

Abstract
Background
Anopheles sinensis is the major malaria vector in China and Southeast Asia. Vector control is one of the most effective measures to prevent malaria transmission. However, there is little transcriptome information available for the malaria vector. To better understand the biological basis of malaria transmission and to develop novel and effective means of vector control, there is a need to build a transcriptome dataset for functional genomics analysis by large-scale RNA sequencing (RNA-seq).
Methods
To provide a more comprehensive and complete transcriptome of An. sinensis, eggs, larvae, pupae, male adults and female adults RNA were pooled together for cDNA preparation, sequenced using the Illumina paired-end sequencing technology and assembled into unigenes. These unigenes were then analyzed in their genome mapping, functional annotation, homology, codon usage bias and simple sequence repeats (SSRs).
Results
Approximately 51.6 million clean reads were obtained, trimmed, and assembled into 38,504 unigenes with an average length of 571 bp, an N50 of 711 bp, and an average GC content 51.26%. Among them, 98.4% of unigenes could be mapped onto the reference genome, and 69% of unigenes could be annotated with known biological functions. Homology analysis identified certain numbers of An. sinensis unigenes that showed homology or being putative 1:1 orthologues with genomes of other Dipteran species. Codon usage bias was analyzed and 1,904 SSRs were detected, which will provide effective molecular markers for the population genetics of this species.
Conclusions
Our data and analysis provide the most comprehensive transcriptomic resource and characteristics currently available for An. sinensis, and will facilitate genetic, genomic studies, and further vector control of An. sinensis.
Keywords: Anopheles sinensis, Transcriptome, RNA-Seq, Codon usage bias, Simple sequence repeat, Malaria, Vector control
Background
The Anopheles sinensis Wiedemann is a major malaria vector in China and other southeastern Asia countries with wide distribution from Afghanistan to northern China, Korea, Japan, Taiwan, and southward into western Indonesia [1-5]. An investigation of mosquito abundance in Yancheng, Jiangsu, China showed that An. sinensis accounted for 60.6% of the total number collected in the Rare Birds National Nature Reserve [6]. From the early 21st century, malaria re-emerged in central China. A total of 64,178 malaria cases and 52,082 suspected cases with 38 deaths were reported in 917 counties of 23 Provinces in 2006, 66.4% of which came from Huang-Huai Plain [7]. Malaria outbreaks and re-emergences appeared only in the areas with An. sinensis in recent years, and resulted from the extensive distribution, high population density and increasing vectorial capacity of An. sinensis in China [8-11]. In addition to its principal role in the transmission of malaria, this mosquito also plays a role in the transmission of filarial nematodes [12,13]. Mosquito vector control is one of most effective measures to prevent malaria transmission and relies primarily on the use of pyrethroids through insecticide-impregnated bednets and indoor residual spraying at present [14]. However, high levels of resistance to pyrethroids have been reported in An. sinensis from China and Korea [15-17]. To better understand the biological basis of An. sinensis, especially the molecular mechanisms of malaria transmission and insecticide resistance, there is a need to explore the transcriptomic biology of this mosquito species. Earlier, Jo et al. (2011) sequenced and analyzed two cDNA libraries from An. sinensis challenged with and without actinomycin-D; however, the overall transcriptomic information of An. sinensis is still very limited [18]. As far as March 2013, only 573 nucleotide sequences, 5 expressed sequence tags (ESTs), and 64 protein sequences from An. sinensis have been available in GenBank.
Advances in next-generation sequencing (NGS) and assembly algorithm rapidly promote the development of transcriptome sequencing (RNA-seq), which can reconstruct the entire transcriptome in a selected species of interest and generate quantitative expression scores for each transcript [19]. This transcriptome analysis will likely replace large-scale microarray approaches [20,21], as it’s lower cost, greater sequence yield, and higher sensitivity in detection of low abundant and novel transcripts allows the measurement of transcriptome composition to address the quantitative survey of RNA expression patterns in comparative genomic-levels [19] and develop molecular markers [22]. In the past few years, RNA-seq has been used for a number of genomes investigated, covering bacteria, Archaea and lower eukaryotes to higher eukaryotes. It is particularly effective when a reference genome is not available [23]. NGS transcriptome sequencing has also been successfully applied to mosquito species Anopheles gambiae[11,24], Aedies aegypti[25,26], Culex quinquefasciatus[27], Anopheles funestus[28] and Anopheles albimanus[29], which clearly demonstrated its utility for functional and evolutionary studies [30,31].
The study aims to construct a reference transcriptome of An. sinensis sampled from different developmental stages of egg, larva, pupa, and adult using the Illumina Hiseq2000 sequencing platform, and to present a comprehensive analysis of the de novo transcriptome sequencing result. As a result, a total of 38,504 unigenes were assembled and identified, among them 26,650 were annotated. Codon usage bias analysis revealed the characters of codon usage in this species. Moreover, a large number of simple sequence repeats (SSRs) were determined. To our knowledge, this is the first report on the complete transcriptome and characterization of An. sinensis, and will facilitate future genetic and genomic studies on this species.
Methods
Mosquito culture and total RNA preparation
The colony of An. sinensis was reared in the Institute of Entomology and Molecular Biology, Chongqing Normal University, China at 26 ± 1°C with 75 ± 5% relative humidity. Eggs (3 samples at the age of 0, 1 and 2 days of development, respectively), larvae (4 samples at 1st, 2nd, 3rd and 4th instar, respectively), pupae (6 samples 1, 2 and 3 days old, with both male and female, respectively), male adults (7 samples with 1, 2, 3, 4, 5, 6 and 7 days old), female adults (10 samples 1, 2, 3, 4 and 5 days old, with both before blood meal and after blood meal, respectively) were collected, snap frozen in liquid nitrogen, and then stored at −80°C prior to RNA extraction.
Total RNA was separately extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. To eliminate genomic DNA, the RNA samples were treated with RNase-Free DNase I according to manufacturer’s protocol (Qiagen, USA). The RNA integrity was confirmed using the Agilent 2100 Bioanalyzer with a minimum integrity number value of 7. Ten percent, 30%, 20%, 20% and 20% of total RNA quantity separately from eggs, larvae, pupae, male adults and female adults RNA sample were pooled together for cDNA preparation.
mRNA purification, cDNA synthesis and Illumina sequencing
Beads with Oligo (dT) were used to isolate poly (A) mRNA from total RNA extracted. Fragmentation buffer was added for interrupting mRNA to short fragments. Taking these short fragments as templates, random hexamer-primers were used to synthesize the first-strand cDNA. The second-strand cDNAs were synthesized using buffer, dNTPs, RNaseH and DNA polymerase I. Short fragments were purified with QiaQuick PCR extraction kit (Qiagen, USA) and resolved with EB buffer for end reparation and tailing A. The short fragments were then connected with sequencing adapters. After the agarose gel electrophoresis, the ligated products were purified and amplified with PCR to create the final cDNA library. Finally, the cDNA library was sequenced using Illumina HiSeq™ 2000 in Beijing Genomics Institute (BGI)-Shenzhen, China, according to manufacturer’s instructions.
