

Abstract
Candida albicans is one of the most prevalent fungal pathogens, causing both mucosal candidiasis and invasive candidemia. Antimicrobial peptides (AMPs), part of the human innate immune system, have been shown to exhibit antifungal activity but have not been effective as pharmaceuticals because of low activity and selectivity in physiologically relevant environments. Nevertheless, studies on α-peptide AMPs have revealed key features that can be designed into more stable structures, such as the 14-helix of β-peptide-based oligomers. Here, we report on the ways in which two of those features, hydrophobicity and helicity, govern the activity and selectivity of 14-helical β-peptides against C. albicans and human red blood cells. Our results reveal both antifungal activity and hemolysis to correlate to hydrophobicity, with intermediate levels of hydrophobicity leading to high antifungal activity and high selectivity toward C. albicans. Helical structure-forming propensity further influenced this window of selective antifungal activity, with more stable helical structures eliciting specificity for C. albicans over a broader range of hydrophobicity. Our findings also reveal cooperativity between hydrophobicity and helicity in regulating antifungal activity and specificity. The results of this study provide critical insight into the ways in which hydrophobicity and helicity govern the activity and specificity of AMPs and identify criteria that may be useful for the design of potent and selective antifungal agents.
Candida albicans is a commensal organism and the most common fungal pathogen in humans, causing both mucosal candidiasis and invasive candidemia.1 As an opportunistic pathogen, C. albicans can cause life-threatening invasive infections in immunocompromised individuals such as organ recipients, cancer patients, and human immunodeficiency virus (HIV)-infected patients.1−10 The mortality rate of systemic Candida infection is approximately 30–50%.11,12 Candidemia, a disease in which Candida spp. are detected in the bloodstream, is also often associated with biofilm formation on indwelling medical devices such as central venous or urinary catheters, joint prostheses, dialysis access, cardiovascular devices, and central nervous system devices.13 The resistance of C. albicans within biofilms to antifungal drugs such as fluconazole, amphotericin B, flucytosine, itraconazole, and ketoconazole has been reported to be 30–2000 times greater than in planktonic cells.14 Clearly, more active and specific classes of antifungal compounds are needed to reduce the severity of antifungal infection, develop effective treatment protocols, and reduce mortality in affected patients.
Antimicrobial peptides (AMPs) are components of the innate host defense system15−17 and possess activity against bacteria, fungi, viruses, and tumors.18−20 More than 2,000 AMPs are listed in the Antimicrobial Peptide Database,21 and 35% of these have been reported to possess some degree of antifungal activity. However, AMPs have several limitations as therapeutics, including low stability and activity in physiological media, low specificity toward fungal cells, and susceptibility to proteolysis in vivo.22−26 While naturally occurring AMPs may not be well suited for use as antifungal therapeutics for these and other reasons, they have nevertheless provided key molecular-level insights into structural features and functional behaviors that confer antimicrobial activity. For example, AMPs have been demonstrated to induce membrane lysis in target cells via carpet or pore formation.27−29 As a result, the development of target cell resistance to AMPs, a problem that is understood to occur upon the use of conventional antifungal drugs, has been suggested to be low.30−32 Insights and key principles gleaned from studies investigating physicochemical interactions between AMPs and cell targets may facilitate design of chemical compounds that display antifungal activity and may possess more suitable pharmacokinetic and pharmacodynamic properties than AMPs.
