abnormal fibrinogenesis could result in zonular dysplasia caused by the FBN1 mutations. The ADAMTS protein promotes fibrillin-1 assembly including ADAMTSL6, ADAMTS10, and ADAMTSL4 [16], whereas homocysteine reduces the deposition of the fibrillin-1 network; thus, mutations in the ADAMTS and CBS genes might manifest as EL [15]. In addition, microfibrils regulate the bioavailability of TGFβ superfamily growth factors, such as TGFBR2, that are involved in this signaling pathway and rarely result in EL [17].
The Denmark survey for the nosologic characteristics of congenital EL showed that MFS accounted for 68.2% of patients who had an established clinical diagnosis, EL et pupillae for 21.2%, simple dominant EL for 8.0%, HCU for 1.1%, and SOD and WMS for 0.7% each [2]. However, the systemic evaluation of variants in these genes in a cohort of patients is rare, particularly in China. In our current study, five candidate genes were analyzed in 40 probands with congenital EL to identify the spectrum and frequency of candidate genes in Chinese patients with congenital EL.
Methods
Patients
Forty consecutive and unrelated congenital EL patients were collected from the Pediatric and Genetic Clinic of Zhongshan Ophthalmic Center between June 1997 and August 2013. Written informed consent conforming to the tenets of the Declaration of Helsinki was obtained from the participants or their guardians before the study. Ethical approval was obtained from the Institutional Review Board of Zhongshan Ophthalmic Center. An ophthalmological examination including visual acuity assessment, intraocular pressure (IOP), slit-lamp biomicroscopy, ophthalmoscopic observation, and retinoscopy with cycloplegia was performed. The systemic examination included measurements of arm span and height and a skeletal examination. For each patient and affected relative, an echocardiography screen to detect cardiovascular diseases was suggested. Patients with traumatic and idiopathic lenticular dislocation were excluded.
Mutation screening
Genomic DNA was extracted from the leukocytes of a peripheral blood sample from each participant as previously described [18]. Briefly, Genomic DNA was prepared from leukocytes of peripheral venous blood by whole blood lysis, followed by phenol-chloroform extraction and ethanol precipitation. The DNA pellet was dissolved in TE buffer (pH 8.0).The candidate genes in our analysis were evaluated in the following order: FBN1 (NM_000138.3), ADAMTS10 (NM_030957.2), ADAMTSL4 (NM_019032.4), TGFBR2 (NM_003242.5), and CBS (NM_000071.2). The primer pairs were designed using the primer3 online tool. The primer sequences of the five genes and their optimal annealing temperatures are listed in Appendix 1. The products from individual exons were sequenced using the ABI BigDye Terminator cycle sequencing kit v3.1 and an ABI 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). The sequencing results were aligned with consensus sequences to identify variations using a sequencing program (Lasergene SeqMan II; DNAStar, Madison, WI). Every variant was confirmed through repeated sequencing, and its frequency was determined from the exome variant server (EVS) and 1000 Genomes database. Any variation detected in our patients was further evaluated as a control by sequencing 96 normal individuals. The effect of a novel missense mutation on the encoded protein was predicted using the PolyPhen-2 [19] and SIFT [20] online tools. Splice-site mutations were predicted using the Splice Site Prediction program by Neural Network [21]. Segregation analyses of mutations were performed on patients with available family members.
Results
Fibrillin-1 mutations
The FBN1 gene screen identified 25 pathogenic variants in 34 of the 40 congenital EL families. Among them, 22 had been previously reported, and three were novel (c.1955G>T, c.2222delA, and c.4381T>C; Appendix 2). Five known variants (c.184C>T, c.364C>T, c.1916G>A, c.1633C>T, and c.5788+5G>A) were found repeatedly in 14 unrelated families. The variants were evaluated using PolyPhen-2 and SIFT, and all were most likely pathogenic (Appendix 2). In addition, the novel variants were not found in the EVS and 1000 Genomes cohorts. The two novel missense mutations, c.1955G>T (p.Cys652Phe) in proband QT1252 and c.4381T>C (p.Cys1461Arg) in proband QT753 resulted in missense mutations, both involving a change in highly conserved cysteine residues that form a disulfide bond in the cbEGF-like domain; these mutated alleles might lead to incorrectly folded monomers. The mother of QT1252 carried this variant and manifested EL without cardiovascular symptoms. The QT753 proband was a sporadic case of EL, and his normal parents lacked this gene variant. When he was eight years old, his echocardiography was normal. However, five years later, echocardiography detected a serious aortic dilation (Figure 1). The other novel mutation, c.2222delA (p.Asn741Thrfs*31) in proband QT1228, was located in exon 18 and resulted in a premature termination codon (PTC) in the next exon after coding 31 amino acids (Appendix 2). Proband QT1228 had only EL with mild valvular regurgitation. His mother, brother, and cousin had no EL but had scoliosis. These four family members had mild regurgitation, but the uncle of the proband had an aortic replacement operation. None of the three novel mutations were present in the 96 normal controls. The pedigrees and cosegregation analyses of the three families with novel mutations are shown in Figure 2A.