De novo transcriptome assembly
The raw reads produced from HiSeq™ 2000 were cleaned by removing adapter sequences, low-quality sequences (reads with ‘N’ larger than 5%), and reads with more than 10% Q < 20 bases. The quality reads were assembled into unigenes using short reads assembling program – Trinity with the fellow parameters: min_glue =2, V = 10, edge-thr =0.05, min_kmer_cov = 2, group_pairs_distance = 250 [32]. Reads that contain a certain length of overlap area were first joined to form longer fragments, which are called contigs without gaps. Then the reads were mapped back to contigs; with paired-end reads it was able to detect contigs from the same transcript as well as the distances between these contigs. Next, Trinity connected the contigs, and obtained sequences that no longer could be extended. Such sequences are defined as unigenes. The assembled sequences less than 200 nt were deleted. At last, unigenes were divided into two classes by gene family clustering. One is clusters: several unigenes with similarity higher than 70% were classified into one cluster with the prefix CL. And the other is singletons with the prefix Unigene. To evaluate the quality of unigenes, all unigenes were realigned onto An. sinensis reference genome [33] using blat [34] with default setting. Fragment per kb per million reads (FPKM) for each gene of each sample was calculated to show the expression quantity of the gene in the sample, thus avoiding the influence of sequencing length and difference [35]. Each FPKM was log10 transformed. The raw reads dataset for the transcriptome of An. sinensis was submitted to the NCBI SRA database (http://www.ncbi.nlm.nih.gov/Traces/sra/) with the accession number SRR851144. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GBEO00000000. The version described in this paper is the first version, GBEO01000000.
Functional annotation
The BLASTX search (with E-value < = 1e-5) of each unigene larger than 200 nt was conducted against Nr, Swiss-Prot, KEGG, COG and GO databases, and the BLASTN search (E-value < = 1e-5) against the Nt to predict the function and metabolic pathways of unigenes. The best aligning result was used to decide sequence direction and the coding sequences (CDs) of unigenes, respectively. If results of these protein databases conflicted with each other, a priority order of Nr, Swiss-Prot, KEGG and COG was applied to decide sequence direction of unigenes. ESTScan [36] was used to predict the sequence direction and CDs when unigenes were unaligned to any of the databases. Gene function classifications with gene ontology (GO) annotations of the unigenes, an international standardized gene functional classification system, were determined by Blast2GO program [37]. Based on GO annotation WEGO software [38] was used to display GO functional classification.
Homology analysis between An. sinensis and other Dipteran genomes
To confirm validly assembled sequences and identify sequences with bioinformatic associations with other species, one-directional BLAST and Bi-directional BLAST were conducted between An. sinensis transcriptome and other Dipteran translated genomes. Diperant genomes of An. gambiae (release 3.6), Ae. aegypti (release 1.3) and Culex quinquefasciatus (release 1.3) from VectorBase (https://www.vectorbase.org/) and Drosophila melanogaster (release r5.47) from Flybase (http://flybase.org/), were downloaded. Peptide sequences equal to or longer than 50 a.a. of these species were employed for the comparisons because 1) amino acid sequences could achieve better hits than nucleotide sequences; 2) using short sequences are not as sensitive as using long sequences, and BLAST parameters need to be changed for short sequences (<50). Firstly, in one-directional BLAST, An. sinensis unigenes were used as queries to compare with these four translated genomes using BLASTX (E-value < = 1e-5), and sequences producing significant alignments were calculated and kept for further analysis. Secondly, to conduct Bi-directional BLAST, we then performed a reciprocal search with TBLASTN (E-value < = 1e-5) using these 4 Dipteran genomes as the queries, respectively. The top hit sequence was determined as one-directional’best-hits’ for each query. Pairs of sequences that were each other’s best hit were identified in both directions and regarded as putative 1:1 ortholog genes between An. sinensis and other Dipteran genomes. Bi-directional best hit has been widely used to identify the orthologous genes between closely related species and this approach has been demonstrated to perform better than more complex orthology identification algorithms.
To conduct comparative GO classification, the GO annotations for genes in An. gambiae genomes were first retrieved from An. gambiae genome (release 3.6). The obtained annotations were analyzed by WEGO software (http://wego.genomics.org.cn/cgi-bin/wego/index.pl) [38] to display the distribution of gene functions and the similarity and difference among An. sinensis and An. gambiae.
Characterization of ORFs and codon usage
The ORFs in each unigene sequence was predicted by BLAST against protein databases using BLASTX (E-value < = 1e-5) in the following order: Nr, SwissProt, KEGG, COG. Sequences with hits in former databases would not go to next round search against later database. The coding regions were then extracted according to the best BLASTX match with a custom perl script. We analyzed GC content and codon usage bias of ORFs longer than 150 nt by CodonW (http://codonw.sourceforge.net/), and processed the output files of Codonw with a perl script. The correlation analysis was performed using SPSS 13.0 statistics software (http://www-01.ibm.com/software/analytics/spss/).
SSR Detection
Unigenes of An. sinensis longer than 1 kb obtained in this study were subjected to the detection of SSRs using a perl script adapted from Simple Sequence Repeat Identification Tool (SSRIT, http://www.gramene.org/db/markers/ssrtool) [39]. The parameters were designed for identifying perfect di-, tri-, tetra-, penta-, and hexa-nucleotide motifs, with minimum thresholds of six, five, four, four and four repeats, respectively. Mononucleotide repeats were not considered due to the possibility of Illumina homopolymer sequencing problem associated with this technology.
Results and discussion
Illumina sequencing and assembly
To obtain a global overview of the An. sinensis transcriptome and gene activity at nucleotide resolution, a mixed cDNA sample representing diverse developmental stages of An. sinensis was prepared and sequenced using the Illumina Genome Analyzer. The sequencing of each cDNA fragment yielded two independent reads, 90-nt from both ends of the fragment. We obtained a total of 60.87 million raw reads. After the removal of adaptor sequences, ambiguous and low-quality reads, we obtained 51.61 million (84.79% raw reads) of clean reads with 4.64 Giga nucleotides (Gnt), 95.92% Q20, 51.26% GC content, and 0.00% unknown nucleotide ‘N’ (Table 1).
Table 1.
Statistics of RNA-seq based sequencing, assembling and functional annotation for An. sinensis
| Sequencing results | Number of total raw reads | 60,866,926 |
|---|---|---|
| |
Number of total clean reads |
51,606,364 |
| |
Number of total clean nucleotides (nt) |
4,644,572,760 |
| |
Q20 percentage of total clean reads |
95.92% |
| |
GC percentage of total clean nucleotides |
51.26% |
| |
N percentage of total clean nucleotides |
0.00% |
|
Assembling results |
Number of unigenes |
38,504 (5,372 into distinct clusters; 33,132 singletons) |
| |
Total length (nt) of total unigenes |
21,977,286 |
| |
Mean length (nt) of total unigenes |
571 |
| |
N50 (nt) of total unigenes |
711 |
|
Annotation |
Unigenes with Nr database |
25,456 (66% of 38,504 unigenes) |
| (E-value < =1e-5) |
Unigenes with Nt database |
20,554 (53%) |
| |
Unigenes with Swiss-Prot database |
17,651 (46%) |
| |
Unigenes with KEGG database |
16,622 (43%), 257 pathways |
| |
Unigenes with COG database |
7,204 (19%), 25 functional categories |
| |
Unigenes with GO database |
16,588 (43%), 62 subcategories grouped to 3 main categories |
| |
Biological process |
27 sub-categories |
| |
Cellular component |
17 sub-categories |
| |
Molecular function |
18 sub-categories |
| Total unigenes annotated | 26,650 (69% of 38,504 unigenes) |
These clean reads were assembled de novo by Trinity, which produced 38,504 unigenes (longer than 200 nt), with an average length of 571 nt and a N50 of 711 nt (Table 1). Out of these unigenes, 5,372 (14.0%) could be classified into distinct clusters, 33,132 (86.0%) were distinct singletons, 14,269 (37.1%) were longer than 500 nt, and 4,921 (12.8%) were longer than 1000 nt. The length distributions of unigenes are shown in Additional file 1.