Naturally occurring AMPs, also known as host defense peptides, can be categorized into 5 major classes: linear cationic α-helical peptides, anionic peptides, specific amino acid-enriched peptides, anionic and cationic disulfide bond-containing peptides, and peptide fragments of large proteins.33 These AMPs display direct antimicrobial activity but often lose antimicrobial cytotoxicity at physiologic pH and ionic strength. Recent studies have demonstrated AMP host immunomodulatory activity, which complements their antimicrobial activity.34
One of the predominant classes of antimicrobial peptide is linear and α-helical in structure, and key properties that confer antimicrobial activity include hydrophobicity, facial amphiphilicity, and helical propensity.27 These structural features have also proven to be important in conferring antimicrobial activity on peptidomimetic oligomers35−41 and polymers,42−47 including β-peptide-based structures composed, either entirely or in part, of β-amino acid residues.48,49 Owing to their folding principles, structural diversity, secondary structure stability,50,51 and resistance to proteolysis,52 β-peptides represent a promising class of compounds to elucidate mechanisms of AMP activity and to template development of new antifungal therapeutics. Antibacterial53−56 and antifungal57,58 activity of β-peptides adopting 14-helical structures and the antibacterial activity of β-peptides adopting 12-helical59−61 and 10/12-helical62 structures have been reported. However, the contributions of different β-peptide structural features to biological activity, including antibacterial and antifungal properties, are not well understood. Thus, we sought to determine how hydrophobicity and helicity govern, either alone or in concert, the antifungal activity and specificity of 14-helical β-peptides.
We synthesized 25 globally amphiphilic 14-helical β-peptides that contain approximately three helical turns. In designing these β-peptides, we varied hydrophobicity and helicity by altering side chain composition and the presence or absence of a helix-stabilizing cyclic aminocyclohexane carboxylic acid (ACHC) side chain. To quantify the hydrophobicity of the β-peptides, we characterized retention times using RP-HPLC, a measure that has been used previously to assess the hydrophobicity of peptides.63−65 Correlations have been reported between antimicrobial peptide activity and retention time of RP-HPLC.66−68 The helicity of β-peptides was characterized using circular dichroism. Circular dichroism titration showed that the helicity differences depended on the presence or absence of a helix-stabilizing cyclic ACHC side chain. Our results also indicate that, at constant helicity, β-peptide hydrophobicity directly correlates with both antifungal and hemolytic activity, but that a window of hydrophobicity exists over which these structures exhibit both high antifungal activity and high selectivity (i.e., low hemolysis at the antifungal minimum inhibitory concentration, MIC). We also demonstrate that β-peptide helicity governs antifungal activity, with more stable β-peptides possessing antifungal activity at lower concentrations than less stable molecules at the same hydrophobicity. Taken together, these results reveal both hydrophobicity and helicity to regulate antifungal activity and hemolysis, and provide design parameters for constructing active and selective antifungal compounds.
Results and Discussion
Design and Synthesis of 14-Helical β-Peptides
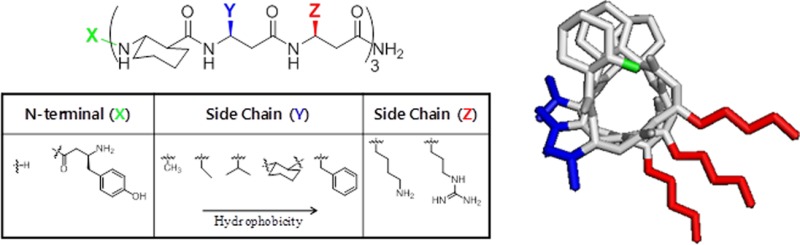
In order to ascertain the structural features of AMPs that confer antifungal and hemolytic activity, we synthesized a set of 25 14-helical β-peptides (peptides 1–25, Figure 1), each 9 or 10 residues long, having a net charge of +4 and designed to adopt globally amphiphilic structures (see Figure 1a–c). These peptides have approximately three residues per helical turn50,51 and were designed to contain two hydrophobic residues and one cationic residue in a repeated trimer, thus generating localized hydrophobic and cationic faces of the helix.
Figure 1.

14-Helical β-peptide design and chemical structures. 3D structures (a–c) were generated on the basis of available crystal structure data85 and then geometry was optimized using Gaussian 03 at the B3LYP/6-31G level. (a) Stick view of β-peptide 4. The N-terminus (green), hydrophobic side chains (blue), and cationic side chains (red) are indicated in color. (b and c) Surface views of β-peptide 4. Surface colors represent atom type H (gray), C (green), O (red), and N (blue). (d) N-Terminus (X) and side chains (Y and Z) were altered as indicated to vary peptide hydrophobicity. (e) Chemical structure of β-peptides containing a helix-stabilizing ACHC residue. (f and g) Chemical structures of β-peptides lacking an ACHC residue. β3-hVal (f) and β3-hIle (g) were incorporated in place of the ACHC residue.
