

Abstract
Objective
This study tests the hypothesis that injury to the somatosensory cortex is associated with periorbital allodynia and increases in nociceptive neuropeptides in the brainstem in a mouse model of controlled cortical impact (CCI) injury.
Methods
Male C57BL/6 mice received either CCI or craniotomy-only followed by weekly periorbital von Frey (mechanical) sensory testing for up to 28 days post-injury. Mice receiving an incision only and naïve mice were included as control groups. Changes in calcitonin gene-related peptide (CGRP) and substance P (SP) within the brainstem were determined using enzyme-linked immunosorbent assay and immunohistochemistry, respectively. Activation of ionized calcium-binding adaptor molecule-1–labeled macrophages/microglia and glial fibrillary acidic protein (GFAP)-positive astrocytes were evaluated using immunohistochemistry because of their potential involvement in nociceptor sensitization.
Results
Incision-only control mice showed no changes from baseline periorbital von Frey mechanical thresholds. CCI significantly reduced mean periorbital von Frey thresholds (periorbital allodynia) compared with baseline and craniotomy-only at each endpoint, analysis of variance P < .0001. Craniotomy significantly reduced periorbital threshold at 14 days but not 7, 21, or 28 days compared with baseline threshold, P < .01. CCI significantly increased SP immunoreactivity in the brainstem at 7 and 14 days but not 28 days compared with craniotomy-only and controls, P < .001. CGRP levels in brainstem tissues were significantly increased in CCI groups compared with controls (incision-only and naïve mice) or craniotomy-only mice at each endpoint examined, P < .0001. There was a significant correlation between CGRP and periorbital allodynia (P < .0001, r = −0.65) but not for SP (r = 0.20). CCI significantly increased the number of macrophage/microglia in the injured cortex at each endpoint up to 28 days, although cell numbers declined over weeks post-injury, P < .001. GFAP+ immunoreactivity was significantly increased at 7 but not 14 or 28 days after CCI, P < .001. Craniotomy resulted in transient periorbital allodynia accompanied by transient increases in SP, CGRP, and GFAP immunoreactivity compared with control mice. There was no increase in the number of macrophage/microglia cells compared with controls after craniotomy.
Conclusion
Injury to the somatosensory cortex results in persistent periorbital allodynia and increases in brainstem nociceptive neuropeptides. Findings suggest that persistent allodynia and increased neuropeptides are maintained by mechanisms other than activation of macrophage/microglia or astrocyte in the injured somatosensory cortex.
Keywords: post-traumatic headache, traumatic brain injury, calcitonin gene-related peptide, trigeminal, allodynia, central sensitization
Long-term disability from traumatic brain injury (TBI) is prevalent occurring in approximately 3.17 million persons on an annual basis.1 Among the long-term consequences of TBI, post-traumatic headache (PTH) disorder represents the most common chronic pain syndrome within this patient population.2–7 PTH disorders are highly prevalent across all grades of TBI severity.8 Mild and moderate TBI is more prevalent compared to severe cases of TBI; however, there is a current controversy surrounding epidemiologic reports that cite a higher incidence of PTH for mild TBI compared with moderate and severe cases. A recent prospective 12-month study of TBI patients provides compelling evidence that the prevalence of PTH is unrelated to injury severity.9
Important aspects of PTH to consider relate to the time-course of this disorder. In many patients, it resolves in 3 months; in others, it persists for much longer. Currently, PTH, as classified by the International Classification of Headache Disorders, is defined as headache secondary to head trauma that develops within 7 days after injury or regaining consciousness. TBI is a risk factor for acute episodic headache (headache occurring less than 15 days/month) persisting less than 3 months transitioning into chronic headache (≥15 days/month).10,11 Clinical evidence suggest that the persistence of PTH beyond the expected tissue-healing time-course would argue for its chronicity, as well as mechanisms of central sensitization.12–14 A recent prospective study found the onset of PTH occurred between 7 and 30 days after injury or later in nearly half of patients.15 A study by Ofek and Defrin found that patients with TBI had a mean onset of chronic pain, including head pain, just over 6 months after injury.14 Headache was reported at 3, 6, and 12 months in 41% of patients with TBI in a study by Hoffman and colleagues.9 Studies using quantitative sensory testing (eg, von Frey mechanical stimuli for allodynia [cutaneous sensitivity to mechanical stimuli that are innocuous under normal conditions]) show that 40% of patients with TBI experience chronic head and face pain after head injury.14,16 Significant reductions in pressure-pain thresholds were found at least 1 year after mild-to-moderate TBI.16 To date, investigations into the proposed mechanisms and treatments for PTH have been impeded by a lack of preclinical models. Therefore, a research initiative implemented by our laboratory is to study mechanisms of PTH and its chronification using a well-known animal model of TBI, controlled cortical impact (CCI) injury.
Headache pain, whether acute or chronic, involves abnormal activation of the trigeminovascular system. Using a model of diffuse TBI, Hall and Lifshitz have characterized hypersensitivity of the whiskers to be associated with neuroplasticity in the cortical barrel circuit and thalamus.17 Peripheral axons of the trigeminal ganglion innervate the peri-orbital skin, facial whiskers, anterior scalp, meninges, and cerebral vasculature. Periorbital and facial allodynia have been well-documented in rodent migraine models following infusion of an inflammatory soup over the meninges.18–20 Afferent fibers of the trigeminal ganglia relay nociceptive information to the trigeminal nuclei in which the neuropeptides, calcitonin gene-related peptide (CGRP) and substance P (SP), play important roles. CGRP and SP are also important in normal physiologic and pathologic function including cerebrovascular regulation and the development of neurogenic inflammation.21–26 CGRP has been particularly well-studied for its role in primary headache disorders such as migraine.27,28 To date, preclinical studies examining allodynia and the potential role of CGRP and SP in PTH are nonexistent. This study aimed to characterize periorbital sensory changes to mechanical stimuli (allodynia) and neuroplasticity in the brainstem in a mouse model of TBI, as this has not yet been reported in any model of TBI.
The present study tests the hypothesis that focal injury to the somatosensory cortex will be associated with increased neuropeptides within the brainstem trigeminal nucleus caudalis and periorbital allodynia in a mouse model of CCI injury. The time-course for macrophage/microglial and astrocyte responses in the somatosensory cortex after CCI were examined because these cell populations are a potential cellular source of mediators (eg, cytokines, nitric oxide [NO], excitatory amino acids) of nociceptor sensitization known to contribute to behavioral morbidity. The pattern of periorbital allodynia over 4 weeks in mice with CCI and those undergoing a craniotomy only was characterized, while changes in the nociceptive neuropeptides, CGRP, and SP within the brainstem were determined. The temporal relationships between periorbital sensory changes (allodynia), central nociceptive neuropeptides, and gliosis (macrophage/microglia and astrocyte activation) were examined.
EXPERIMENTAL PROCEDURES
Experimental Design, Animal Care, and Anesthesia
Prior to initiating any research, the Thomas Jefferson University Institutional Animal Care and Use Committee reviewed and approved the research protocol and approved the use of male C57BL/6 mice from Charles River as experimental animals. Animal care and use was monitored by the University Animal Care and Use Committee to assure compliance with the provisions of Federal Regulations and the National Institutes of Health (NIH) “Guide for the Care and Use of Laboratory Animals.” All animals were housed in the Thomas Jefferson University Laboratory Animal Services Facility that is accredited by the American Association for the Accreditation of Laboratory Animal Care and complies with NIH standards.
The animals were deeply sedated with isoflurane throughout the surgical procedures and injury. Eighty-four male C57BL/6 mice (approximately 8 weeks of age and weighing approximately 22–24 g) were randomly divided into CCI (n = 38), craniotomy-only (n = 36), or control (n = 10) groups. CCI and craniotomy mice were further divided into 7-day (n = 14/group), 14-day (n = 12/group), and 28 day (10–12/group) subgroups. Craniotomy-only mice received the same surgical procedures as CCI without the cortical impact. Two control groups included naïve mice (n = 6) that did not receive an incision, craniotomy, or CCI (naïve group), and incision-only mice (n = 4) that received a skin incision only without craniotomy or CCI (incision-only group). All mice were allowed to recover from anesthesia and kept in separate cages postoperatively with unrestricted food and water. Animals were euthanized with a lethal dose of pentobarbital (120 mg/kg, intraperitoneal [I.P.]) for post-mortem histological analysis.