The An. sinensis reference genome [33] was used to evaluate the assembled transcripts. As a result, about 98.4% (37,884/38,504) of unigenes could be mapped onto the reference genome (Additional file 2). Among them, 17,362 unigenes mapped with 1 block might have 1 exon, or be non-coding RNAs [40] (Figure 1A). The uncertainty could be due to low expression of transcript or assembly issue. The remaining 20,522 unigenes were mapped with at least 2 blocks onto the reference genome, which suggest at least 2 exons. 34,306 (90.6% of 37,884) mapped unigenes could be realigned onto the genome with more than 90% coverage, which supported the quality of the assembled unigenes (Figure 1B).
Figure 1.
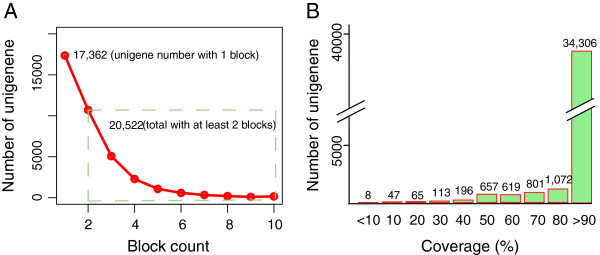
Distribution of block count (A) and coverage (B) of alignment between An. sinensis transcriptome and reference genome. Coverage is the ratio of match-to-length for each unigene.
Functional annotation
BLASTX search for each unigene sequence was conducted against several protein databases, including NCBI Non-redundant protein database (Nr), NCBI Non-redundant nucleotide database (Nt), Swiss-Prot, the Kyoto Encyclopedia of Genes and Genomes (KEGG), Cluster of Orthologous Groups (COG) databases and Gene Ontology (GO) databases, with an E-value threshold of 1e-5. The results indicated that out of 38,504 unigenes, a total of 25,456 (66%) unigenes were annotated against Nr, 20,554 (53%) against Nt, 17,651 (46%) against Swiss-Prot, 16,622 (43%) against KEGG, 7,204 (19%) against COG, and 16,588 (43%) against GO database (Table 1). Altogether, BLAST searches against Nr, Nt, Swiss-Prot, KEGG, COG and GO databases showed that a total of 26,650 (69% of 38,504 unigenes) identified unigenes could be annotated with known biological functions (Table 1).
The E-value distribution, similarity distribution and species distribution against the Nr database were then analyzed (Figure 2). For the E-value distribution of the predicted proteins, 46.5% of the mapped sequences had significant hits with a stringent threshold of less than 1e-45, and 53.5% of the mapped sequences with thresholds from 1e-45 to 1e-5 (Figure 2A). For the similarity distribution of the predicted proteins, 46.1% of the sequences had hits with similarity higher than 80% against the Nr database, and 74.5% of the sequences with similarity higher than 60% (Figure 2B). The species distribution showed that 94.8% of An. sinensis unigenes matched to 5 mosquito species, and only 5.2% of unigenes matched other species. Among these 5 mosquito species, 66.9% of the unigenes matched to An. gambiae str. PEST, followed by An. darlingi with 20.3% matches, Aedes aegypti with 3.4% matches, Culex quinquefasciatus with 3.2% matches and An. gambiae with only 1.0% matches (Figure 2C).
Figure 2.
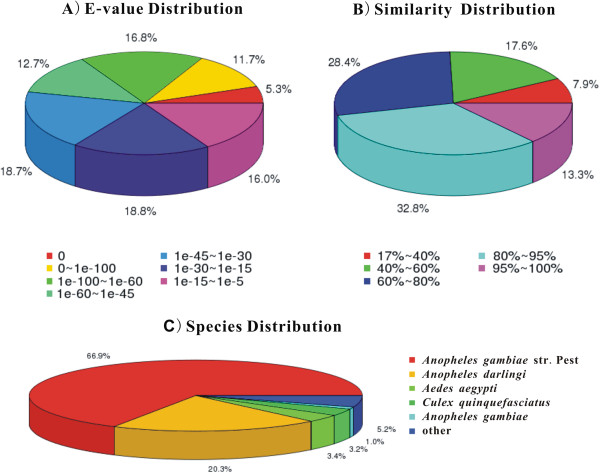
NR classification of An. sinensis unigenes. A) E-value distribution; B) Similarity distribution. C) Species distribution.
Based on GO annotation, 16,588 unigenes were each assigned a gene ontology term and categorized into 62 subcategories belonging to 3 main categories, including biological process (27), cellular component (17), and molecular function (18) (Table 1, Figure 3). In the category of molecular function, the GO term quantity in binding activity and catalytic activity was obviously larger than others, and in the categories of biological processes the subcategories of cellular process and metabolic process appeared somewhat dominant and in cellular component the subcategories of cell and cell part showed a little prevailing (Figure 3). This GO assignment result is similar to the transcriptome earlier sequenced in Taenia multiceps, in which binding, cell and metabolic processes were the three largest groups [41]. These GO annotations demonstrated that expressed genes in An. sinensis encoded diverse structural, regulatory, metabolic and transporter proteins.As a result of the search of 38,504 unigenes against COG database for orthologous genes, 7,204 unigenes were finally predicted and classified into 25 functional categories. The largest category was “General function prediction only” with 2,537 unigenes (35.2%) that were associated with basic physiological and metabolic functions, followed by “Carbohydrate transport and metabolism” (1,494 unigenes, 20.7%), “Transcription” (1,394, 19.4%), “Translation, ribosomal structure and biogenesis” (1,281, 17.8%) and “Posttranslational modification, protein turnover, chaperones” (1,175, 16.3%). The “RNA processing and modification” (73, 1.0%), “Extracellular structures” (31, 0.4%) and “Nuclear structure” (6, 0.08%) represented the smallest groups of unigenes (Figure 4).
Figure 3.
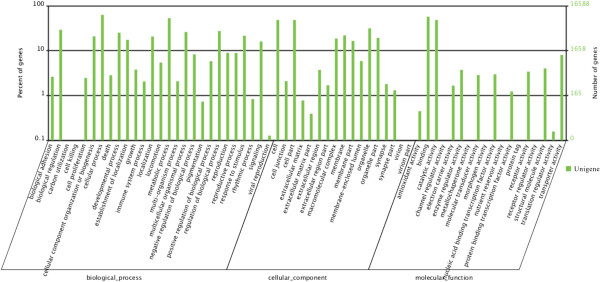
GO function classification of An. sinensis unigenes.
Figure 4.