In this study, we systematically investigated the influence of two AMP properties, hydrophobicity and helicity, on the antifungal activity and selectivity of 14-helical β-peptides. To probe the influence of these properties, we synthesized peptides having different structural features that influence hydrophobicity and stability, including (i) the presence (peptides 1–16, Figure 1e) or absence (peptides 17–25, Figure 1f,g) of the helix-stabilizing ACHC residue, (ii) addition of an N-terminal β3-hTyr residue (Figure 1d), and (iii) variations in the hydrophobic Y and cationic Z position side chains in the helical repeat (Figure 1d).
β-Peptides were produced by microwave-assisted Fmoc synthesis at 20–40 μmol scales and purified by RP-HPLC using a C18 column. MALDI mass spectrometry was used to validate the mass of each peptide (Supplementary Table S1). Retention times determined by C18 RP-HPLC were used as a measure of the relative hydrophobicity of the peptides (Table 1, Supplementary Figure S1). To quantify the helicity of the β-peptides, we characterized CD in solvents containing different fractions of PBS and methanol.
Table 1. β-Peptide Retention Time, Minimum Inhibitory Concentrations (MIC) against C. albicans, and % Hemolysis at the Antifungal MIC.
| β-peptide | tRa (min ± SD) | MICb (μg/mL) | % hemolysis at MICc ± SD |
|---|---|---|---|
| 1 | 19.3 ± 0.1 | >128 | 2.6 ± 0.9* |
| 2 | 22.5 ± 0.2 | 64 | 3.0 ± 2.4 |
| 3 | 23.2 ± 0.1 | 32 | 1.1 ± 2.7 |
| 4 | 24.5 ± 0.2 | 8 | 2.3 ± 0.7 |
| 5 | 25.4 ± 0.1 | 8 | 1.6 ± 0.3 |
| 6 | 23.1 ± 0.2 | 16 | 0.3 ± 1.7 |
| 7 | 23.8 ± 0.1 | 16 | 3.0 ± 2.3 |
| 8 | 26.2 ± 0.2 | 8 | 36.4 ± 6.0 |
| 9 | 20.4 ± 0.2 | 128 | 1.4 ± 0.6 |
| 10 | 23.5 ± 0.1 | 16 | 9.4 ± 9.3 |
| 11 | 24.2 ± 0.1 | 16 | 7.5 ± 5.2 |
| 12 | 25.7 ± 0.1 | 8 | 37 ± 15 |
| 13 | 26.5 ± 0.2 | 8 | 39.8 ± 2.7 |
| 14 | 24.0 ± 0.2 | 16 | 9.5 ± 2.2 |
| 15 | 24.6 ± 0.2 | 16 | 11.6 ± 2.1 |
| 16 | 27.4 ± 0.2 | 4 | 72 ± 14 |
| 17 | 22.5 ± 0.1 | 128 | 2.8 ± 0.1 |
| 18 | 23.5 ± 0.1 | 64 | 0.9 ± 1.9 |
| 19 | 22.7 ± 0.2 | 128 | 3.2 ± 2.9 |
| 20 | 24.3 ± 0.2 | 32 | 8.8 ± 3.6 |
| 21 | 25.2 ± 0.2 | 16 | 4.2 ± 2.0 |
| 22 | 22.8 ± 0.2 | >128 | 3.1 ± 4.4* |
| 23 | 23.8 ± 0.1 | 128 | 4.5 ± 3.2 |
| 24 | 24.6 ± 0.1 | 32 | 7.2 ± 5.0 |
| 25 | 25.7 ± 0.2 | 16 | 7.2 ± 3.4 |
The average value obtained from three independent analytical RP-HPLC measurements.
The value obtained from an average of three independent experiments with triplicate measurements.