TBI
TBI was produced using a CCI injury model, as described previously by our laboratory in mice.29–32 Mice were anesthetized with isoflurane (5% induction; 2% maintenance). Previous studies by our laboratory demonstrate that the well-established model of CCI is a valid, accurate model to study PTH. Features of the CCI injury model in rodents include a reproducible ipsilateral contusion to the somatosensory cortex with regions of diffuse axonal injury, all of which have been well-described.33–39 To ensure aseptic techniques, heads were shaved using electric clippers and prepped with betadine and alcohol prior to incision. Thermistors (Physiotemp, Clifton, NJ, USA) were implanted into the temporalis muscle and rectum to monitor and maintain respective brain and core temperatures at 37 + 0.5°C.The head was fixed in a stereotaxic frame, and a high-speed drill was used to create a right-sided 4-mm craniotomy positioned at Bregma exposing the somatosensory cortex. Care was taken to leave the dura intact, and the bone flap was removed. TBI was induced using an electromagnetic stereotaxic impactor (Leica Biosystems Richmond formerly MyNeuroLab, Richmond, IL, USA) consisting of a 3-mm diameter, rounded aluminum tip. CCI was induced using the parameters of 1.0-mm depth, 3.0 m/second velocity, and 100-millisecond contact time at an impact angle perpendicular to the cortical surface. The injury cavity is located between 0 and −1.0 mm anterior-posterior (AP) to Bregma (Fig. 1).40,41 The approximate location of the focal cavity core is −1.0 mm AP to Bregma and includes the lateral primary motor cortex and primary sensory hind-limb and fore-limb regions but not the primary sensory barrel field (Fig. 1b).41 Following injury, the bone flap was replaced and sealed with permanent cyanoacrylate-based fast-acting adhesive closures, and the skin sutured closed with 6.0 black silk sutures. The use of the adhesive allows the bone flap to be reintegrated with the existing calvarium, prevents the generation of bleeding during closure, and eliminates the risk of additional brain injury once mice awake from anesthesia that may occur from leaving a piece of the bone plate out. A single dose of short-acting buprenorphine (0.05 mg/kg; subcutaneous injection) was administered before removal from isoflurane to facilitate acute postoperative recovery.
Fig 1.
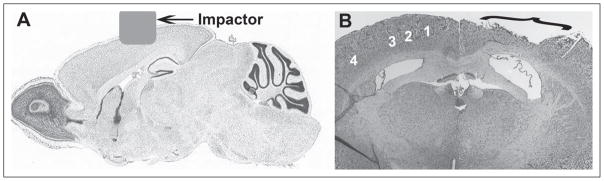
Images depict a sagittal (A) and coronal (B) view of a mouse cortical impact injury. Image A shows the location of the impactor tip over the sensory cortex on a sagittal mouse brain section adapted from the Allen Mouse Brain Atlas (http://mouse.brain-map.org).40 Image B shows a nissl stained brain section at approximately −1.0 posterior to Bregma showing the location of the cavitation core in the somatosensory cortex after impact injury. The somatosensory cortex regions according to The Mouse Brain Atlas by Franklin and Paxinos41 are indicated by numbers 1–4 including primary motor cortex (1), primary sensory cortex hind-limb region (2), primary sensory cortex forelimb region (3), and primary sensory cortex barrel field (4).41 Gross morphologic damage after cortical impact injury induced by our laboratory includes primary motor and sensory hind limb and fore limb but not the barrel field.
Periorbital von Frey Sensory Testing
von Frey mechanical sensory testing was performed for incision-only control (n = 4), naïve control (n = 6), craniotomy-only (n = 36), and CCI (n = 38) mice. Mice were tested during the daylight portion of their circadian cycle using a universal plastic tube restraint designed for rodents up to 40 g (inner diameter 3.5 cm, length 10 cm). Mice enter the restraint device uncoaxed. Restraint is performed without force, tight restraint or change in the degree of restraint for a short time, less than 15 minutes; mice acclimate to the restrainer for 5–10 minutes to find their positioning plus no longer than 5 minutes of testing. The restrainer is inverted at a 30-degree angle incline, as mice prefer an upward facing position that facilitates rapid entry into testing position and prevents mice from turning around during testing (Fig. 2). Baseline, 7-, 14-, 21-, and 28-day post-tests were conducted on all mice. von Frey monofilaments (North Coast Medical, Inc., Morgan Hill, CA, USA) with calibrated bending forces according to the size of the filament were used to test for periorbital sensory changes (periorbital allodynia). Allodynia refers to an abnormal response to a stimulus that would be innocuous under normal conditions. von Frey sensory testing is a well-known validated method to test for allodynia in rodents and evaluate sensory changes after central nervous system (CNS) injury.42 von Frey sensory testing was adapted from established protocols used by the laboratories of Oshinsky for periorbital testing18,43 and Winkelstein for forepaw testing44–46 in rats. Specifically, von Frey forces were modified to take into account the lower thresholds typically observed in normal control mice (<2.0 g) rather than the higher forces typically used for testing rats (up to 10 g).47,48 Mechanical thresholds were determined by applying the von Frey monofilaments to the periorbital region on the right and left side of the face over the rostral portion of the eye and distal from the suture site. In order for a stimulation to elicit a positive response, the filament must make firm perpendicular contact with the skin, causing the filament to bend and thereby producing a precise bending force that was previously calibrated by the manufacturer. A positive response was characterized by the following criteria: mouse vigorously stroked its face with the forepaw, head withdrawal from the stimulus, or head shaking. During our preliminary studies, the von Frey stimuli were presented in a sequential ascending order to determine the range of filament threshold forces in our mouse model (0.008–2.0 g) to be used for all testing. From our preliminary studies, filaments with graded forces (0.008, 0.02, 0.04, 0.07, 0.16 g) were designated as the testing filaments for all baseline measures and post-testing. For baseline and post-test measures, each mouse was stimulated 10 times bilaterally with each filament. Force thresholds were defined as greater than a 50% response frequency for a von Frey stimulus. Baseline threshold values and naïve data showed a mean force threshold to fall between 0.07 and 0.16 g. Periorbital thresholds for each filament are presented as mean threshold (g) ± standard error of the mean (SEM).
Fig 2.

Image of von Frey monofilament and restrainer apparatus used for evaluating periorbital mechanical sensory changes (allodynia). Inset indicates regions where periorbital von Frey stimulus is applied (white x) during testing. C57BL6 black eyes are delineated by white dots to indicate the location relative to stimuli.
Immunohistochemistry
Immunohistochemistry was performed on 6 control (3 naïve controls and 3 incision-only controls), 16 craniotomy-only, and 16 CCI mice to evaluate the changes in SP and the time-course for microglial/macrophage and astrocytes activation. Immunohistochemical techniques were utilized for these analyses in order to characterize both the temporal and spatial changes within the brain regions of interest (ROI), primarily the somatosensory cortex and medullary trigeminal zones. On postoperative days 7, 14, and 28, mice were administered a lethal dose of sodium pentobarbital and underwent cardiac perfusion with heparinized saline, followed by 4% paraformaldehyde. Brains were quickly removed and post-fixed in 4% paraformaldehyde for 2 hours at which point they were transferred to 30% sucrose in PO4 buffer until sinking for cryo-protection. Brains were divided into brainstem and forebrain regions, and sectioned coronally with a cryostat at −24°C (10-μm thick, 1 : 10 series, 100 microns between series), and air-dried overnight.
Immunohistochemical analyses were performed on forebrain (Bregma +1.10 to −2.5 mm) and/or brainstem tissues (Bregma −5.40 to −8.24 mm). Tissues were washed and incubated in 10% normal goat serum in O.3% Triton-100 for 1 hour. Alternate sections were labeled using the following primary rabbit antibodies: (1) SP, a nociceptive neuropeptide (1 : 250; Millipore catalog no. AB1566); (2) ionized calcium-binding adaptor molecule-1 (Iba-1), a cytoplasmic peptide selectively expressed in monocytes and microglia (1 : 250; Wako Pure Chemical Industries, Osaka, Japan); (3) glial fibrillary acidic protein (GFAP) (1 : 500; Millipore no. AB1540) specific to astrocytes. For Iba-1-labeled slides, the nuclear counterstain DAPI (Invitrogen Life Science, Grand Island, NY, USA) was applied for stereological cell counting. After 24 hours of incubation with the primary antibody at room temperature, tissues were incubated with fluorescent secondary antibodies DyLight 488- or 549-conjugated AffiniPure Goat anti-rabbit immunoglobulin G (Jackson ImmunoResearch, West Grove, PA, USA) for 2 hours. Negative control staining was performed by omitting the primary antibodies.
Quantification of SP, GFAP-Labeled Astrocytes, Iba-1-Labeled Macrophage/Microglia
Images were captured using an Olympus BX-51 (Center Valley, PA, USA) and Image capture software, SPOT Advanced (Sterling Heights, MI, USA). Quantification of %SP and GFAP immunoreactivity were performed, as described previously by Elliott and colleagues for 20× red blue green (RBG) images using ImageJ software (version 1.43J, NIH, Bethesda, MD, USA).23,24,26 Although SP immunoreactivity was present in some rostral brainstem sections during our preliminary investigation, majority of the SP immunoreactivity was present in the caudal trigeminal nucleus. Therefore, subsequent quantification of SP was performed for the trigeminal nucleus caudalis region (−7.76 to −8.24 mm Bregma) identified using the mouse brain atlas by Franklin and Paxinos.41 Briefly, quantification of SP immunoreactivity included 3 square ROI (0.146 mm2) used to measure 3 sections within the trigeminal nucleus caudalis per mouse brain. Immunohistochemistry was chosen to evaluate changes in SP in order to determine the anatomic regions of the brainstem that were affected.
Quantification of percent GFAP immunoreactivity on 20× RBG images using ImageJ software included 3 square ROI (0.28 mm2) measured from the preserved tissue adjacent to the injury cavity for 3 sections per brain between −0.5 and −1.0 mm posterior to Bregma (9 measurements per mouse). The color threshold for the RBG image was set to select the green fluorescent GFAP immunoreactive product, and the pixel count was measured and recorded. Using the color-thresholding feature available from ImageJ, the pixel threshold is set so that fine processes of quiescent astrocytes were measured. The total number of pixels was then measured by selecting all the pixels in the region of interest to ensure consistent sampling between specimens and recorded. Subsequently, percent GFAP immunoreactivity is calculated as the measured thresholded immunofluorescent product in pixels divided by the total pixels the region of interest. The percent immunoreactivity for the 6 ROI was averaged and reported as mean ± SEM.