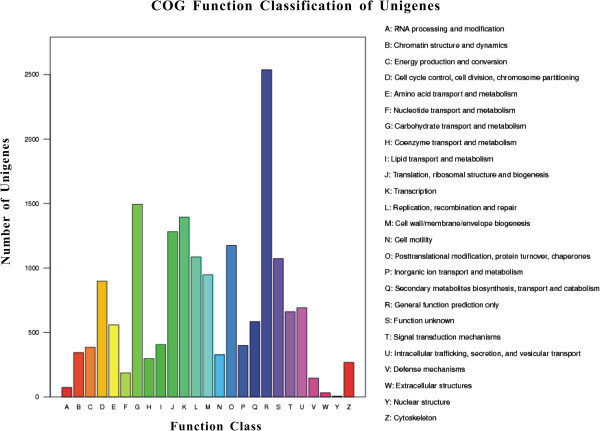
COG classification of An. sinensis unigenes.
The potential involvement in biological pathways of the assembled unigenes was annotated with corresponding Enzyme commission (EC) numbers and pathways from BLASTX searches against the KEGG database [42]. The KEGG database annotation provided the inner-cell metabolic pathways and functions of gene products of unigenes. Out of 38,504 assembled unigenes, a total of 16,622 unigenes were assigned into 257 KEGG pathways (Table 1). The pathways largely represented by unique sequences were metabolic pathways (2,211 unigenes), RNA transport (675), pathways in cancer (599) and amoebiasis (576). These annotations provided us with a valuable resource for the investigation of specific processes, structures, functions and pathways in An. sinensis research.
Homology analysis between An. sinensis and other Dipteran genomes
In order to obtain a more detailed understanding of An. sinensis biology and reveal a potential evolutionary relationship among Dipteran species, orthologous genes shared between An. sinensis and 4 other Dipteran species were compared, including Drosophila melanogaster, An. gambiae, Ae. aegypti and Culex quinquefasciatus. In comparison of the An. sinensis unigene sequences to predicted a.a. sequences of other Dipteran species, 12,973 a.a. sequences (90.7% of 14,296) of An. gambiae showed a significant similarity to those of An. sinensis, followed in decreasing ratio by Ae. aegypti with 11,997 (69.1%), Cx. quinquefasciatus with 11,990 (63.4%) and Dr. melanogaster 10,119 (36.7%) a.a. sequence matches (Table 2 and Figure 5). It was consistent with the expectation that the match ratio of sequences with significant similarity in pairwise comparisons between two species decreased with increasing phylogenetic distance [28]. Bi-directional BLAST was used to identify 1:1 orthologs between An. sinensis and other Dipteran species. Putative 6,586, 6,116, 6,084 and 4,919 pairs were identified between An. sinensis and An. gambiae, Ae. aegypti, Cx. quinquefasciatus, and Dr. melanogaster, respectively (Table 2 and Figure 5). In a previous comparison between An. funestus contigs and An. gambiae protein sequences, Crawford et al. [28] identified 5,434 pairs between An. funestus contigs and An. gambiae using the standard reciprocal best-hit criteria. The result was comparative with our 1:1 orthologues of 6,586 pairs between An. sinensis and An. gambiae. The difference might be due to different phylogenetic distance in Anopheles, assembling methods and the updated version of An. gambiae data.
Table 2.
Homology analysis between An. sinensis and other Dipteran genomes using BLASTX with cut-off E-value of 1E-5
| An. gambiae | Ae. aegypti | Cx quinquefasciatus | Dr. melanogaster | |
|---|---|---|---|---|
| Number of a.a. sequences |
14324 |
17408 |
19018 |
27538 |
| Number of sequences with a.a. > 50 |
14296 |
17345 |
18906 |
27410 |
| Number of one-directional BLAST hits |
12973 |
11997 |
11990 |
10119 |
| Number of Bi-directional BLAST hits |
6586 |
6116 |
6084 |
4919 |
| Genome sequence version |
AgamP3.6 |
AaegL1.3 |
CpipJ1.3 |
r5.47 |
| Source of genome sequence | Vectorbase | Vectorbase | Vectorbase | Flybase |
Figure 5.
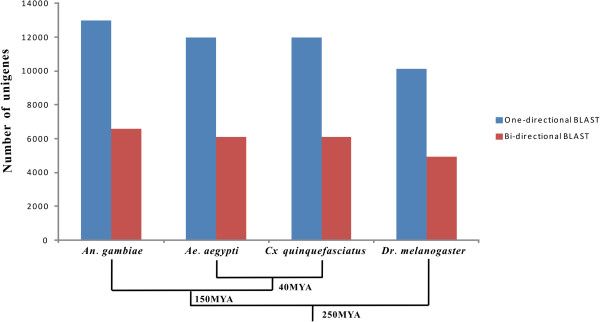
Homologous gene numbers between An. sinensis, and Ae. aegypti, Cx quinquefasciatus and Dr. melanogaster detected by one- and bi-directional BLAST searches. The numbers decreased with the phylogenetic distance between An. sinensis and other Dipteran species (the divergence times were adapted from Grimldi et al. [43].
The GO classification was further used to identify the difference of functional category between An. sinensis unigenes and the genes of An. gambiae. The result of the comparative analysis of GO terms between An. sinensis unigenes and the genes of An. gambiae was shown in Figure 6. In total, 16,588 unigenes of An. sinensis and 9,872 genes of An. gambiae were assigned with one or more GO terms, respectively. The major subcategories in the GO classification were shared by the genes of these 2 species of mosquitoes; however, qualitative and quantitative differences existed. The differences might result from the difference both in species and sample source, and further research is needed to elucidate the hypothesis.
Figure 6.
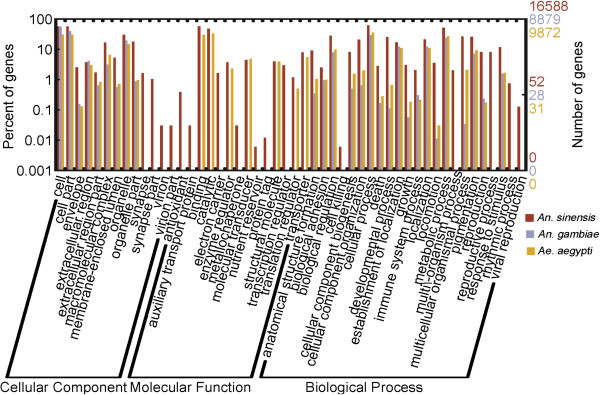
GO terms similarity distribution among An. sinensis and An. gambiae. Bar graph was plotted using a web-based tool, WEGO.
Codon usage bias
Codon usage bias, higher usage frequency of specific condons than other synonymous codons during the translation of genes, can help understand the physiological, biochemical and molecular mechanism as a useful tool in functional genomic research [44]. The extent of codon usage bias may vary within and among species, which can be influenced by various factors, including expression level, GC content, codon position, gene length, environmental stress and population size [45].
In the study, total codon usages were obtained and most frequently, seldom used codons were identified. A total of 24,361 ORFs longer than 150 nt were used in the codon usage bias analyses. A total 4,048,458 counts of codons and the relative synonymous codon usage (RSCU) for these sequences were calculated (Table 3, Additional file 3). Average RSCU values showed that the seven most frequently used codons in An. sinensis are: CUG, CCG, UCG, CGC, GUG, ACG and AUC; the seven seldom used codons in An. sinensis are: UAU, CCU, ACU, UCU, AGA, AGG and UUA without considering stop codons (Additional file 3). The results are very similar to the results obtained from other Dipteran insects based on the genome-specific frequencies of the codons [46].