The average value obtained from three independent experiments with duplicate measurements.
Hemolysis at MIC measured at 128 μg/mL β-peptide. Active (MIC ≤ 16 μg/mL) and selective (hemolysis at MIC ≤ 20%) β-peptides are in bold font.
Characterization of Antifungal and Hemolytic Activities
The antifungal activities of the β-peptides were determined by measuring their minimum inhibitory concentrations (MICs) against C. albicansin vitro. The peptides exhibited a wide range of MIC values, ranging from 4 to >128 μg/mL. As an example, a plot of the concentration-dependent growth inhibition of one of the most active peptides, peptide 5, is shown in Figure 2a, with the MIC of 8 μg/mL indicated by the arrow. The MICs of all of the peptides synthesized in this study are provided in Table 1 and the growth inhibition plots used to identify the MICs are shown in Supplementary Figure S2.
Figure 2.

Examples of measurement of antifungal MIC (a) and hemolysis at the MIC (b) of β-peptide 5. (a) C. albicans cells (103 cells/mL) were incubated with β-peptides for 48 h, and β-peptide susceptibility was assessed using an XTT reduction assay to compare the absorbance at 490 nm for β-peptide-treated samples and untreated samples. Data points are the averages of three independent experiments of three replicates each. (b) β-peptides were incubated with human red blood cells for 1 h, and the absorbance of the supernatant was measured at 405 nm to calculate the percent of red blood cells lysed; 100% hemolysis was determined using a melittin control. Error bars denote standard deviation (n = 3).
To evaluate β-peptide specificity toward fungal cells, we compared their ability to inhibit C. albicans growth with their induction of human red blood cell lysis. As an example, the percent hemolysis of peptide 5 as a function of peptide concentration is indicated in Figure 2b, and results for all other peptides tested are provided in Supplementary Figure S3. The specificity of β-peptides against target fungal cells versus mammalian cells is of particular interest. To provide a measure of this specificity, we also compared percent hemolysis at the MIC for each peptide (Table 1). The percent hemolysis at the MIC ranged from <1% to ∼72% for the various peptides investigated in this study. The most active and selective β-peptides were peptides 4 and 5, each of which had an MIC of 8 μg/mL and less than 5% hemolysis at the MIC.
β-Peptide Hydrophobicity Directly Correlates to Antifungal and Hemolytic Activity
To explore the relationship between β-peptide hydrophobicity and antifungal activity and selectivity in compounds with similar helicity, we varied (i) the N-terminal residue (Figure 1a, green; Figure 1d, X), (ii) the composition of the hydrophobic side groups in the repeating unit (Figure 1a, blue; Figure 1d, Y), and (iii) the composition of the cationic residue in the repeating unit (Figure 1a, red; Figure 1d, Z). All of the β-peptides used in these studies (peptides 1–16) contained an ACHC residue as one of the hydrophobic residues in the repeating unit.
We also added a β3-hTyr residue to the N-terminus, (Figure 1a and 1d, X) to increase the hydrophobicity of β-peptides 1–8. Peptides containing the N-terminal β3-hTyr (peptides 9–16) exhibited a RP-HPLC retention time that was approximately 1.0 ± 0.15 min longer, and are thus regarded as more hydrophobic, than those that did not contain the N-terminal β3-hTyr (peptides 1–8) (Table 1). We also observed that β-peptides with RP-HPLC retention times of approximately 23 min or less exhibited a significant decrease in MIC upon addition of an N-terminal β3-hTyr, but that the addition of an N-terminal β3-hTyr had no effect on the retention times of more hydrophobic β-peptides (Supplementary Figure S4a; Table 1). Addition of an N-terminal β3-hTyr to relatively hydrophobic β-peptides with RP-HPLC retention times of 25 min or more increased hemolysis at MIC but had no effect on hemolysis at the MIC of more hydrophilic β-peptides (Supplementary Figure S4b; Table 1).