Unbiased stereological analyses for cell counting was performed using similar methodology as previously described by our laboratory.49 Using the Spot Advanced image capture software and NIH ImageJ 1.43, Iba-1 cells are counted in 5 non-overlapping, high-power, rectangular counting frames (high-power field [HPF], 300 μm × 220 μm) at 400× magnification selected from preserved perilesional tissue for 3 sections per brain. HPFs in naïve animals were selected from tissue sections corresponding to the location of the lesion in injured animals. Within each field, only those positively staining cells with a nucleus in the focal plane were counted.
Cell-specific components of the neuroinflammatory response to injury were evaluated for Iba-1-labeled macrophages/microglia and GFAP-labeled astrocytes utilizing estimated cell counts and percent immunoreactivity, respectively. Different methodology was used for the assessment of glial responses because these individual glial cell populations undergo different morphologic changes after injury. In particular, reactive astrocytes hypertrophy and elongate their processes, whereas microglia retract their processes. Although both increase the production of the proteins of interest, the hypertrophic and elongated processes characteristic of a reactive astrocyte phenotype are more suited for quantifying the percent immunoreactivity and allows for a more rapid means to quantify changes; cell counts were utilized to evaluate the macrophage/microglial component of the cellular neuroinflammatory response because infiltrating macrophages in addition to migrating and proliferating microglia are known to contribute to the increase in the number of Iba-1-labeled cells. Circulating macrophages and resident activated microglia are not distinguishable by our immunohistochemical techniques.
Enzyme-Linked Immunosorbent Assay for CGRP
After mice (6 control, 17 craniotomy, and 20 CCI) were euthanized with a lethal dose of sodium pentobarbital (120 mg/kg, I.P.), brains were rapidly dissected, and brainstem tissues were flash frozen in liquid nitrogen and stored at 80°C. Brainstem tissues were sectioned according to the mouse brain atlas by Franklin and Paxinos (−5.40 to −8.24 mm Bregma) to include the beginning of the spinal trigeminal tract to the most caudal section of the spinal trigeminal nucleus (ie, trigeminal nucleus caudalis).41 Thus, any detection of CGRP will most likely be released from the central terminals of the trigeminal ganglion at the medullary trigeminal nuclei, although additional sources of CGRP from other sensory afferents were not excluded from our samples.50 All brainstem samples were homogenized unpooled with ethylene-diamine tetraacetic acid-free complete protease inhibitor cocktail tablets using 50 μL/10 mg of tissue (Roche Diagnostics, GMPH, Basel Switzerland). Homogenates were centrifuged at 14,000 rpm for 15 minutes at 4°C. Total protein content for each sample was determined using BCA-200 protein assays (Bicin Choninic Acid, Pierce, Rockford, IL, USA). Tissue lysates were analyzed for CGRP according to the manufacturer’s protocol (CGRP enzyme-linked immunosorbent assay [ELISA] Kit; Cayman Chemicals, Ann Arbor, MI, USA; cat# 589001). Each sample was run in duplicate, and data (pg CGRP protein) were normalized to mg total protein.
Statistical Analyses
To determine the effects of CCI and craniotomy on ipsilateral and contralateral periorbital mechanical sensory responses, 2-way analysis of variances (ANOVAs) were used with the factors: time (baseline, 7, 14, 21, and 28) and group (naïve, incision-only, craniotomy-only, and CCI). ANOVAs were followed by Bonferroni post-hoc tests for multiple comparisons with adjusted P values. Single-factor bonferroni post-hoc analyses were also performed to evaluate within group comparisons to compare each week to baseline data and at matching temporal endpoints for between group comparisons. To analyze group differences for SP, Iba-1, and GFAP immunohistochemical and CGRP ELISA data, group comparisons were made using single univariate ANOVA, followed by Bonferroni post-hoc comparisons with an adjusted P value. The following group comparisons were performed: (1) 7-, 14-, and 28-day craniotomy-only compared with control; (2) 7-, 14-, and 28-day CCI mice compared with control; (3) craniotomy-only compared with CCI groups at matched 7-, 14- and 28-day endpoints. A Pearson’s correlation analysis was used to examine the relationships of CGRP and SP with periorbital allodynia. All data were analyzed using the GraphPad Prism 5 statistical program (La Jolla, CA, USA). Significance levels were set at P < .05 for all statistical analyses, adjusted P values are reported, and data are reported as the mean and SEM.
RESULTS
Periorbital von Frey Thresholds
Daily von Frey testing was performed to measure baseline thresholds and determine whether there was any effect of repeated temporary (>15 minutes) restraint over time in 4 naïve mice. Mean baseline threshold was 0.11 ± 03 g in naïve mice over time. Baseline, right vs left von Frey thresholds were not significantly different, P = .22. Repeated, daily restraint did not alter right and left baseline von Frey thresholds, P = .31 and .53, respectively. von Frey sensory testing for incision-only mice showed no changes from baseline periorbital mechanical thresholds for up to 14 days after incision, P = .97 (Fig. 3A). Mean ipsilateral peri-orbital thresholds showed significant differences by group (P < .0001 and F = 30.19) and time (P < .0001 and F = 9.031), and a significant interaction (P < .04 and F = 2.649). CCI resulted in reductions in mean periorbital thresholds (mean threshold range 0.01–0.03 g) that were consistently lower (nearly 10-fold) compared with craniotomy-only mean thresholds (0.05–0.09 g). Post-hoc analyses showed that mean ipsilateral periorbital threshold was significantly reduced (periorbital allodynia) in CCI mice compared with baseline at 7 days (P < .001), 14 days (P < .001), 21 days (P < .001), and 28 days (P < .001) after injury (Fig. 3B). Mean ipsilateral periorbital thresholds were significantly reduced in CCI mice compared with craniotomy-only mice at 7 days (P < .001), 14 days (P < .01), 21 days (P < .001), and 28 days (P < .001) after injury. Mean ipsilateral threshold was significantly reduced in the craniotomy-only group compared with baseline at 14 days (P < .001). Mean ipsilateral thresholds for 7-, 21-, and 28-day craniotomy mice were not statistically different from baseline threshold. ANOVA results for mean contralateral thresholds showed significant differences by group (P < .003 and F = 9.049) and time (P < .002 and F = 4.584) and no significant interaction (P = .57 and F = 0.7262). Post-hoc analyses reveal that mean contralateral threshold was significantly reduced in CCI groups compared with baseline at 7 (P < .01), 14 (P < .001), 21 (P < .001), and 28 days (P < .01) (Fig. 3C). Mean contralateral threshold was significantly reduced in CCI groups compared with craniotomy at 21 and 28 days after injury (P < .05). Mean contralateral threshold in craniotomy-only mice was significantly reduced compared with baseline at 14 days (P < .001). Mean contralateral thresholds in craniotomy mice at 7, 21, and 28 days were not statistically different from baseline threshold. There were no significant group differences for contralateral thresholds between CCI and craniotomy at 7 and 14 days after injury in which were contralateral thresholds generally higher (less sensitive) than ipsilateral thresholds.
Fig 3.

Mean periorbital threshold (g) in response to von Frey (mechanical) stimuli for an incision-only, controlled cortical impact (CCI) and craniotomy-only groups, n = 8–12/group. Mean periorbital threshold for incision-only mice showed no changes from baseline periorbital mechanical thresholds over 2 weeks (A). Mean ipsilateral periorbital threshold was significantly reduced in CCI mice compared with baseline at 7 (***P < .001), 14 (***P < .001), 21 (***P < .001), and 28 days (***P < .001) after injury (B). Mean ipsilateral periorbital thresholds was significantly reduced in CCI mice at 7 (###P < .001), 14 (##P < .01), 21 (###P < .001), and 28 days (###P < .001) after injury compared with craniotomy-only mice. Mean ipsilateral threshold was significantly reduced in the craniotomy-only mice at 14 days compared with baseline (***P < .001). Mean ipsilateral thresholds for 7-, 21-, and 28-day craniotomy mice were not statistically different from baseline threshold. (C) Mean contralateral threshold was significantly reduced in CCI groups compared with baseline at 7(**P < .01), 14 (***P < .001), 21 (***P < .001), and 28 days (**P < .01). Mean contralateral threshold was significantly reduced in CCI groups at 21 and 28 days after injury compared with craniotomy (#P < .05). Mean contralateral threshold in craniotomy-only mice at 14 days was significantly reduced compared with baseline (***P < .001). Mean thresholds in craniotomy mice at 7, 21, and 28 days were not statistically different from baseline threshold. There were no significant group differences for contralateral thresholds between CCI and craniotomy at 7 and 14 days after injury.
Neuropeptides
There were no differences found for any of the cellular responses or neuropeptides of interest between naïve and incision-only control mice. Therefore, the naïve control and incision-only control groups were combined and presented as a control group (control) for all post-mortem immunohistochemical and ELISA analyses.