Figure 1.
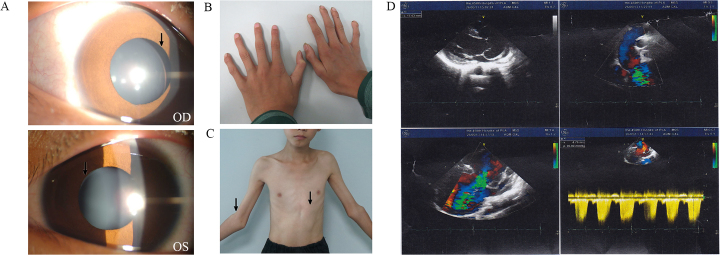
Clinical phenotype in a proband with MFS. The picture shows the ocular, skeletal and cardiovascular features of the proband in family QT753. (A) shows the ectopia lentis. (B) shows the arachnodactyly. (C) show the cubitus valgus and the pectus carinatum. D: The echocardiography of this patient was normal when he was 8 years old. But five years later, when the he was 13 years old, echocardiography showed an aortic diameter of 5.8 cm at the sinuses of Valsalva with severe aortic valve insufficiency.
Figure 2.

Novel mutations identified in FBN1 and ADAMTS10 genes. A: Sequence chromatography of the novel mutation in FBN1 and the corresponding normal sequence is shown in the left column; the genotype analysis of the pedigree is shown in the right column. B: Sequence changes of QT401 in ADAMTS10 are shown in the left column; the genotype analysis of the pedigree is shown in the right column.
Other gene mutations
The ADAMTS10 gene screen revealed a novel compound heterozygous variant, c.1586G>A (p.Gly529Glu) and c.2485T>A (p.Trp829Arg) in a family with WMS. Bioinformatic analysis demonstrated that both variants are pathogenic and that neither is present in EVS and 1000 Genomes. The mutation cosegregated with the disease, as shown in Figure 2B. No variations were observed in the normal controls with direct sequencing. The clinical phenotype of the family showed that the proband is a 14-year-old boy who was only 1.36 m tall, and his affected sister is 12 years old and only 1.25 m tall. Both siblings have microspherophakia, brachydactyly, and elbow joint stiffness. However, his normal brother is 1.43 m tall at 10 years old and lacks these abnormities. The parents of the proband have brachydactyly without other abnormities (Figure 3). For the remaining five patients, no pathogenic variants were detected in the FBN1, ADAMTS10, ADAMTSL4, TGFBR2, and CBS genes (Appendix 3).
Figure 3.

Clinical phenotype of the Weill-Marchesani syndrome family. Clinical phenotype of family QT401 with Weill-Marchesani syndrome (A) ectopia lentis in QT401 II:1; (B) normal finger in QT401II3 and brachydactyly in QT401I:1, I:2, II:1, and II:2; (C) microspherophakia in QT401 II:1 and II:2 as shown by anterior segment optical coherence tomography; (D) short stature in QT401 II:1.
Clinical data
Detailed family histories and clinical data were analyzed for the 40 unrelated patients with congenital EL, showing that 27 had a family history, and 13 were sporadic cases. More than half of the probands (21/40) had poor vision from early childhood or under the age of 5 years, and all of the patients exhibited bilateral congenital EL (Appendix 2). The mean age of referral for ophthalmology, genetics consultation, and EL diagnosis in our patient group was approximately 8.0 years old (ranging from 2 to 33 years). When the FBN1 gene mutation is considered [22], 34 patients might be diagnosed with MFS, including five mutation-negative patients; three patients with c.188A>G or c.1916G>A were isolated EL, two patients with c.1955G>T or c.3083A>G were incomplete MFS, and one was WMS (Appendix 2 and Appendix 3).