To broaden the spectrum of hydrophobicity in the β-peptide series, we introduced aliphatic β-amino acids of varying side chain carbon number and also incorporated an aromatic β-amino acid (β3-hPhe). The RP-HPLC retention times of 14-helical β-peptides with different residues in the middle position of the repeat exhibited the same trend (β3-hPhe > β3-hVal > ACHC > β3-Et > β3-hAla) in β-peptides both containing and lacking an N-terminal β3-hTyr residue (Table 1). The antifungal activity and selectivity upon varying side chain carbon number exhibited the same trends as observed upon N-terminal β3-hTyr modification. The antifungal MICs increased as retention times dropped below 23 min (Supplementary Figure S4c), and the percent hemolysis at the MIC generally increased as β-peptide retention time surpassed 25 min (Supplementary Figure S4d). These trends were observed for β-peptides with different N-termini and cationic side chains, including (i) X = H, Z = β3-hLys (peptides 1, 2, 4, 6, 8), (ii) X = β3-hTyr, Z = β3-hLys (peptides 9, 10, 12, 14, 16), (iii) X = H, Z = β3-hArg (peptides 3, 5, 7), and (iv) X = β3-hTyr, Z = β3-hArg (peptides 11, 13, 15) (Supplementary Figure S4c,d).
We also varied β-peptide hydrophobicity by changing the identity of the cationic residue in the repeating trimer. Naturally occurring AMPs, including magainin 2,69 cecropin 2,70 dermaseptin B2,71 mellitin,72 SMAP-29,73 MBP-1,74 and melamine75 contain lysines and arginines as cationic residues. Thus, we compared the antifungal activity and hemolysis of β-peptides containing β3-hLys and β3-hArg. Peptides containing β3-hArg exhibited RP-HPLC retention times 0.7 ± 0.14 min longer than those of analogous peptides containing β3-hLys (Table 1). The changes in antifungal activity upon varying the cationic side chain exhibited similar trends as were observed upon adding an N-terminal β3-hTyr and varying the hydrophobic side chain carbon number (discussed above). The MICs increased as β-peptide retention time decreased below 23 min and did not change upon cationic side chain modification for more hydrophobic β-peptides (Supplementary Figure S4e). However, the hemolysis at the MIC did not change upon substitution of β3-hArg for β3-hLys at any of the retention times. All peptides, regardless of substitution of β3-hArg for β3-hLys, with retention times greater than 25.5 min exhibited elevated hemolysis at the MIC (Supplementary Figure S4f).
To visualize the relationships between peptide hydrophobicity and antifungal and hemolytic activity, we plotted the MIC and the percent hemolysis at the MIC as a function of peptide retention time for the series of ACHC-containing β-peptides (peptides 1–16). Figure 3a reveals a strong correlation between β-peptide hydrophobicity and β-peptide antifungal and hemolytic activity as the N-terminus, hydrophobic side chain, and cationic side chain residues are varied. The MIC decreased, and hemolysis at the MIC increased, as the retention time increased. β-Peptides with retention times between 23 and 25.5 min, indicated in the box in Figure 3a and in bold text in Table 1, exhibited an MIC of 16 μg/mL or less and less than 20% hemolysis at the MIC. At retention times below 23 min, antifungal activity was low but specificity was high, while at retention times above 25.5 min antifungal activity was high but specificity was low. These key results reveal a window of hydrophobicity, with a retention time from 23 and 25.5 min under the conditions used in this study, that results in high β-peptide antifungal activity and selectivity, and suggest that variations in the N-terminus, hydrophobic side chain structure, and cationic side chain structure strongly influence MIC and hemolysis through their influence on β-peptide hydrophobicity.
Figure 3.