Table 3.
GC content and codon bias in predicted ORFs of unigenes in An. sinensis .
| Total number of examined ORFs | 24,361 |
|---|---|
| % GC of 24,361 ORFs |
55.25% |
| GC3 (% GC at 3rd codon position) |
65.40% |
| Nc (Effective number of condons) |
46.71 |
| Total number of codons | 4,048,458 |
The average GC content of 24,361 ORFs was 55.25% and the average GC content of all 38,504 unigenes in An. sinensis was 51.26% (Table 3). The unigene GC percentage was similar to the genome GC percentage in An. gambiae (55.8%) and Dr. melanogaster (53.9%) [46]. The GC content at the third codon position (GC3) was considered to largely decide genome base composition, which varies in different species. Our results showed the average GC3 was 65.40% in An. sinensis (Table 3, Additional file 4). To our knowledge, it is the first time the GC3 content in mosquitoes has been reported.
Effective number of codons (Nc), a measure quantifing how far the codon usage of a gene departs from average usage of synonymous codons in study of the state of codon usage bias in genes and genomes [47], has been reported in a range from 20 (maximally bias) to 61 (unbiased) [48]. The mean Nc value of An. sinensis was calculated to be 46.71 (Table 3), a medium value in the range, which suggested a medium extent of codon preference in An. sinensis[49,50]. A plot of Nc versus GC3 (Nc plot) has been widely used to study the codon usage variation among genes in different organisms [51]. If the codon usage variation among the genes was only determined by variation in GC3 content, then the values of Nc would fall on the continuous curve between Nc and GC3. The Nc plot for An. sinensis showed that most of the genes fall within a restricted cloud, at GC3 between 0.013 and 1, and Nc values between 21.15 and 61 (Additional file 4 and 5). Most genes had an Nc value lower than the expected value located on the curve, suggesting that the codon usage of a large number of An. sinensis genes are not only determined by GC3.
Commonly, highly expressed genes (such as ribosomal protein genes) tend to reflect greater codon bias than lowly expressed genes [52]. A significant positive correlation between codon usage, bias and gene-expression levels determined from microarray data have been demonstrated in Ae. aegypti and An. gambiae[53]. To test the effect of codon bias on gene expression in An. sinensis, we compared the codon bias index (CBI) values with the observed gene expression levels (log10 transformed FPKM) for 24,361 ORFs as determined from whole transcriptome data in An. sinensis. We observed a significantly positive correlation between the CBI and gene expression levels in An. sinensis (Pearson Correlation: r = 0.037, P =1.04e-8) (Additional files 4 and 6).
In our study, total codon usages were obtained, and most frequently, seldom used codons were identified. The Nc plot showed that most genes have an Nc value lower than the expected value located on the curve, suggesting that the codon usage of a large number of An. sinensis genes are subject to other factors. We also tested the influence of GC3 and expression levels on codon usage bias in An. sinensis. A significant correlation between codon usage and expression levels was observed in An. sinensis (Additional file 6). Knowledge of the codon usage pattern in An. sinensis can provide a basis for understanding the mechanisms of codon usage bias and for selecting appropriate host expression systems to improve the expression of exogenous genes in An. sinensis and thus facilitate vector control.
SSR discovery
To explore SSR profiles in the An. sinensis transcriptome, 4,921 unigene sequences longer than 1 kb were searched for SSRs. As a result, 1,223 sequences containing a total of 1,904 and 307 kinds of SSRs were identified, with 681 of the sequences containing more than 1 SSR. The frequency of SSR in An. sinensis transcriptome was 1 per 11.5 kilobases (Table 4). The tri-nucleotide repeat motif was the most abundant, accounting for 57.7%, followed by di-nucleotide repeat motif (36.2%), tetra-nucleotide (5.3%), penta-nucleotide (0.5%), and hexa-nucleotide (0.3%) repeat units (Table 5). The largest abundance of tri-nucleotide repeat motifs is consistent with the result from another insect species Spodoptera exigua[54]. The frequencies of SSRs with different numbers of tandem repeats were also calculated and shown in Table 5. SSRs with five tandem repeats (occupying 37.3%) were the most common, followed by six tandem repeats (30.0%), seven tandem repeats (15.5%), eight tandem repeats (6.9%), and four tandem repeats (4.3%). A detailed list of SSRs identified was shown in Additional file 7.
Table 4.
Features of SSRs identified in the An. sinensis transcriptome
| Total number of examined unigenes | 38,504 |
|---|---|
| Number of unigenes longer than 1 kb |
4,921 |
| Total nucleotides screened (knt) |
21,977 |
| Number of unigenes containing SSRs |
1,223 |
| Number of identified SSRs |
1,904 |
| Kinds of identified SSRs |
307 |
| Number of unigenes containing more than 1 SSRs |
681 |
| Frequency of SSR in transcriptome | 1/11.5Kb |
Table 5.
Frequency of SSRs in An. sinensis transcriptome
|
Number of nucleotides |
Number of motif repeat |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | 5 | 6 | 7 | 8 | 9 | 10 | >10 | Total | % | |
| Di |
- |
- |
297 |
168 |
111 |
58 |
24 |
31 |
689 |
36.2 |
| Tri |
- |
685 |
263 |
126 |
18 |
1 |
1 |
4 |
1098 |
57.7 |
| Tetra |
70 |
22 |
4 |
1 |
3 |
- |
- |
2 |
102 |
5.3 |
| Penta |
7 |
2 |
1 |
- |
- |
- |
- |
- |
10 |
0.5 |
| Hexa |
4 |
1 |
- |
- |
- |
- |
- |
- |
5 |
0.3 |
| Total |
81 |
710 |
565 |
295 |
132 |
59 |
25 |
37 |
1904 |
|
| % | 4.3 | 37.3 | 30.0 | 15.5 | 6.9 | 3.1 | 1.3 | 1.9 | ||
SSRs are valuable genetic markers used in population genetic analysis and genetic mapping [55]. The traditional isolation of genomic DNA-derived SSRs is difficult and costly. However, large-scale transcriptome sequencing programs based on NGS methods produced large amounts of sequence data, from which the isolation and identification of genome-wide and gene-based SSRs become easier and cheaper. More importantly, SSR markers identified from transcriptome sequences can be used as a tool for identifying associations with functional genes, and therefore with phenotypes [56]. The SSR markers derived from transcriptome databases should be of more benefit, because they should mainly occur in the protein-coding regions of annotated unigenes, which represents genes of known or predicted identity and function.
As the majority of SSR markers derived from transcriptome databases should occur in the protein-coding sequences of annotated unigenes, representing genes of known or predicted identity and function. Efficient discovery of SSR loci based on transcriptome data have been demonstrated in many organisms, such as in Phlebotomus papatasi[57], Sp. exigua[54]. To our knowledge, this is the first work to identify the SSRs from transcriptome database for mosquitoes, although SSRs in Cx quinquefasciatus[58], An. gambiae[59] and An. sinensis[60] were reported earlier from genomic DNA data. 1,904 SSRs reported herein from An. sinensis was much over the number 21 earlier reported from the species [60]. The samples for An. sinensis transcriptomes were collected for every developmental stage. Therefore, the 1,904 SSRs should have covered most, if not all, of SSRs in the An. sinensis transcriptome, which will contribute to the genome mapping of this species as well as of closely related species, and also assist in the mapping and identification of important functional genes.