Discussion
In this study, we detected 34 FBN1 mutations in 40 congenital EL families, including 22 known and three novel mutations. Thirty-four of the 40 patients had MFS, two patients manifested incomplete MFS, and three patients had isolated EL. Interestingly, the c.2413T>C mutation was reported to be associated with incomplete MFS and to manifest only as an aortic aneurysm [23]. However, the clinical phenotype in our study found that the mutation involved the eyes and skeleton but not the cardiovascular system and that the diagnosis could be classical MFS. This finding suggests that there is high variability among different families. According to the revised Ghent nosology, a diagnosis of MFS could be obtained in 71% of patients after the mutation was identified in the FBN1 gene; in contrast, only 59% of patients were diagnosed using the older criteria [24]. Our study showed that most of congenital EL was FBN1-associated (34/40) and fulfilled the revised Ghent criteria (34/40) in Chinese patients. This percentage is much higher than in Caucasian individuals [2,9]. In our study, the genetic screening was performed with Sanger sequencing, and therefore, large-scale genomic rearrangements could not be excluded due to the limitations of this method, so the FBN1 gene mutation is not excluded in the patients with FBN1-negative EL. In addition, we found a novel compound heterozygous mutation (c.1586G>A and c.2485T>A) in the ADAMTS10 gene. The family members QT401 I 1 and QT401 I 2, who carried only a single heterozygous mutation, presented with certain mild clinical manifestations of WMS such as brachydactyly but lacked other significant abnormalities. Before the ADAMTS10 mutation was identified, there had been reports that some heterozygotes with recessive WMS presented with certain mild clinical manifestations of the disease [25], although no pedigrees with a definite gene mutation have been reported. Because ADAMTS10-associated WMS is inherited in an autosomal-recessive manner, the brachydaktly in the affected individual in the parental generation is probably a phenocopy.
The fibrillin-1 gene contains 65 exons encoding a glycoprotein of 2871 amino acids. To date, almost 1,500 mutations in the FBN1 gene have been included in HGMD (The Human Gene Mutation Database). Epidermal growth factor-like (EGF) domains, the most frequent motif in fibrillin-1, have six highly conserved cysteine residues that form disulfide bonds with each other. Creating or eliminating these cysteine residues leads to incorrect folding of the EGF-like domain structure [26]. An international study of 1,013 MFS cases with known FBN1 mutations revealed an association between cysteine mutations and EL. Moreover, missense mutations in the 5′ region had a higher probability of combined EL. Premature termination codon mutations are more frequent in patients with major skeletal involvement [27]. In a summary of the FBN1 mutations identified in our study, loci in exons 1–21 accounted for 60.0% (15/25), loci in EGF domains for 72.0% (18/25), and cysteine changes for 76.0% (19/25). Our results are in line with earlier reports and may point an important role of the cysteine residues in maintaining the function of the suspensory ligaments. The clinical features of family QT1228, which has a PTC mutation, confirm that PTC mutations are mainly associated with skeletal involvement and suggest high variability even within the same family. Various mutations have been shown to cause a phenotypic continuum. For example, patients with isolated EL or EL syndrome (ELS) have milder findings. Phenotype–genotype correlations regarding FBN1 mutations would be helpful in determining the appropriate medical management.
The diagnosis of MFS in young children is difficult, especially in sporadic cases, because of the great variability in the expression of the disease and because the phenotype evolves over the lifetime. EL, the only ocular manifestation considered a major diagnostic criterion, is present in 60% of patients with MFS. Lens dislocation usually occurs early in life with MFS and is best assessed by an ophthalmologist with a slit-lamp examination with dilated pupils. Our study shows that more than half of patients with MFS acquired EL before the age of 5 or during early childhood and that all of these patients visited our clinic with poor vision as the initial symptom. The detection of FBN1 mutations in these patients was 85.0% (34/40), much higher than in patients with aortic aneurysms or dissections [28]. A previous study of the MFS phenotype evolution during childhood showed that the prevalence of EL remained stable from early childhood and that aortic root dilatation remained stable when receiving β-blocker therapy, whereas the prevalence of skeletal features changed with age [29]. These findings suggested that EL has a stronger correlation with FBN1 mutations than aortic diseases in young patients with MFS. Therefore, congenital EL as one of the primary symptoms has important significance for diagnosing MFS. In addition, the early diagnosis of MFS is essential to establish specific management.