Immunohistochemical examination of brainstem tissues showed SP immunoreactivity throughout the length of the trigeminal tract and nuclei (rostral to caudal, −5.4 to −8.0 mm posterior to bregma), with the greatest immunoreactive product appearing bilaterally in the most caudal part of the brainstem, the trigeminal nucleus caudalis. There was no difference in naïve and incision-only controls that show a low level of SP immunoreactivity, and therefore, groups were combined under 1 control group for analysis (Fig. 4). High-power micrographs show SP immunoreactive product appearing in a punctuate pattern in the trigeminal nucleus caudalis (Fig. 4D,E). SP immunoreactivity in the trigeminal nucleus caudalis was significantly increased in CCI mice at 7 and 14 days, and in craniotomy-only mice at 7 days compared with control mice (ANOVA P = .0001, and F = 19.16 and 25.62). Post-hoc comparisons show that SP immunoreactivity was significantly increased in CCI mice at 7 (P < .001 and P < .01) and 14 days (P < .05) compared with control and craniotomy mice. SP immunoreactivity was also transiently increased in craniotomy mice on postoperative day 7 compared with controls, P < .001. SP immunoreactivity in CCI mice at 28 days after injury was not statistically different compared with control mice. There were also no significant increases in SP immunoreactivity in craniotomy-only mice at 14 and 28 days compared with control mice. Pearson’s correlation showed that there was no significant correlation between SP and periorbital von Frey thresholds, r = −0.20.
Fig 4.

Percent substance P (SP) immunoreactivity in the spinal trigeminal nucleus in controlled cortical impact (CCI) and craniotomy mice at 7, 14, and 28 days after injury, n = 6/group. SP immunoreactivity was increased in CCI mice compared with control mice at 7 (***P < .001) and 14 days (*P < .05). SP immunoreactivity was increased in CCI mice compared with craniotomy mice at 7 (##P < .01) and 14 days (#P < .05). SP immunoreactivity was increased in 7-day craniotomy-only mice compared with control (***P < .001). SP immunoreactivity in 28-day CCI, 14-day craniotomy-only, and 28-day craniotomy-only mice were not significantly different compared with control mice. SP immunofluorescent images of incision control (A and D), craniotomy-only (B), and CCI mice (C and E). Note: SP immunoreactivity is not visible in control brains under low 4× magnification (A) though a low level of staining is visible under higher 60× magnification (D). Increases in SP immunoreactivity is visible in craniotomy-only mice (arrowheads, B) and CCI mice (arrowheads, C). Note the absence of SP immunostaining in the middle region of the caudal trigeminal nucleus in craniotomy mice (B), whereas increased SP is shown throughout the entire dorsal-to-ventral regions of the caudal trigeminal nucleus in CCI mice (C). High-power micrographs showing SP immunoreactive product appearing in a punctuate pattern in incision control (D) and increased in CCI (E) caudal trigeminal nucleus. White scale bars = 100 μm. Color images are available online.
CGRP levels for naïve and incision-only mice were not significantly different and were reported as controls (Fig. 5). CCI mice had a significant increase in CGRP levels in brainstem tissues compared with control and craniotomy mice (ANOVA P < .0001, and F = 14.39 and 14.38). CGRP levels were significantly increased in CCI mice at 7 (P < .01), 14 (P < .05), and 28 days (P < .01) after injury compared with control mice (Fig. 5). CGRP levels were significantly increased in CCI mice compared with craniotomy-only mice at 7 (P < .05) and 28 days (P < .001) after injury (F = 19.58). CGRP levels were significantly increased in craniotomy-only mice at postoperative days 7 and 14 (P < .05 and P < .001, respectively) but not statistically different at 28 days compared with control mice. Increased CGRP levels was significantly correlated with decreased periorbital mechanical thresholds, P < .0001 and Pearson’s r = −0.65.
Fig 5.

Calcitonin gene-related peptide (CGRP) levels (pg/mg of protein) in the brainstem measured using enzyme-linked immunosorbent assay. CGRP levels were significantly increased in controlled cortical impact (CCI) mice compared with control mice at 7 (**P < .01), 14 (*P < .05), and 28 days (**P < .01) after injury (A). CGRP levels were significantly increased in CCI mice compared with craniotomy-only mice at 7 (#P < .05) and 28 days (###P < .001) after injury. CGRP levels were significantly increased in 7- and 14-day craniotomy-only compared with control mice (*P < .05 and ***P < .001, respectively). CGRP levels were not significantly different between 28-day craniotomy-only and control mice. Increases in CGRP levels were significantly correlated with decreases in periorbital von Frey thresholds, P < .0001 and Pearson’s r = −0.65 (B).
Macrophage/Microglia and Astrocyte Responses
The number of Iba-1-labeled macrophage/ microglia in the somatosensory cortex for naïve and incision-only mice was not statistically different, in which group cell counts were combined and reported as controls (Fig. 6). There were significant increases in the number of perilesional Iba-1 immunoreactive macrophage/microglia at 7, 14, and 28 days after CCI compared with control mice (ANOVA P < .0001 and F = 27.17). The greatest number of Iba-1 cells was found at 7 days after CCI, in which there was a progressive decline in numbers at days 14 and 28 (Figs. 6 and 8). Post-hoc results show that the number of perilesional Iba-1 cells was significantly increased in CCI mice at 7 (P < .001), 14 (P > .001), and 28 days (P < .05) compared with control mice. Also, the number of perilesional Iba-1 cells was significantly increased in CCI compared with craniotomy-only at 7 and 14 days after injury, P < .001 and P < .01, respectively (Fig. 6). There was no significant increase in the number of Iba-1 cells in the contralateral cortex or in craniotomy-only mice at any time point. Although the number of macrophage/microglia were not significantly increased in craniotomy-only mice at any time point examined, increased Iba-1 macrophage/ microglial immunoreactivity was noted in the area that delineates the craniotomy site depicted in Figure 6, which was completely resolved by 28 days after craniotomy. It should be noted that a methodological limitation of this study is that our cell count technique does not include analysis of the morphological changes that microglia undergo during various stages of activation.
Fig 6.

Ionized calcium-binding adaptor molecule-1 (Iba-1+) microglial/macrophage cell counts in the right somatosensory cortex of controlled cortical impact (CCI) and craniotomy groups at 7, 14, and 28 days after injury and control mice, n = 6/group. The number of perilesional microglia/macrophage cells was significantly increased in CCI compared with control mice at 7 (***P < .001), 14 (***P < .001), and 28 days (**P < .01) after injury. The number of perilesional microglia/ macrophage cells was significantly increased in CCI compared with craniotomy mice at 7 (###P < .001) and 14 days (##P < .01). Low-power images showing Iba-1 immunoreactivity in the somatosensory cortex region of interest captured with a 4× objective (A–D). Although the number of macrophage/ microglia was not significantly increased in craniotomy-only mice compared with controls at any time point examined, a slight increase in Iba-1 immunoreactivity is shown at 7 days in craniotomy-only mice under the craniotomy site (arrowheads) (A), which was completely resolved by 28-day post-craniotomy (B). Iba-1-labeled macrophage/microglial activation at 7 days (A, C) compared with 28 days (B, D) in both CCI and craniotomy groups, which was not completely resolved in the CCI group at 28 days (arrow). White scale bar = 100 μm. Color images are available online.
Fig 8.
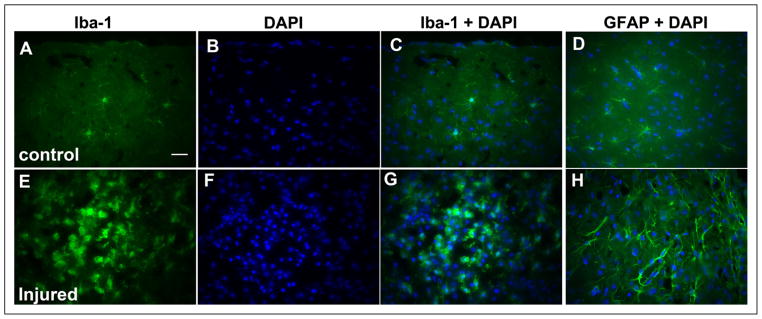
High-power micrographs (40×) showing immunohistochemical labeling of ionized calcium-binding adaptor molecule-1 (Iba-1) and glial fibrillary acidic protein (GFAP) with DAPI nuclear double labeling from the somatosensory cortex of control (top A–D) and controlled cortical impact (CCI)-injured (lower E–H) mice at postoperative day 7. Resting microglia with thin, branched processes labeled with antibodies against Iba-1 (A), DAPI nuclear stain (B), merged double labeling with anti-Iba-1, and DAPI (C) in an uninjured mouse brain. Resting astrocytes double-labeled with anti-GFAP and DAPI in an uninjured mouse brain (D). Anti-Iba-1 and anti-GFAP immunoreactivity, and the number of immunoreactive cells are increased in the perilesional zone after CCI (E–H). Activated macrophage/microglia cells showing retracted processes with amoeboid-like soma, increased anti-Iba-1 immunoreactivity, and cell proliferation (E), DAPI staining (F), and merged double labeling with anti-Iba-1 and DAPI (G). Activated astrocytes showing increased GFAP immunoreactivity with elongated, hypertrophied processes, and proliferation double-labeled with DAPI (H). White scale bar = 25 μm.
Astrocyte GFAP immunoreactivity in the somatosensory cortex for naïve and incision-only mice was not statistically different and percent GFAP immunoreactivity values were combined and reported as controls (Fig. 7). ANOVA revealed that GFAP immunoreactivity in the somatosensory cortex was significantly increased in 7-day CCI mice, and 7- and 14-day craniotomy-only mice (ANOVA P < .0001; F = 19.37 and 26.15 for CCI and craniotomy, respectively) compared with controls. Post-hoc comparisons showed increased GFAP immunoreactivity in the ipsilateral cortex in CCI mice at 7 days compared with controls (P < .001). There was no significant increase in GFAP immunoreactivity in the somatosensory cortex in CCI mice at 14 and 28 days post-injury compared with controls (Figs. 7 and 8). GFAP immunoreactivity in craniotomy-only was significantly increased on postoperative days 7 and 14 but not 28 days compared with control mice (P < .001 and P < .05, respectively). There was no significant increase in GFAP immunoreactivity in the contralateral cortex compared with controls at any time point (data not shown).