(a) Antifungal and hemolytic activities correlate with β-peptide RP-HPLC retention times in β-peptides containing an ACHC. The active (MIC ≤ 16 μg/mL) and selective (hemolysis at MIC ≤ 20%) β-peptides are indicated in the box. MIC was determined by incubating C. albicans cells (103 cells/mL) with β-peptides for 48 h, and β-peptide susceptibility was assessed using an XTT reduction assay to compare the absorbance at 490 nm for β-peptide-treated samples and untreated samples. Data points are the averages of three independent experiments of three replicates each. Hemolysis at the MIC was determined by incubating β-peptides with human red blood cells for 1 h, and the absorbance of the supernatant was measured at 405 nm to calculate the percent of red blood cells lysed; 100% hemolysis was determined using a melittin control. (b) The relationships between antifungal activity and RP-HPLC retention time in β-peptides containing (1–16) and lacking (17–25) an ACHC residue. The ACHC was replaced with β3-hVal in peptides 17–21 and β3-hIle in peptides 22–25. Error bars denote standard deviation (n = 3).
We next investigated the influence of hydrophobicity on the activity and specificity of β-peptides 17–25 lacking the helix-stabilizing ACHC side chain (Figure 1f and 1g). The ACHC-containing β-peptides adopted a helical structure at physiologic pH and ionic strength. However, ACHC-lacking β-peptides exhibited 10–20% helicity at physiological pH and ionic strength (Supplementary Figure S6). To vary the hydrophobicity of β-peptides lacking ACHC residues, we incorporated β3-hVal or the less hydrophobic β3-Et in the middle (Y) position of each repeating trimer. β-Peptides lacking an ACHC exhibited increased antifungal activity as hydrophobicity increased, similar to β-peptides containing the ACHC residue. However, the β-peptides lacking an ACHC required a greater degree of hydrophobicity to exhibit significant antifungal activity. For example, whereas β-peptides containing an ACHC possessed MICs of 16 μg/mL or lower at a retention times above 23 min, the β-peptides lacking an ACHC did not exhibit MICs of 16 μg/mL until retention times exceeded 25 min (Figure 3b). These results indicate that hydrophobicity alone does not govern antifungal activity and suggest that helicity also plays a key role. β-Peptides lacking ACHC did not exhibit the increase in hemolysis at retention times greater than 25 min that β-peptides containing an ACHC demonstrated (Supplementary Figure S5). Thus, the hydrophobic (tR >25 min) β-peptides lacking an ACHC possessed both antifungal activity (MIC of 16 μg/mL or lower) and specificity (<10% hemolysis at the MIC).
Effect of Cooperativity of Peptide Helicity and Hydrophobicity on Antifungal Activity
Our results demonstrate a strong correlation between β-peptide hydrophobicity and antifungal activity and hemolysis. To elucidate the relationship between helicity and antifungal activity and selectivity, we replaced ACHC with β3-hVal (peptides 17–21, Figure 1f) or β3-hIle (peptides 22–25, Figure 1g) in the first residue of the repeating trimer. Figure 3b illustrates the nature of this correlation and the ranges of retention times that provide antifungal activity, namely, that antifungal activity also depends on β-peptide properties other than hydrophobicity, including the presence of an ACHC side chain. We hypothesized that helicity also affects antifungal activity. To investigate this possibility, we compared the helical stabilities of three β-peptides with high antifungal activities (MIC = 16 μg/mL; β-peptides 11, 21, 25), three β-peptides with intermediate antifungal activities (MIC = 32 μg/mL; β-peptides 3, 20, 24), and three β-peptides with low antifungal activities (MIC = 128 μg/mL; β-peptides 9, 17, 23) in solvents containing various ratios of methanol and water. As expected, β-peptides containing the ACHC side chain (3, 9, 11) maintained helicity as the methanol fraction decreased to zero, while the β-peptides lacking an ACHC lost 14-helical structure (Supplementary Figure S6). β-Peptides containing β3-hVal in place of ACHC (17, 20, 21) exhibited slightly greater helicity than β-peptides containing β3-hIle (23, 24, 25).
Figure 4 illustrates the relationship between the antifungal MIC and β-peptide helicity and hydrophobicity. In comparing β-peptides with the same MIC, greater helicity corresponded to a lower retention time. For example, β-peptides 11, 21, and 25 all exhibited an MIC of 16 μg/mL. β-Peptide 11 demonstrated the greatest helicity and lowest retention time, while β-peptide 25 formed the least stable helix and possessed the greatest retention time. Similar trends existed at the 32 and 128 μg/mL MIC values. Thus, helicity and hydrophobicity collectively govern antifungal activity.