In summary, we analyzed five candidate genes in Chinese congenital EL patients and expanded the mutation spectrum of these genes. We found that most congenital EL cases are associated with FBN1 and could be diagnosed as MFS. Congenital EL remains stable from early childhood and is strongly associated with FBN1 mutations, which strongly supports the MFS diagnosis and can be used to provide preventive treatment for atypical and childhood patients with MFS.
Acknowledgments
We thank the patients and family members for their participation. This study was supported in part by the National Natural Science Foundation of China (81170847; to X. G).
Appendix 1.
Primers used for amplification and sequencing. To access the data, click or select the words “Appendix 1.”
Appendix 2.
Summary of clinical findings and Mutations in patients with FBN1 and ADAMTS10 mutations [23,30-43]. To access the data, click or select the words “Appendix 2.”
Appendix 3.
Clinical features of the five probands without mutation. To access the data, click or select the words “Appendix 3.”
References
- 1.Hing S, Speedwell L, Taylor D. Lens surgery in infancy and childhood. Br J Ophthalmol. 1990;74:73–7. doi: 10.1136/bjo.74.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fuchs J, Rosenberg T. Congenital ectopia lentis. A Danish national survey. Acta Ophthalmol Scand. 1998;76:20–6. doi: 10.1034/j.1600-0420.1998.760105.x. [DOI] [PubMed] [Google Scholar]
- 3.Nelson LB, Maumenee IH. Ectopia lentis. Surv Ophthalmol. 1982;27:143–60. doi: 10.1016/0039-6257(82)90069-8. [DOI] [PubMed] [Google Scholar]
- 4.Maslen CL, Glanville RW. The molecular basis of Marfan syndrome. DNA Cell Biol. 1993;12:561–72. doi: 10.1089/dna.1993.12.561. [DOI] [PubMed] [Google Scholar]
- 5.Maumenee IH. The eye in the Marfan syndrome. Trans Am Ophthalmol Soc. 1981;79:684–733. [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang L, Gao LG, Zhang M, Zhou XL. Genotype-phenotype analysis of F-helix mutations at the kinase domain of TGFBR2, including a type 2 Marfan syndrome familial study. Mol Vis. 2012;18:55–63. [PMC free article] [PubMed] [Google Scholar]
- 7.Ahram D, Sato TS, Kohilan A, Tayeh M, Chen S, Leal S, Al-Salem M, El-Shanti H. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J Hum Genet. 2009;84:274–8. doi: 10.1016/j.ajhg.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aragon-Martin JA, Ahnood D, Charteris DG, Saggar A, Nischal KK, Comeglio P, Chandra A, Child AH, Arno G. Role of ADAMTSL4 mutations in FBN1 mutation-negative ectopia lentis patients. Hum Mutat. 2010;31:E1622–31. doi: 10.1002/humu.21305. [DOI] [PubMed] [Google Scholar]
- 9.Chandra A, Aragon-Martin JA, Hughes K, Gati S, Reddy MA, Deshpande C, Cormack G, Child AH, Charteris DG, Arno G. A genotype-phenotype comparison of ADAMTSL4 and FBN1 in isolated ectopia lentis. Invest Ophthalmol Vis Sci. 2012;53:4889–96. doi: 10.1167/iovs.12-9874. [DOI] [PubMed] [Google Scholar]
- 10.Adès LC, Holman KJ, Brett MS, Edwards MJ, Bennetts B. Ectopia lentis phenotypes and the FBN1 gene. Am J Med Genet A. 2004;126A:284–9. doi: 10.1002/ajmg.a.20605. [DOI] [PubMed] [Google Scholar]
- 11.Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Beroud G, Boileau C, Munnich A, Cormier-Daire V. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40:34–6. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Megarbane A, Alswaid A, Dollfus H, Alembik Y, Munnich A, Legeai-Mallet L, Cormier-Daire V. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75:801–6. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kraus JP. Komrower Lecture. Molecular basis of phenotype expression in homocystinuria. J Inherit Metab Dis. 1994;17:383–90. doi: 10.1007/BF00711354. [DOI] [PubMed] [Google Scholar]