Fig 7.

The percentage of astrocytes-specific anti-glial fibrillary acidic protein (GFAP) immunoreactivity in the right somatosensory cortex of controlled cortical impact (CCI) and craniotomy groups at 7, 14, and 28 days after injury and in controls, n = 6/group. GFAP immunoreactivity was increased CCI mice compared with controls at 7 days (***P < .001) and compared with craniotomy-only at 7 days (###P < .001). GFAP immunoreactivity was increased in craniotomy-only mice compared with controls at 7 and 14 days after surgery, ***P < .001 and *P < .05, respectively. Images taken at 10× show GFAP-immunoreactive astrocytes in the somatosensory cortex region of interest (A–D). Immunofluorescent images show activated astrocytes at 7 days (A, C) compared with 28 days (B, D) in both CCI and craniotomy groups, which was completely resolved at 28 days after craniotomy. Increased GFAP immunoreactivity in craniotomy-only mice is shown at 7 days in craniotomy-only mice under the craniotomy site (A). Astrocytic glial scarring (arrow) and necrotic tissue (arrowhead) is shown at 28 days after CCI (D). White scale bar = 100 μm. Color images are available online.
DISCUSSION
Cortical Injury Induces Glial Activation in the Somatosensory Cortex, Increased Central Neuropeptides, and Sensory Changes That Are Distinct From Craniotomy-Only
Important findings from the present study are the significant group differences between CCI and craniotomy-only groups for all outcome measures. In many preclinical studies of TBI, craniotomized animals have been used as sham controls; however, recent studies show tissue trauma in the brain and behavioral abnormalities after craniotomy.18,20,51 It is important to note the finding of mild, transient allodynia, changes in neuropeptides and gliosis in our craniotomy-only group. The presence of mild periorbital allodynia in the brainstem in craniotomized mice is not surprising given the common finding of chronic headache in patients (50% of patients) undergoing craniotomy for pathology other than direct head trauma (eg, elective aneurysm clipping and tumor resection).52 A study by Oshinksy et al also reported that rats receiving saline through a cannula had a small reduction in periorbital thresholds at 2 weeks post-craniotomy. A key difference between our study and the study by Oshinsky et al is rats with thresholds that did not return to baseline at 7 days post-craniotomy were excluded as controls from the Oshinsky study. Conversely, reports of increased cytokines in the cerebral cortex and meninges of craniotomized control rats accompanied by deficits on neurological and acoustic startle tests demonstrate that craniotomy induces a form of mild tissue trauma in the brain.20,51 Proposed mechanisms for post-craniotomy headache include trauma to the pericranial muscles and periosteum, dural irritation, reduced volume of cerebrospinal fluid, and aseptic meningitis.53 Even slight glial activation may produce mediators such as pro-inflammatory cytokines, inducible NO, or excessive glutamate that can transiently stimulate dural nociceptors. In addition, Stokely et al reported a significant increase in activated dural mast cells after craniotomy that was accompanied by increases in histamine and blood-brain-barrier permeability changes in the cerebral cortex.54 Histamine release by activated mast cells may be an additional source of transient sensory changes induced by craniotomy in our model.
Periorbital Mechanical Allodynia Persists After Cortical Impact
Mean baseline periorbital von Frey mechanical threshold was 0.11 ± 0.5 g (individual thresholds ranged between 0.07 and 0.6 g) in a mouse model of CCI. Baseline von Frey thresholds were within the reported threshold range (0.04–2.0 g) for paw or trunk von Frey testing in murine models of neuropathic pain.47,48,55 Overall, CCI resulted in reductions in mean periorbital thresholds (mean threshold range 0.01–0.03 g) that were consistently lower compared with baseline (0.07–0.6 g) and craniotomy-only thresholds (0.05–0.09 g). Similar sensory changes were demonstrated in a mouse model of spinal contusion in which an increased response frequency was reported for a 0.02-g von Frey stimulus applied to the trunk.47 Reductions in von Frey thresholds in our mouse CCI model are also comparable with the reductions noted in rat neuropathic pain models; however, thresholds for rats are typically on a higher scale compared with mice. In rats, periorbital or forepaw baseline thresholds ranged between 8 and 10 g or higher, and were reduced to <2 g in models of migraine or cervical nerve compression.18,25,46,56
A significant reduction in periorbital thresholds in response to von Frey mechanical stimuli (periorbital allodynia) was sustained up to 4 weeks after CCI injury. Our finding of allodynia after CCI is consistent with studies in headache models, in which chemical stimulation of the dura resulted in periorbital allodynia lasting weeks after the final infusion.18,57 Although periorbital and facial allodynia have been shown in migraine models and human migraine patients, this is the first study to report persistent periorbital allodynia in a model of TBI.18–20 An intriguing finding is that periorbital allodynia persisted over 4 weeks, while macrophage/microglia and astrocyte responses were declined. Because microglial and astrocyte responses are near resolution by 4 weeks, the finding that sensory changes are sustained leads to the hypothesis that additional mechanisms maintain the observed behavior. Pro-inflammatory mediators and/or excitatory neurotransmitters released transiently in the traumatized brain may repeatedly stimulate meningeal sensory afferents, activate the trigeminal nociceptive pathway, and in turn induce central sensitization and elicit abnormal sensory responses.
Sensory changes have been reported in studies using a midline fluid percussion model of TBI.17,58 Persistent sensitivity to whisker stimulation after fluid percussion injury was associated with synaptic plasticity within the cortical barrel field.58 Peripheral sensory afferents of the trigeminal ganglia that supply the periorbital cutaneous skin also innervate the rodent facial whisker pad, as well as the meninges and cerebral vasculature. A combination of inflammation, hyperexcitation, and disrupted circuitry in the thalamus and cortex were proposed mechanisms for increased neuronal activation in the thalamus and whisker hypersensitivity after midline fluid percussion injury.17,58,59 Although there is no gross morphologic damage or glial activation within the cortical barrel field in our injury model, changes in synaptic function, molecular inflammatory mediators, and neuronal activation in this area along with other sensory cortices cannot be ruled out and may explain, in part, sensory changes observed in our study. In sum, we propose that allodynia in our model may be due to either repeated activation of meningeal nociceptors and central sensitization, as well as possibly enhanced activation of cortical sensory neurons.
There are a number of excellent reviews describing the potential mechanisms of migraine, such as nociceptor and central sensitization, which may contribute to the periorbital allodynia observed in our model of TBI.60–63 Central sensitization may result from repeated nociceptor sensitization or independent mechanisms. Central sensitization refers to a heightened sensitivity (ie, lowered activation threshold and increased responsiveness to synaptic input) of higher order neurons.13,57 Upon central sensitization of the trigeminovascular system, the function of neurons in the medullary brainstem, upper cervical spinal cord dorsal horn, or in the thalamus and cortex may be altered. According to Woolf and Salter, long-lasting alterations in the expression of transmitters, receptors, or ion channels may alter the normal response characteristics to innocuous stimuli, in turn eliciting allodynia.13 Other post-traumatic disturbances such as sensitization of cortical sensory neurons, loss of inhibitory interneurons or synapses, and cortical spreading depression are potential contributors to a prolonged reduction in periorbital threshold in our CCI model. The precise mechanism of allodynia in our model remains elusive, although nociceptor sensitization is evidenced by increases in CGRP and SP. A limitation of our study is that the nociceptive neuropeptide changes in the dura and trigeminal ganglia after TBI were not determined. In addition, the effects of injury on other central components of the trigeminovascular pain circuitry, such as thalamic and cortical neurons, warrant further examination.
On a final note with regards to allodynia, the presence of bilateral allodynia in the absence of bilateral glial responses in the uninjured somatosensory cortex in our CCI model requires further investigation that is beyond the scope of our study. It is plausible that bilateral allodynia elicited in our model may be due to bilateral changes in pro-inflammatory or excitatory sensitizers known to stimulate nociceptors or possibly alter the excitability of cortical sensory neurons or another unidentified mechanism.
Increases in Central Neuropeptide Levels Persist After Cortical Impact
Our data show increased SP immunoreactivity within the trigeminal nucleus caudalis to persist for 14 days after CCI compared with craniotomy-only; however, SP immunoreactivity was back to naïve levels by 28 days post-injury. A previous study by our laboratory showed elevated SP immunoreactivity within the trigeminal nucleus caudalis 2 days after CCI.29 Although our previous findings indicate that SP is involved in the acute responses to TBI, present findings for changes in SP in the brainstem did not persist nor did they correlate with periorbital allodynia, as was found for CGRP. Combined, our past and present findings suggest that SP may be a therapeutic target for the acute neurogenic inflammatory-related effects of TBI; however, findings point to other targets for prolonged sensory changes.