Conclusions
An. sinensis is a major malaria vector in China and Southeast Asia; however, the lack of transcriptome data has seriously hindered the research of biology, ecology and molecular mechanisms regarding malaria transmission and control of the mosquito species. To establish a transcriptome resource that will facilitate future genomic level studies in this species, we used the Illumina Hiseq2000 sequencing platform for An. sinensis transcriptome sequencing, and produced 38,504 assembled unigenes with 25,456 (66%) annotated. This study dramatically increased the CDs number of genes from An. sinensis. To our knowledge, our results represented approximately 70-fold more genes than all An. sinensis genes deposited in GenBank (as of Jan, 2013). Certain numbers of them showed homology or could be identified as 1:1 orthologues with other Dipteran genomes, which provided us with significances in evolutionary studies of Dipteran insects. Knowledge of the codon usage pattern in An. sinensis provides the basis to understand the mechanisms of codon usage bias and would be useful for future transgenic manipulation in mosquitoes. Moreover, the SSR markers identified may serve as potential marker selection in the mapping and identification of disease-related genes and contribute to the genome mapping and mosquito population genetics research. We believe that results obtained from this study will also serve as a useful genomic dataset to accelerate research of disease transmission mechanisms, resistance mechanisms, and functional genomics in An. sinensis. It is possible that some of the results we presented might become useful in the future vector control.
Abbreviations
CBI: Codon bias index; CDs: coding sequences; COG: Cluster of Orthologous Groups databases; FPKM: Fragment per kilobase per million reads; GC3: GC content at the third codon position; GO: Gene ontology; KEGG: The Kyoto Encyclopedia of Genes and Genomes; Nc: Effective number of codons; Nc plot: A plot of Nc versus GC3; NGS: Next-generation sequencing; Nr: NCBI Non-redundant protein database; Nt: NCBI Non-redundant nucleotide database; ORFs: Open reading frames; RNA-seq: Transcriptome sequencing; RSCU: Synonymous codon usage; SSRs: Simple sequence repeats.
Competing interests
All authors declare that they have no competing interests.
Authors’ contributions
BC conceived and designed the study, jointly performed data analysis and drafted the manuscript. YJZ jointly contributed to data analysis and manuscript drafting. WL helped in performing dada analysis. ZH, FS, YT, QH, LQ, ZY, WF and YC conducted sample collecting and experiments. All authors read and approved the final manuscript.
Supplementary Material
Length distribution of unigenes on An. sinensis reference genome.
{kind=link}
Alignment between An. sinensis transcriptome and reference genome. Only longest alignment was kept in the cases that a unigene was returned with multiple blat hits.
Total codon usage and codon usage bias in An. sinensis transcriptome.
The ID and FPKM of unigenes, and the CBI, Nc and GC3 of ORF in each unigene in An. sinensis.
A plot of Nc versus GC3 (Nc-plot) for An. sinensis ORFs. The pink dotted curve represents the expected curve between GC3 and Nc under random codon usage. Blue dot each indicates a corresponding ORF of each unigene (totally 24,361 unigenes).
{kind=link}
Relationship between CBI (codon bias index) and expression level (Log10 (FPKM)) of all An. sinensis transcriptome unigenes.
{kind=link}
SSR identification of An. sinensis unigenes.
Contributor Information
Bin Chen, Email: c_bin@hotmail.com.
Yu-Juan Zhang, Email: yujuan.zhang418@gmail.com.
Zhengbo He, Email: zhengbohe83@aliyun.com.
Wanshun Li, Email: wanshun.Li@bgitechsolutions.com.
Fengling Si, Email: sifengling1217@gmail.com.
Yao Tang, Email: 312734210@qq.com.
Qiyi He, Email: hqy7171@163.com.
Liang Qiao, Email: qiaoliangswu@163.com.
Zhentian Yan, Email: yzt318@163.com.
Wenbo Fu, Email: fuice@126.com.
Yanfei Che, Email: 1806136063@qq.com.
Acknowledgements
We appreciate the technical support for Illumina sequencing and initial data analysis provided by Beijing Genome Institute at Shenzhen, China. This work was supported by Par-Eu Scholars Program, and grants from The National Natural Science Foundation of China (31372265 and 31200947), Coordinated Research Project of the International Atomic Energy Agency (18268/R0), National Institute of Health (R01 AI095184), Key Scientific and Technological Project of Chongqing (CSTC2012GG-YYJSB80002), Natural Science Foundation Project of Chongqing (CSTC2013JCYJA00013), Science and Technology Project of Chongqing Municipal Education Commission (No. KJ130629).
References
- Chareonviriyaphap T, Bangs MJ, Ratanatham S. Status of malaria in Thailand. Southeast Asian J Trop Med Public Health. 2000;31(2):225–237. [PubMed] [Google Scholar]
- Sinka ME, Bangs MJ, Manguin S, Chareonviriyaphap T, Patil AP, Temperley WH, Gething PW, Elyazar IR, Kabaria CW, Harbach RE, Hay SI. The dominant Anopheles vectors of human malaria in the Asia-Pacific region: occurrence data, distribution maps and bionomic precis. Parasit Vectors. 2011;4:89. doi: 10.1186/1756-3305-4-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goo YK, Seo EJ, Choi YK, Shin HI, Sattabongkot J, Ji SY, Chong CK, Cho SH, Lee WJ, Kim JY. First characterization of Plasmodium vivax liver stage antigen (PvLSA) using synthetic peptides. Parasit Vectors. 2014;7:64. doi: 10.1186/1756-3305-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu G, Xia H, Zhou H, Li J, Lu F, Liu Y, Cao J, Gao Q, Sattabongkot J. Susceptibility of Anopheles sinensis to Plasmodium vivax in malarial outbreak areas of central China. Parasit Vectors. 2013;6(1):176. doi: 10.1186/1756-3305-6-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toma T, Miyagi I, Tamashiro M, Tsuzuki A. Susceptibility of the mosquitoes Anopheles minimus, An. sinensis, and An. saperoi (Diptera: Culicidae) from the Ryukyu Archipelago, Japan, to the rodent malaria Plasmodium yoelii nigeriense. J Med Entomol. 2002;39(1):146–151. doi: 10.1603/0022-2585-39.1.146. [DOI] [PubMed] [Google Scholar]
- Wu ZM, Zhu HM, Chang TX, SC L. [Investigation of mosquito abundance and composition around the Rare Birds National Nature Reserve of Yancheng, Jiangsu Province] Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 2007;25(4):310–313. [PubMed] [Google Scholar]
- Zhou SS, Huang F, Wang JJ, Zhang SS, Su YP, Tang LH. Geographical, meteorological and vectorial factors related to malaria re-emergence in Huang-Huai River of central China. Malar J. 2010;9:337. doi: 10.1186/1475-2875-9-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh AC, Liu XL, Jackson S, Li P, Shang LY. Resurgence of vivax malaria in Henan Province, China. Bull World Health Organ. 1998;76(3):265–270. [PMC free article] [PubMed] [Google Scholar]