- 14.Hubmacher D, Apte SS. Genetic and functional linkage between ADAMTS superfamily proteins and fibrillin-1: a novel mechanism influencing microfibril assembly and function. Cellular and molecular life sciences. Cell Mol Life Sci. 2011;68:3137–48. doi: 10.1007/s00018-011-0780-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadiq MA, Vanderveen D. Genetics of ectopia lentis. Semin Ophthalmol. 2013;28:313–20. doi: 10.3109/08820538.2013.825276. [DOI] [PubMed] [Google Scholar]
- 16.Gabriel LA, Wang LW, Bader H, Ho JC, Majors AK, Hollyfield JG, Traboulsi EI, Apte SS. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. 2012;53:461–9. doi: 10.1167/iovs.10-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet. 2003;33:331–2. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- 18.Li L, Xiao X, Li S, Jia X, Wang P, Guo X, Jiao X, Zhang Q, Hejtmancik JF. Detection of variants in 15 genes in 87 unrelated Chinese patients with Leber congenital amaurosis. PLoS ONE. 2011;6:e19458. doi: 10.1371/journal.pone.0019458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomarkers. 2010;14:533–7. doi: 10.1089/gtmb.2010.0036. [DOI] [PubMed] [Google Scholar]
- 21.Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-Berhouet S, d'Enghien CD, Lauge A, Castera L, Gauthier-Villars M, Stoppa-Lyonnet D. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29:975–82. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- 22.Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476–85. doi: 10.1136/jmg.2009.072785. [DOI] [PubMed] [Google Scholar]
- 23.Sakai H, Suzuki S, Mizuguchi T, Imoto K, Yamashita Y, Doi H, Kikuchi M, Tsurusaki Y, Saitsu H, Miyake N, Masuda M, Matsumoto N. Rapid detection of gene mutations responsible for non-syndromic aortic aneurysm and dissection using two different methods: resequencing microarray technology and next-generation sequencing. Hum Genet. 2012;131:591–9. doi: 10.1007/s00439-011-1105-7. [DOI] [PubMed] [Google Scholar]
- 24.Laffargue F, Lienhardt-Roussie A, Lacombe D, Delrue MA. Clinical signs of Marfan syndrome in children under 10 years of age. Arch Pediatr. 2013;2013;2020:1193–200. 1193–200. doi: 10.1016/j.arcped.2013.08.009. [DOI] [PubMed] [Google Scholar]
- 25.Faivre L, Dollfus H, Lyonnet S, Alembik Y, Megarbane A, Samples J, Gorlin RJ, Alswaid A, Feingold J, Le Merrer M, Munnich A, Cormier-Daire V. Clinical homogeneity and genetic heterogeneity in Weill-Marchesani syndrome. Am J Med Genet A. 2003;123A:204–7. doi: 10.1002/ajmg.a.20289. [DOI] [PubMed] [Google Scholar]
- 26.Whiteman P, Hutchinson S, Handford PA. Fibrillin-1 misfolding and disease. Antioxid Redox Signal. 2006;8:338–46. doi: 10.1089/ars.2006.8.338. [DOI] [PubMed] [Google Scholar]
- 27.Faivre L, Collod-Beroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Beroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Ades LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–66. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waldmuller S, Muller M, Warnecke H, Rees W, Schols W, Walterbusch G, Ennker J, Scheffold T. Genetic testing in patients with aortic aneurysms/dissections: a novel genotype/phenotype correlation? European Association for Cardio-thoracic Surgery. 2007;31:970–5. doi: 10.1016/j.ejcts.2007.02.027. [DOI] [PubMed] [Google Scholar]
- 29.Stheneur C, Tubach F, Jouneaux M, Roy C, Benoist G, Chevallier B, Boileau C, Jondeau G. Study of phenotype evolution during childhood in Marfan syndrome to improve clinical recognition. Genet Med. 2014;16:246–50. doi: 10.1038/gim.2013.123. [DOI] [PubMed] [Google Scholar]
- 30.Katzke S, Booms P, Tiecke F, Palz M, Pletschacher A, Turkmen S, Neumann LM, Pregla R, Leitner C, Schramm C, Lorenz P, Hagemeier C, Fuchs J, Skovby F, Rosenberg T, Robinson PN. TGGE screening of the entire FBN1 coding sequence in 126 individuals with marfan syndrome and related fibrillinopathies. Hum Mutat. 2002;20:197–208. doi: 10.1002/humu.10112. [DOI] [PubMed] [Google Scholar]