Increases in CGRP levels in the brainstem persist for 4 weeks after CCI. Increases in CGRP were significantly correlated with periorbital allodynia in our CCI model. Examination of CGRP in the brainstem was based on evidence that the trigeminal ganglion is known to be the primary source of CGRP, whereby CGRP is released in the brainstem from the central terminals of the ganglion.64,65 CGRP immunoreactivity limited to sensory fibers has been reported in the superficial laminae of the spinal trigeminal nuclei and extending into the spinal trigeminal tract in rats.66 In a study of diffuse TBI, CGRP in serum was increased for up to 7 days after injury.67 Increases in nociceptive neuropeptides, CGRP, and SP found by our laboratory and others are in contrast with the acute reductions in CGRP reported by Armstead and colleagues in which these differences may be explained by the time point examined.29,68–70 A series of studies by Armstead and colleagues show that decreases in CGRP in the injured brain during the acute injury phase, within hours after TBI, contributes to impaired cerebral pressure autoregulation.69,70 To the best of our knowledge, our study is the first to report CGRP changes in the brainstem over weeks after injury in a model of TBI.
Sensory afferents of the trigeminal ganglion relay nociceptive information to the brainstem trigeminal complex by releasing CGRP and SP.18,58,71,72 A proposed mechanism driving the initial increases in SP and CGRP after CCI and craniotomy may be the early glial-derived neuroinflammatory cascades acting to stimulate and sensitize nociceptors within the meninges. Inflammatory mediators facilitate nociceptor neurotransmission through directly stimulating trigeminal nociceptors via the transient receptor potential cation channel subfamily V member 1 (TRPV1) receptor or lowering the threshold of trigeminal afferents.63 For example, cytokines, prostanoids, bradykinin, and protons and products of mast-cell degranulation (eg, serotonin, prostaglandin I2, and histamine) released during inflammation are potential sources of nociceptor sensitization after TBI. If the activation threshold of sensory neurons is lowered due to sensitization, subsequent mechanical stimulation of sensory afferents of the trigeminal ganglia using von Frey filaments may trigger increases in nociceptive neuropeptides.73 In sum, we propose that increases in SP and CGRP suggest injury to cortex induces sensitization of nociceptors.
Glial Activation Progressively Declines Over Time After Cortical Impact
In the present study, we found that a significant increase in the number of macrophage/microglia in the injured somatosensory cortex was greatest at 7 days followed by progressive declines at 14 and 28 days, although numbers remained significantly increased above control levels. In our previous study, acute increases in the number of macrophage/microglial activation at 2 days after CCI were at even higher numbers (31% more) compared with our present results at 7 days.29 This decline in the number of Iba-1-labeled cells over time is mostly likely due to a reduction in the number of infiltrating macrophages; however, the different cell types were not distinguishable in the present study. In contrast with macrophage/microglia responses, a significant increase in astrocyte GFAP immunoreactivity was found only at 7 days after CCI. Studies by other laboratories using the CCI injury model report localized microglial and astrocyte activation in the injured cortex and hippocampus to peak around 3 days, persist up to 14 days, with subsequent resolution by 28 days post-injury.34,74,75 Findings that the macrophage/microglial response precedes and outlasts the astrocyte response to TBI has been previously reported.75 Results that macrophage/microglial activation lasts longer compared with astrocyte reactivity suggest that these glial populations make unique contributions to the proposed meningeal nociceptor sensitization or activation of cortical sensory neurons. We propose that transient macrophage/ microglia and astrocyte activation in the somatosensory cortex over weeks after injury may contribute to the early events involved in sensitization of dural nociceptors, as evidenced by increases in nociceptive neuropeptides. More importantly, glial activation in the cortex did not correlate with CGRP or periorbital allodynia because both were sustained when gliosis in the cortex declined over time.
Glial activation in the CNS contributes to the initiation and maintenance of chronic pain states.76,77 Activation of microglia has been implicated in the pathophysiologic mechanisms of both migraine and chronic pain.76,78 Macrophage/microglia are considered to be the predominant source of pro-inflammatory cytokines and inducible nitric oxide synthase (iNOS) after TBI, although astrocytes and neurons are also cellular sources.79 Pro-inflammatory cytokines directly sensitize nociceptors and promote pro-nociceptive pathways such as cyclooxygenase-2-mediated prostanoid release.80,81 NO and constituitive isoforms of NOS, endothelial and neuronal NOS, contribute to the pathology of migraine and TBI; however, the role of iNOS is unclear.82,83 In mice, infusion of an NO donor induced hindpaw allodynia along with increased neuronal activation in the trigeminal nucleus caudalis and upper cervical spinal cord.84
In addition, astrocytes have important functions in the neurovascular unit, as well as glutamate uptake in which reactive astrocytes, as found in our injured brain, may contribute to altered neuronal function. Reactive and/or swollen astrocytes after TBI may have a reduced capacity to clear glutamate, in turn contributing to hyperexcitatory conditions.85 Although cortical glial-derived mediators would be expected to promote increases in excitability, it remains to be determined whether there are significant changes in the excitability of sensory cortical neurons that contribute to allodynia in our injury model. Whether altered microglia and astrocyte function occurs in other regions of the trigeminal sensory pathway aside from the somatosensory cortex remains to be determined. In sum, our findings that glial activation is outlived by increases in CGRP, as well as allodynia indicate additional mechanisms maintain these changes. Additional, more extensive analysis of candidate molecular mediators, particularly in the dura, trigeminal ganglia, and caudal brainstem regions, is warranted.
CONCLUSIONS AND FUTURE DIRECTIONS
Injury to the somatosensory cortex induces increases in CGRP levels in the brainstem and bilateral periorbital allodynia persisting up to 4 weeks after injury. SP immunoreactivity was also transiently increased after cortical injury. Combined, changes in central neuropeptides and sensory behavior in a mixed focal and diffuse cortical injury model of TBI are indicative of abnormally sustained activation of the trigeminovascular system. The increases in CGRP and allodynia outlast the increases in macrophage/ microglial and astrocytes activation. These findings provide evidence for the chronification of TBI-induced sensory alterations in which local glial activation in the somatosensory cortex may not be required for maintenance of these changes. This work provides a foundation upon which to build future TBI studies investigating mechanisms of PTH. The current range of TBI models including CCI allow for studies of preserved neuronal circuits of interest, such as those along the trigeminovascular pain pathway that may contribute to PTH. Adaptations to the CCI model to a milder, diffuse TBI without a focal injury or closed head injury may be used for comparative studies to understand the differences in PTH pathology between different types of injuries.
Acknowledgments
Experiments were conducted by Dr. Melanie B. Elliott, Dr. Peter S. Amenta, Olatilewa O. Awe, and Karena Browne. Data interpretation and manuscript preparation were done by Drs. Melanie Elliott, Michael L. Oshinsky (support by R01-NS061571), and Jack I. Jallo. Thank you to Dr. Elisabeth Van Bockstaele for contributing microscopy equipment for our histological analyses and Karena Browne for contributing to the behavioral testing. We would also like to thank Dr. Mary Barbe for reviewing and providing valuable feedback on this manuscript.
Footnotes
Conflict of Interest: There are no conflicts of interest to report for any of the authors.