- Pan JY, Zhou SS, Zheng X, Huang F, Wang DQ, Shen YZ, Su YP, Zhou GC, Liu F, Jiang JJ. Vector capacity of Anopheles sinensis in malaria outbreak areas of central China. Parasit Vectors. 2012;5:136. doi: 10.1186/1756-3305-5-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XB, Liu QY, Guo YH, Jiang JY, Ren DS, Zhou GC, Zheng CJ, Zhang Y, Liu JL, Li ZF, Chen Y, Li HS, Morton LC, Li HZ, Li Q, Gu WD. The abundance and host-seeking behavior of culicine species (Diptera: Culicidae) and Anopheles sinensis in Yongcheng city, people’s Republic of China. Parasit Vectors. 2011;4:221. doi: 10.1186/1756-3305-4-221. Available: http://www.parasitesandvectors.com/content/4/1/221 Accessed 2013 March 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, Afrane Y, Dunn WA, Atieli FK, Zhou G, Zhong D, Li J, Githeko A, Yan G. Comparative transcriptome analyses of deltamethrin-resistant and -susceptible anopheles gambiae mosquitoes from Kenya by RNA-Seq. PLoS One. 2012;7(9):e44607. doi: 10.1371/journal.pone.0044607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QJ, Duan JH, Hu GL, Yu LR, Yang WQ, Li LZ, Ouyang HK. [Epidemiological characteristics and control of filariasis in Hunan Province] Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong Bing Za Zhi. 1990;8(2):134–137. [PubMed] [Google Scholar]
- Jin LZ, Xu JJ. Quantitative studies on the development of inoculated Brugia malayi microfilariae in Anopheles sinensis and Culex quinquefasciatus. Southeast Asian J Trop Med Public Health. 1990;21(3):418–423. [PubMed] [Google Scholar]
- WHO. Global Malaria Program: Use of Indoor Residual Spraying for Scalling up Global Malaria Control and Elimination. Geneva: World Health Organization; (WHO Position Statement). Available at: http://whqlibdoc.who.int/hq/2006/WHO_HTM_MAL_2006.1112_eng.pdf Accessed April 6, 2009 2006. [Google Scholar]
- Cui F, Raymond M, Qiao CL. Insecticide resistance in vector mosquitoes in China. Pest Manag Sci. 2006;62:1013–1022. doi: 10.1002/ps.1288. [DOI] [PubMed] [Google Scholar]
- Kim H, Baek JH, Lee WJ, Lee SH. Frequency detection of pyrethroid resistance allele in Anopheles sinensis populations by real-time PCR amplification of specific allele (rtPASA) Pestic Biochem Physiol. 2007;87:54–61. [Google Scholar]
- Zhong D, Chang X, Zhou G, He Z, Fu F, Yan Z, Zhu G, Xu T, Bonizzoni M, Wang MH, Cui L, Zheng B, Chen B, Yan G. Relationship between knockdown resistance, metabolic detoxification and organismal resistance to pyrethroids in anopheles sinensis. PLoS One. 2013;8(2):e55475. doi: 10.1371/journal.pone.0055475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo YH, Lee YS, Kang SW, Kho WG, Park HS, Choi SH, Kim YJ, Hong YS, Noh MY, Oh SH, Kim I, Han YS. Bioinformatic analysis and annotation of expressed sequence tags (ESTs) generated from Anopheles sinensis mosquitoes challenged with apoptosis-inducing chemical, actinomycin-D. Entomological Research. 2011;41(2):53–59. [Google Scholar]
- Wilhelm BT, Landry JR. RNA-Seq-quantitative measurement of expression through massively parallel RNA-sequencing. Methods. 2009;48(3):249–257. doi: 10.1016/j.ymeth.2009.03.016. [DOI] [PubMed] [Google Scholar]
- Marioni JC, Mason CE, Mane SM, Stephens M, Gilad Y. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008;18(9):1509–1517. doi: 10.1101/gr.079558.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon SA, Zhai J, Nandety RS, McCormick KP, Zeng J, Mejia D, Meyers BC. Short-read sequencing technologies for transcriptional analyses. Annu Rev Plant Biol. 2009;60:305–333. doi: 10.1146/annurev.arplant.043008.092032. [DOI] [PubMed] [Google Scholar]
- Zhang YJ, Hao Y, Si F, Ren S, Hu G, Shen L, Chen B. The de novo transcriptome and its analysis in the worldwide vegetable pest, Delia antiqua (diptera: anthomyiidae) G3 (Bethesda) 2014;4(5):851–859. doi: 10.1534/g3.113.009779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Wang Z. Next-generation transcriptome assembly. Nat Rev Genet. 2011;12(10):671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- Pitts RJ, Rinker DC, Jones PL, Rokas A, Zwiebel LJ. Transcriptome profiling of chemosensory appendages in the malaria vector Anopheles gambiae reveals tissue- and sex-specific signatures of odor coding. BMC Genomics. 2011;12:271. doi: 10.1186/1471-2164-12-271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, Dunn WA, Campbell CL, Olson KE, Dimon MT, Marinotti O, James AA. RNA-seq analyses of blood-induced changes in gene expression in the mosquito vector species, Aedes aegypti. BMC Genomics. 2011;12:82. doi: 10.1186/1471-2164-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, Dunn WA, Campbell CL, Olson KE, Marinotti O, James AA. Complex modulation of the aedes aegypti transcriptome in response to dengue virus infection. PLoS One. 2012;7(11):e50512. doi: 10.1371/journal.pone.0050512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid WR, Zhang L, Liu F, Liu N. The transcriptome profile of the mosquito Culex quinquefasciatus following permethrin selection. PLoS One. 2012;7(10):e47163. doi: 10.1371/journal.pone.0047163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JE, Guelbeogo WM, Sanou A, Traore A, Vernick KD, Sagnon N, Lazzaro BP. De novo transcriptome sequencing in Anopheles funestus using Illumina RNA-seq technology. PLoS One. 2010;5(12):e14202. doi: 10.1371/journal.pone.0014202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Barnetche J, Gomez-Barreto RE, Ovilla-Munoz M, Tellez-Sosa J, Lopez DE, Dinglasan RR, Mohien CU, MacCallum RM, Redmond SN, Gibbons JG, Rokas A, Machado CA, Cazares-Raga FE, Gonzalez-Ceron L, Hernandez-Martinez S, Rodriguez Lopez MH. Transcriptome of the adult female malaria mosquito vector Anopheles albimanus. BMC Genomics. 2012;13:207. doi: 10.1186/1471-2164-13-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons JG, Janson EM, Hittinger CT, Johnston M, Abbot P, Rokas A. Benchmarking next-generation transcriptome sequencing for functional and evolutionary genomics. Mol Biol Evol. 2009;26(12):2731–2744. doi: 10.1093/molbev/msp188. [DOI] [PubMed] [Google Scholar]