- 31.Comeglio P, Johnson P, Arno G, Brice G, Evans A, Aragon-Martin J, da Silva FP, Kiotsekoglou A, Child A. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat. 2007;28:928. doi: 10.1002/humu.9505. [DOI] [PubMed] [Google Scholar]
- 32.Loeys B, Nuytinck L, Delvaux I, De Bie S, De Paepe A. Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin-1 gene FBN1 because of suspected Marfan syndrome. Arch Intern Med. 2001;161:2447–54. doi: 10.1001/archinte.161.20.2447. [DOI] [PubMed] [Google Scholar]
- 33.Karttunen L, Raghunath M, Lonnqvist L, Peltonen L. A compound-heterozygous Marfan patient: two defective fibrillin alleles result in a lethal phenotype. Am J Hum Genet. 1994;55:1083–91. [PMC free article] [PubMed] [Google Scholar]
- 34.Loeys B, De Backer J, Van Acker P, Wettinck K, Pals G, Nuytinck L, Coucke P, De Paepe A. Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Hum Mutat. 2004;24:140–6. doi: 10.1002/humu.20070. [DOI] [PubMed] [Google Scholar]
- 35.Kilpatrick MW, Lembessis P, Rose E, Tsipouras P. A novel G to A substitution at nucleotide 1734 of the FBN1 gene predicting a C534Y mutation responsible for marfan syndrome. Hum Hered. 1999;49:176–7. doi: 10.1159/000022867. [DOI] [PubMed] [Google Scholar]
- 36.Hayward C, Porteous ME, Brock DJ. Mutation screening of all 65 exons of the fibrillin-1 gene in 60 patients with Marfan syndrome: report of 12 novel mutations. Hum Mutat. 1997;10:280–9. doi: 10.1002/(SICI)1098-1004(1997)10:4<280::AID-HUMU3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 37.Howarth R, Yearwood C, Harvey JF. Application of dHPLC for mutation detection of the fibrillin-1 gene for the diagnosis of Marfan syndrome in a National Health Service Laboratory. Genet Test. 2007;11:146–52. doi: 10.1089/gte.2006.0514. [DOI] [PubMed] [Google Scholar]
- 38.Stheneur C, Collod-Beroud G, Faivre L, Buyck JF, Gouya L, Le Parc JM, Moura B, Muti C, Grandchamp B, Sultan G, Claustres M, Aegerter P, Chevallier B, Jondeau G, Boileau C. Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. European journal of human genetics. Eur J Hum Genet. 2009;17:1121–8. doi: 10.1038/ejhg.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Robinson PN, Booms P, Katzke S, Ladewig M, Neumann L, Palz M, Pregla R, Tiecke F, Rosenberg T. Mutations of FBN1 and genotype-phenotype correlations in Marfan syndrome and related fibrillinopathies. Hum Mutat. 2002;20:153–61. doi: 10.1002/humu.10113. [DOI] [PubMed] [Google Scholar]
- 40.Arbustini E, Grasso M, Ansaldi S, Malattia C, Pilotto A, Porcu E, Disabella E, Marziliano N, Pisani A, Lanzarini L, Mannarino S, Larizza D, Mosconi M, Antoniazzi E, Zoia MC, Meloni G, Magrassi L, Brega A, Bedeschi MF, Torrente I, Mari F, Tavazzi L. Identification of sixty-two novel and twelve known FBN1 mutations in eighty-one unrelated probands with Marfan syndrome and other fibrillinopathies. Hum Mutat. 2005;26:494. doi: 10.1002/humu.9377. [DOI] [PubMed] [Google Scholar]
- 41.Villamizar C, Regalado ES, Fadulu VT, Hasham SN, Gupta P, Willing MC, Kuang SQ, Guo D, Muilenburg A, Yee RW, Fan Y, Towbin J, Coselli JS, LeMaire SA, Milewicz DM. Paucity of skeletal manifestations in Hispanic families with FBN1 mutations. Eur J Med Genet. 2010;53:80–4. doi: 10.1016/j.ejmg.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halliday D, Hutchinson S, Kettle S, Firth H, Wordsworth P, Handford PA. Molecular analysis of eight mutations in FBN1. Hum Genet. 1999;105:587–97. doi: 10.1007/s004399900190. [DOI] [PubMed] [Google Scholar]
- 43.Nijbroek G, Sood S, McIntosh I, Francomano CA, Bull E, Pereira L, Ramirez F, Pyeritz RE, Dietz HC. Fifteen novel FBN1 mutations causing Marfan syndrome detected by heteroduplex analysis of genomic amplicons. Am J Hum Genet. 1995;57:8–21. [PMC free article] [PubMed] [Google Scholar]