References
- 1.Zaloshnja E, Miller T, Langlois JA, Selassie AW. Prevalence of long-term disability from traumatic brain injury in the civilian population of the United States, 2005. J Head Trauma Rehabil. 2008;23:394–400. doi: 10.1097/01.HTR.0000341435.52004.ac. [DOI] [PubMed] [Google Scholar]
- 2.Linder SL. Post-traumatic headache. Curr Pain Headache Rep. 2007;11:396–400. doi: 10.1007/s11916-007-0223-3. [DOI] [PubMed] [Google Scholar]
- 3.Gironda RJ, Clark ME, Ruff RL, et al. Traumatic brain injury, polytrauma, and pain: Challenges and treatment strategies for the polytrauma rehabilitation. Rehabil Psychol. 2009;54:247–258. doi: 10.1037/a0016906. [DOI] [PubMed] [Google Scholar]
- 4.Nampiaparampil DE. Prevalence of chronic pain after traumatic brain injury: A systematic review. JAMA. 2008;300:711–719. doi: 10.1001/jama.300.6.711. [DOI] [PubMed] [Google Scholar]
- 5.Riggio S, Wong M. Neurobehavioral sequelae of traumatic brain injury. Mt Sinai J Med. 2009;76:163–172. doi: 10.1002/msj.20097. [DOI] [PubMed] [Google Scholar]
- 6.Silver JM, McAllister TW, Arciniegas DB. Depression and cognitive complaints following mild traumatic brain injury. Am J Psychiatry. 2009;166:653–661. doi: 10.1176/appi.ajp.2009.08111676. [DOI] [PubMed] [Google Scholar]
- 7.Vaishnavi S, Rao V, Fann JR. Neuropsychiatric problems after traumatic brain injury: Unraveling the silent epidemic. Psychosomatics. 2009;50:198–205. doi: 10.1176/appi.psy.50.3.198. [DOI] [PubMed] [Google Scholar]
- 8.Seifert TD, Evans RW. Posttraumatic headache: A review. Curr Pain Headache Rep. 2010;14:292–298. doi: 10.1007/s11916-010-0117-7. [DOI] [PubMed] [Google Scholar]
- 9.Hoffman JM, Lucas S, Dikmen S, et al. Natural history of headache after traumatic brain Injury. J Neurotrauma. 2011;28:1719–1725. doi: 10.1089/neu.2011.1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theeler BJ, Flynn FG, Erickson JC. Headaches after concussion in US soldiers returning from Iraq or Afghanistan. Headache. 2010;50:1262–1272. doi: 10.1111/j.1526-4610.2010.01700.x. [DOI] [PubMed] [Google Scholar]
- 11.Lenaerts ME, Couch JR. Posttraumatic headache. Curr Treat Options Neurol. 2004;6:507–517. doi: 10.1007/s11940-004-0008-5. [DOI] [PubMed] [Google Scholar]
- 12.Walker WC, Seel RT, Curtiss G, Warden DL. Headache after moderate and severe traumatic brain injury: A longitudinal analysis. Arch Phys Med Rehabil. 2005;86:1793–1800. doi: 10.1016/j.apmr.2004.12.042. [DOI] [PubMed] [Google Scholar]
- 13.Woolf CJ, Salter MW. Neuronal plasticity: Increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- 14.Ofek H, Defrin R. The characteristics of chronic central pain after traumatic brain injury. Pain. 2007;131:330–340. doi: 10.1016/j.pain.2007.06.015. [DOI] [PubMed] [Google Scholar]
- 15.Martins HA, Ribas VR, Martins BB, de Ribas RM, Valenca MM. Post-traumatic headache. Arq Neuropsiquiatr. 2009;67:43–45. doi: 10.1590/s0004-282x2009000100011. [DOI] [PubMed] [Google Scholar]
- 16.Defrin R, Gruener H, Schreiber S, Pick CG. Quantitative somatosensory testing of subjects with chronic post-traumatic headache: Implications on its mechanisms. Eur J Pain. 2010;14:924–931. doi: 10.1016/j.ejpain.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 17.Hall KD, Lifshitz J. Diffuse traumatic brain injury initially attenuates and later expands activation of the rat somatosensory whisker circuit concomitant with neuroplastic responses. Brain Res. 2010;6:161–173. doi: 10.1016/j.brainres.2010.01.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oshinsky ML, Gomonchareonsiri S. Episodic dural stimulation in awake rats: A model for recurrent headache. Headache. 2007;47:1026–1036. doi: 10.1111/j.1526-4610.2007.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Burstein R, Yamamura H, Malick A, Strassman AM. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964–982. doi: 10.1152/jn.1998.79.2.964. [DOI] [PubMed] [Google Scholar]
- 20.Wieseler J, Ellis A, Sprunger D, et al. A novel method for modeling facial allodynia associated with migraine in awake and freely moving rats. J Neurosci Methods. 2010;185:236–245. doi: 10.1016/j.jneumeth.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA. Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med. 2002;8:136–142. doi: 10.1038/nm0202-136. [DOI] [PubMed] [Google Scholar]
- 22.Burstein R, Jakubowski M. Unitary hypothesis for multiple triggers of the pain and strain of migraine. J Comp Neurol. 2005;493:9–14. doi: 10.1002/cne.20688. [DOI] [PubMed] [Google Scholar]
- 23.Elliott MB, Barr AE, Barbe MF. Spinal substance P and neurokinin-1 increase with high repetition reaching. Neurosci Lett. 2009;454:33–37. doi: 10.1016/j.neulet.2009.01.037. [DOI] [PubMed] [Google Scholar]
- 24.Elliott MB, Barr AE, Clark BD, Amin M, Amin S, Barbe MF. High force reaching task induces widespread inflammation, increased spinal cord neuro-chemicals and neuropathic pain. Neuroscience. 2009;158:922–931. doi: 10.1016/j.neuroscience.2008.10.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elliott MB, Barr AE, Clark BD, Wade CK, Barbe MF. Performance of a repetitive task by aged rats leads to median neuropathy and spinal cord inflammation with associated sensorimotor declines. Neuroscience. 2010;170:929–941. doi: 10.1016/j.neuroscience.2010.07.041. [DOI] [PubMed] [Google Scholar]
- 26.Elliott MB, Barr AE, Kietrys DM, Al-Shatti T, Amin M, Barbe MF. Peripheral neuritis and increased spinal cord neurochemicals are induced in a model of repetitive motion injury with low force and repetition exposure. Brain Res. 2008;1218:103–113. doi: 10.1016/j.brainres.2008.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Recober A, Kuburas A, Zhang Z, Wemmie JA, Anderson MG, Russo AF. Role of calcitonin gene-related peptide in light-aversive behavior: Implications for migraine. J Neurosci. 2009;29:8798–8804. doi: 10.1523/JNEUROSCI.1727-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6:573–582. doi: 10.1038/nrneurol.2010.127. [DOI] [PubMed] [Google Scholar]
- 29.Elliott MB, Tuma RF, Amenta PS, Barbe MF, Jallo JI. Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a murine model of traumatic brain injury. J Neurotrauma. 2011;28:973–981. doi: 10.1089/neu.2010.1672. [DOI] [PubMed] [Google Scholar]
- 30.Elliott MB, Jallo JJ, Barbe MF, Tuma RF. Hypertonic saline attenuates tissue loss and astrocyte hypertrophy in a model of traumatic brain injury. Brain Res. 2009;1305:183–191. doi: 10.1016/j.brainres.2009.09.104. [DOI] [PubMed] [Google Scholar]
- 31.Elliott MB, Jallo JJ, Gaughan JP, Tuma RF. Effects of crystalloid-colloid solutions on traumatic brain injury. J Neurotrauma. 2007;24:195–202. doi: 10.1089/neu.2006.0094. [DOI] [PubMed] [Google Scholar]
- 32.Elliott MB, Jallo JJ, Tuma RF. An investigation of cerebral edema and injury volume assessments for controlled cortical impact injury. J Neurosci Methods. 2008;168:320–324. doi: 10.1016/j.jneumeth.2007.10.019. [DOI] [PubMed] [Google Scholar]
- 33.Cernak I. Animal models of head trauma. NeuroRx. 2005;2:410–422. doi: 10.1602/neurorx.2.3.410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen S, Pickard JD, Harris NG. Time course of cellular pathology after controlled cortical impact injury. Exp Neurol. 2003;182:87–102. doi: 10.1016/s0014-4886(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 35.Hall ED, Bryant YD, Cho W, Sullivan PG. Evolution of post-traumatic neurodegeneration after controlled cortical impact traumatic brain injury in mice and rats as assessed by the de Olmos silver and fluorojade staining methods. J Neurotrauma. 2008;25:235–247. doi: 10.1089/neu.2007.0383. [DOI] [PubMed] [Google Scholar]
- 36.Lighthall JW. Controlled cortical impact: A new experimental brain injury model. J Neurotrauma. 1988;5:1–15. doi: 10.1089/neu.1988.5.1. [DOI] [PubMed] [Google Scholar]
- 37.Lighthall JW, Dixon CE, Anderson TE. Experimental models of brain injury. J Neurotrauma. 1989;6:83–97. doi: 10.1089/neu.1989.6.83. [DOI] [PubMed] [Google Scholar]
- 38.Saatman KE, Feeko KJ, Pape RL, Raghupathi R. Differential behavioral and histopathological responses to graded cortical impact injury in mice. J Neurotrauma. 2006;23:1241–1253. doi: 10.1089/neu.2006.23.1241. [DOI] [PubMed] [Google Scholar]
- 39.Vink R, Bullock MR. Traumatic brain injury: Therapeutic challenges and new directions. Neurotherapeutics. 2010;7:1–2. doi: 10.1016/j.nurt.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allen Institute for Brain Science. Allen Mouse Brain Atlas [Internet] Seattle, WA: Allen Institute for Brain Science; 2009. Available from: http://mouse.brain-map.org. [Google Scholar]
- 41.Franklin KB, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. New York: Academic Press; 2008. [Google Scholar]
- 42.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 43.Oshinsky ML, Luo J. Neurochemistry of trigeminal activation in an animal model of migraine. Headache. 2006;46(Suppl 1):S39–S44. doi: 10.1111/j.1526-4610.2006.00489.x. [DOI] [PubMed] [Google Scholar]
- 44.Winkelstein BA, Santos DG. An intact facet capsular ligament modulates behavioral sensitivity and spinal glial activation produced by cervical facet joint tension. Spine. 2008;33:856–862. doi: 10.1097/BRS.0b013e31816b4710. [DOI] [PubMed] [Google Scholar]
- 45.Rothman SM, Kreider RA, Winkelstein BA. Spinal neuropeptide responses in persistent and transient pain following cervical nerve root injury. Spine. 2005;30:2491–2496. doi: 10.1097/01.brs.0000186316.38111.4b. [DOI] [PubMed] [Google Scholar]