- Zhao L, Zhang N, Ma PF, Liu Q, Li DZ, Guo ZH. Phylogenomic analyses of nuclear genes reveal the evolutionary relationships within the BEP clade and the evidence of positive selection in Poaceae. PLoS One. 2013;8(5):e64642. doi: 10.1371/journal.pone.0064642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, di Palma F, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29(7):644–652. doi: 10.1038/nbt.1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhang D, Ding G, Shi L, Hou Q, Ye Y, Xu Y, Zhou H, Xiong C, Li S, Yu J, Hong S, Yu X, Zou P, Chen C, Chang X, Wang W, LV Y, Sun Y, Ma L, Shen B, Zhu C. Genome sequence of Anopheles sinensis provides insight into genetics basis of mosquito competence for malaria parasites. BMC Genomics. 2014;15:42. doi: 10.1186/1471-2164-15-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Methods. 2008;5(7):621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- Iseli C, Jongeneel CV, Bucher P. ESTScan: a program for detecting, evaluating, and reconstructing potential coding regions in EST sequences. Proc Int Conf Intell Syst Mol Biol. 1999;7:138–148. [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21(18):3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L. WEGO: a web tool for plotting GO annotations. Nucleic Acids Res. 2006;34(Web Server issue):W293–W297. doi: 10.1093/nar/gkl031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temnykh S, DeClerck G, Lukashova A, Lipovich L, Cartinhour S, McCouch S. Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential. Genome Res. 2001;11(8):1441–1452. doi: 10.1101/gr.184001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Li W, Wu Y, Chen C, Lei J. De novo transcriptome assembly in chili pepper (Capsicum frutescens) to identify genes involved in the biosynthesis of capsaicinoids. PLoS One. 2013;8(1):e48156. doi: 10.1371/journal.pone.0048156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Fu Y, Yang D, Zhang R, Zheng W, Nie H, Xie Y, Yan N, Hao G, Gu X, Wang S, Peng X, Yang G. Detailed transcriptome description of the neglected cestode taenia multiceps. PLoS One. 2012;7(9):e45830. doi: 10.1371/journal.pone.0045830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004;32(Database issue):D277–D280. doi: 10.1093/nar/gkh063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimaldi D, Engel MS. Evolution of the Insects. Cambridge, UK: Cambridge University Press; 2005. [Google Scholar]
- Angov E. Codon usage: nature’s roadmap to expression and folding of proteins. Biotechnol J. 2011;6(6):650–659. doi: 10.1002/biot.201000332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behura SK, Severson DW. Codon usage bias: causative factors, quantification methods and genome-wide patterns: with emphasis on insect genomes. Biol Rev Camb Philos Soc. 2012;88(1):49–61. doi: 10.1111/j.1469-185X.2012.00242.x. [DOI] [PubMed] [Google Scholar]
- Behura SK, Severson DW. Comparative analysis of codon usage bias and codon context patterns between dipteran and hymenopteran sequenced genomes. PLoS One. 2012;7(8):e43111. doi: 10.1371/journal.pone.0043111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuglsang A. The ‘effective number of codons’ revisited. Biochem Biophys Res Commun. 2004;317(3):957–964. doi: 10.1016/j.bbrc.2004.03.138. [DOI] [PubMed] [Google Scholar]
- Wright F. The’effective number of codons’ used in a gene. Gene. 1990;87(1):23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Lithwick G, Margalit H. Hierarchy of sequence-dependent features associated with prokaryotic translation. Genome Res. 2003;13:2665–2673. doi: 10.1101/gr.1485203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHardy A, Puhler A, Kalinowski J, Meyer F. Comparing expression level-dependent features in codon usage with protein abundance: an analysis of’ predictive proteimics’. Proteomics. 2004;4:46–58. doi: 10.1002/pmic.200300501. [DOI] [PubMed] [Google Scholar]
- Fuglsang A. Impact of bias discrepancy and amino acid usage on estimates of the effective number of codons used in a gene, and a test for selection on codon usage. Gene. 2008;410(1):82–88. doi: 10.1016/j.gene.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Hershberg R, Petrov DA. Selection on codon bias. Annu Rev Genet. 2008;42:287–299. doi: 10.1146/annurev.genet.42.110807.091442. [DOI] [PubMed] [Google Scholar]
- Behura SK, Severson DW. Coadaptation of isoacceptor tRNA genes and codon usage bias for translation efficiency in Aedes aegypti and Anopheles gambiae. Insect Mol Biol. 2011;20(2):177–187. doi: 10.1111/j.1365-2583.2010.01055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual L, Jakubowska AK, Blanca JM, Canizares J, Ferre J, Gloeckner G, Vogel H, Herrero S. The transcriptome of Spodoptera exigua larvae exposed to different types of microbes. Insect Biochem Mol Biol. 2012;42(8):557–570. doi: 10.1016/j.ibmb.2012.04.003. [DOI] [PubMed] [Google Scholar]
- Luikart G, England PR, Tallmon D, Jordan S, Taberlet P. The power and promise of population genomics: from genotyping to genome typing. Nat Rev Genet. 2003;4(12):981–994. doi: 10.1038/nrg1226. [DOI] [PubMed] [Google Scholar]
- Zalapa JE, Cuevas H, Zhu H, Steffan S, Senalik D, Zeldin E, McCown B, Harbut R, Simon P. Using next-generation sequencing approaches to isolate simple sequence repeat (SSR) loci in the plant sciences. Am J Bot. 2012;99(2):193–208. doi: 10.3732/ajb.1100394. [DOI] [PubMed] [Google Scholar]
- Hamarsheh O, Amro A. Characterization of simple sequence repeats (SSRs) from Phlebotomus papatasi (Diptera: Psychodidae) expressed sequence tags (ESTs) Parasit Vectors. 2011;4:189. doi: 10.1186/1756-3305-4-189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickner PV, Debruyn B, Lovin DD, Mori A, Saski CA, Severson DW. Enhancing genome investigations in the mosquito Culex quinquefasciatus via BAC library construction and characterization. BMC Res Notes. 2011;4:358. doi: 10.1186/1756-0500-4-358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Xia Q, Lu C, Zhou Z, Xiang Z. Analysis on frequency and density of microsatellites in coding sequences of several eukaryotic genomes. Genomics Proteomics Bioinformatics. 2004;2(1):24–31. doi: 10.1016/S1672-0229(04)02004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzoni M, Bourjea J, Chen B, Crain BJ, Cui L, Fiorentino V, Hartmann S, Hendricks S, Ketmaier V, Ma X, Muths D, Pavesi L, Pfautsch S, Rieger MA, Santonastaso T, Sattabongkot J, Taron CH, Taron DJ, Tiedemann R, Yan G, Zheng B, Zhong D. Permanent genetic resources added to molecular ecology resources database 1 April 2011–31 May 2011. Mol Ecol Resour. 2011;11(5):935–936. doi: 10.1111/j.1755-0998.2011.03046.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Length distribution of unigenes on An. sinensis reference genome.
Alignment between An. sinensis transcriptome and reference genome. Only longest alignment was kept in the cases that a unigene was returned with multiple blat hits.
Total codon usage and codon usage bias in An. sinensis transcriptome.
The ID and FPKM of unigenes, and the CBI, Nc and GC3 of ORF in each unigene in An. sinensis.
A plot of Nc versus GC3 (Nc-plot) for An. sinensis ORFs. The pink dotted curve represents the expected curve between GC3 and Nc under random codon usage. Blue dot each indicates a corresponding ORF of each unigene (totally 24,361 unigenes).
Relationship between CBI (codon bias index) and expression level (Log10 (FPKM)) of all An. sinensis transcriptome unigenes.
SSR identification of An. sinensis unigenes.