- 46.Rothman SM, Nicholson KJ, Winkelstein BA. Time-dependent mechanics and measures of glial activation and behavioral sensitivity in a rodent model of radiculopathy. J Neurotrauma. 2010;27:803–814. doi: 10.1089/neu.2009.1045. [DOI] [PubMed] [Google Scholar]
- 47.Kerr BJ, David S. Pain behaviors after spinal cord contusion injury in two commonly used mouse strains. Exp Neurol. 2007;206:240–247. doi: 10.1016/j.expneurol.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 48.Narita M, Usui A, Niikura K, et al. Protease-activated receptor-1 and platelet-derived growth factor in spinal cord neurons are implicated in neuropathic pain after nerve injury. J Neurosci. 2005;25:10000–10009. doi: 10.1523/JNEUROSCI.2507-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elliott MB, Tuma RF, Amenta PS, Barbe MF, Jallo JI. Acute effects of a selective cannabinoid-2 receptor agonist on neuroinflammation in a murine model of traumatic brain injury. J Neurotrauma. 2011;28:973–981. doi: 10.1089/neu.2010.1672. [DOI] [PubMed] [Google Scholar]
- 50.Eftekhari S, Edvinsson L. Possible sites of action of the new calcitonin gene-related peptide receptor antagonists. Ther Adv Neurol Disord. 2010;3:369–378. doi: 10.1177/1756285610388343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Cole JT, Yarnell A, Kean WS, et al. Craniotomy: True sham for traumatic brain injury, or a sham of a sham. J Neurotrauma. 2011;28:359–369. doi: 10.1089/neu.2010.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Flexman AM, Ng JL, Gelb AW. Acute and chronic pain following craniotomy. Curr Opin Anaesthesiol. 2010;23:551–557. doi: 10.1097/ACO.0b013e32833e15b9. [DOI] [PubMed] [Google Scholar]
- 53.Kosaras B, Jakubowski M, Kainz V, Burstein R. Sensory innervation of the calvarial bones of the mouse. J Comp Neurol. 2009;515:331–348. doi: 10.1002/cne.22049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Stokely ME, Bhat MA, Koulen P. Microfluorimetry defines early axonal damage in a rat model of optic neuritis: A novel method targeting early CNS autoimmunity. J Neurosci Methods. 2007;166:217–228. doi: 10.1016/j.jneumeth.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 55.Olechowski CJ, Truong JJ, Kerr BJ. Neuropathic pain behaviours in a chronic-relapsing model of experimental autoimmune encephalomyelitis (EAE) Pain. 2009;141:156–164. doi: 10.1016/j.pain.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 56.Lee KE, Winkelstein BA. Joint distraction magnitude is associated with different behavioral outcomes and substance P levels for cervical facet joint loading in the rat. J Pain. 2009;10:436–445. doi: 10.1016/j.jpain.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 57.Burstein R, Jakubowski M, Garcia-Nicas E, et al. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol. 2010;68:81–91. doi: 10.1002/ana.21994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McNamara KC, Lisembee AM, Lifshitz J. The whisker nuisance task identifies a late-onset, persistent sensory sensitivity in diffuse brain-injured rats. J Neurotrauma. 2010;27:695–706. doi: 10.1089/neu.2009.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kelley BJ, Lifshitz J, Povlishock JT. Neuroinflammatory responses after experimental diffuse traumatic brain injury. J Neuropathol Exp Neurol. 2007;66:989–1001. doi: 10.1097/NEN.0b013e3181588245. [DOI] [PubMed] [Google Scholar]
- 60.Levy D, Strassman AM, Burstein R. A critical view on the role of migraine triggers in the genesis of migraine pain. Headache. 2009;49:953–957. doi: 10.1111/j.1526-4610.2009.01444.x. [DOI] [PubMed] [Google Scholar]
- 61.Olesen J, Burstein R, Ashina M, Tfelt-Hansen P. Origin of pain in migraine: Evidence for peripheral sensitisation. Lancet Neurol. 2009;8:679–690. doi: 10.1016/S1474-4422(09)70090-0. [DOI] [PubMed] [Google Scholar]
- 62.Eikermann-Haerter K, Ayata C. Cortical spreading depression and migraine. Curr Neurol Neurosci Rep. 2010;10:167–173. doi: 10.1007/s11910-010-0099-1. [DOI] [PubMed] [Google Scholar]
- 63.Meents JE, Neeb L, Reuter U. TRPV1 in migraine pathophysiology. Trends Mol Med. 2010;16:153–159. doi: 10.1016/j.molmed.2010.02.004. [DOI] [PubMed] [Google Scholar]
- 64.Jenkins DW, Langmead CJ, Parsons AA, Strijbos PJ. Regulation of calcitonin gene-related peptide release from rat trigeminal nucleus caudalis slices in vitro. Neurosci Lett. 2004;366:241–244. doi: 10.1016/j.neulet.2004.05.067. [DOI] [PubMed] [Google Scholar]
- 65.Amrutkar DV, Ploug KB, Olesen J, Jansen-Olesen I. Role for voltage gated calcium channels in calcitonin gene-related peptide release in the rat trigeminovascular system. Neuroscience. 2011;172:510–517. doi: 10.1016/j.neuroscience.2010.10.032. [DOI] [PubMed] [Google Scholar]
- 66.Eftekhari S, Edvinsson L. Calcitonin gene-related peptide (CGRP) and its receptor components in human and rat spinal trigeminal nucleus and spinal cord at C1-level. BMC Neurosci. 2011;12:112–130. doi: 10.1186/1471-2202-12-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hang CH, Shi JX, Li JS, Wu W, Li WQ, Yin HX. Levels of vasoactive intestinal peptide, cholecystokinin and calcitonin gene-related peptide in plasma and jejunum of rats following traumatic brain injury and underlying significance in gastrointestinal dysfunction. World J Gastroenterol. 2004;10:875–880. doi: 10.3748/wjg.v10.i6.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Donkin JJ, Turner RJ, Hassan I, Vink R. Substance P in traumatic brain injury. Prog Brain Res. 2007;161:97–109. doi: 10.1016/S0079-6123(06)61007-8. [DOI] [PubMed] [Google Scholar]
- 69.Armstead WM, Kiessling JW, Bdeir K, Kofke WA, Vavilala MS. Adrenomedullin prevents sex-dependent impairment of autoregulation during hypotension after piglet brain injury through inhibition of ERK MAPK upregulation. J Neurotrauma. 2010;27:391–402. doi: 10.1089/neu.2009.1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Armstead WM, Kiessling JW, Kofke WA, Vavilala MS. Impaired cerebral blood flow autoregulation during posttraumatic arterial hypotension after fluid percussion brain injury is prevented by phenyl-ephrine in female but exacerbated in male piglets by extracellular signal-related kinase mitogen-activated protein kinase upregulation. Crit Care Med. 2010;38:1868–1874. doi: 10.1097/CCM.0b013e3181e8ac1a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Goadsby PJ. Calcitonin gene-related peptide receptor antagonists and triptans: A tale of two treatments. Neurology. 2010;75:1494–1495. doi: 10.1212/WNL.0b013e3181f9631e. [DOI] [PubMed] [Google Scholar]
- 72.Goadsby PJ, Charbit AR, Andreou AP, Akerman S, Holland PR. Neurobiology of migraine. Neuroscience. 2009;161:327–341. doi: 10.1016/j.neuroscience.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 73.Woolf CJ, Ma Q. Nociceptors – noxious stimulus detectors. Neuron. 2007;55:353–364. doi: 10.1016/j.neuron.2007.07.016. [DOI] [PubMed] [Google Scholar]
- 74.Myer DJ, Gurkoff GG, Lee SM, Hovda DA, Sofroniew MV. Essential protective roles of reactive astrocytes in traumatic brain injury. Brain. 2006;129:2761–2772. doi: 10.1093/brain/awl165. [DOI] [PubMed] [Google Scholar]
- 75.Sandhir R, Onyszchuk G, Berman NE. Exacerbated glial response in the aged mouse hippocampus following controlled cortical impact injury. Exp Neurol. 2008;213:372–380. doi: 10.1016/j.expneurol.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Watkins LR, Milligan ED, Maier SF. Glial activation: A driving force for pathological pain. Trends Neurosci. 2001;24:450–455. doi: 10.1016/s0166-2236(00)01854-3. [DOI] [PubMed] [Google Scholar]
- 77.Wieseler-Frank J, Maier SF, Watkins LR. Glial activation and pathological pain. Neurochem Int. 2004;45:389–395. doi: 10.1016/j.neuint.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 78.Grinberg YY, Milton JG, Kraig RP. Spreading depression sends microglia on Levy flights. PLoS ONE. 2011;6:e19294. doi: 10.1371/journal.pone.0019294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Morganti-Kossmann MC, Satgunaseelan L, Bye N, Kossmann T. Modulation of immune response by head injury. Injury. 2007;38:1392–1400. doi: 10.1016/j.injury.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 80.Sommer C, Kress M. Recent findings on how pro-inflammatory cytokines cause pain: Peripheral mechanisms in inflammatory and neuropathic hyperalgesia. Neurosci Lett. 2004;361:184–187. doi: 10.1016/j.neulet.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 81.Zhang XC, Kainz V, Burstein R, Levy D. Tumor necrosis factor-alpha induces sensitization of meningeal nociceptors mediated via local COX and p38 MAP kinase actions. Pain. 2011;152:140–149. doi: 10.1016/j.pain.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cherian L, Hlatky R, Robertson CS. Nitric oxide in traumatic brain injury. Brain Pathol. 2004;14:195–201. doi: 10.1111/j.1750-3639.2004.tb00053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Olesen J. The role of nitric oxide (NO) in migraine, tension-type headache and cluster headache. Pharmacol Ther. 2008;120:157–171. doi: 10.1016/j.pharmthera.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 84.Bates EA, Nikai T, Brennan KC, et al. Sumatriptan alleviates nitroglycerin-induced mechanical and thermal allodynia in mice. Cephalalgia. 2010;30:170–178. doi: 10.1111/j.1468-2982.2009.01864.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stover JF, Schoning B, Beyer TF, Woicie-chowsky C, Unterberg AW. Temporal profile of cerebrospinal fluid glutamate, interleukin-6, and tumor necrosis factor-alpha in relation to brain edema and contusion following controlled cortical impact injury in rats. Neurosci Lett. 2000;288:25–28. doi: 10.1016/s0304-3940(00)01187-3. [DOI] [PubMed] [Google Scholar]