Figure 4.

Relationship between helicity and HPLC retention times of β-peptides exhibiting antifungal MICs of 16, 32, and 128 ug/mL. β-Peptides 11, 3, and 9 contain an ACHC residue, while β-peptides 21, 20, and 17 contain a β3-hVal and β-peptides 25, 24, and 23 contain a β3-hIle in place of the ACHC. Percent helicity in 100% PBS normalized to helicity in 100% methanol was determined by circular dichroism. Error bars denote standard deviation (n = 3).
Comparing β-peptides with similar helical stabilities, increasing retention time correlated with increasing antifungal activity (Supplementary Figure S7). The antifungal MIC of β-peptides possessing similarly high levels of helicity (e.g., Figure 1e series: 3, 9, 11) decreased with increasing retention time. Similar trends existed in the β-peptides with lower helicity in PBS (Figure 1f series: 17, 20, 21; and Figure 1g series: 23, 24, 25).
Comparing β-peptides with similar retention times (e.g., peptides 11, 20, 23) illustrates that antifungal activity increased as 14-helicity increased (Supplementary Figure S8). The ACHC-containing β-peptide 11 had an MIC of 16 μg/mL, while the β3-hVal series peptide 20 and β3-hIle series peptide 23 exhibited reduced antifungal activity. Thus, helicity appears to regulate β-peptide antifungal activity independently of retention time. Taken together, these results indicate that helicity and hydrophobicity cooperatively regulate β-peptide antifungal activity.
Structural Features That Govern Antifungal Activity and Specificity of AMPs and Their Analogues
The finding that helicity and hydrophobicity collectively control β-peptide antifungal activity is consistent with reports of α-peptide activity against microbes and mammalian cells. For example, Dathe and co-workers studied antibacterial activity of magainin 2 amide (M2a).76 They reported that the most hydrophobic analogue of M2a exhibited the greatest activity against E. coli (MIC 2.4 μg/mL). The activity of that analogue (I6A8L15I17) was 16-fold greater than the activity of M2a, and hemolytic activity also increased by about 13-fold. In addition, Hodges and co-workers68 studied the effects of hydrophobicity on the antimicrobial activity of analogues of D-V13K77 derived from V681.78 In Gram-negative bacteria and zygomycota fungi, increasing hydrophobicity decreased activity, but in Gram-positive bacteria, ascomycota fungi, and red blood cells activity increased with hydrophobicity. These results are consistent with the general view that the cell lytic activity of AMPs increases with hydrophobicity but suggest that effects also depend on the specific target organism, perhaps as a result of differences in cell membrane composition.
Helicity has also been shown to affect antibacterial activity and selectivity of α-peptide AMPs. For example, a Gly to Ala substitution in magainin II was reported to increase the helicity, antimicrobial activity, and hemolysis compared to magainin I and II.79 In addition, Pro, which is generally considered to be a helix-disrupting amino acid, has been introduced into antimicrobial peptides. The Pro-free antibacterial peptide V681 exhibited superior antibacterial activity against S. typhimurium and higher hemolytic activity than peptides containing one or two proline residues.80 Similarly, the helicity of temporin L, an antimicrobial peptide that exhibits high hemolytic activity, was varied by incorporating Pro and corresponding d-isomers.81 Increasing temporin L helicity increased both antibacterial and antifungal activity. Thus, our results suggest that helical β-peptides represent structural models for understanding mechanisms of AMP activity and specificity and provide design parameters for developing activity and selective antifungal agents.
Summary
In conclusion, our results demonstrate that 14-helical β-peptide antifungal activity and hemolysis are regulated by hydrophobicity and helicity. We also identified a hydrophobicity range that confers selective antifungal activity to β-peptides. Finally, we identified a cooperative relationship between hydrophobicity and helicity in controlling antifungal and hemolytic activities of β-peptides. These results provide insight into mechanisms of action of AMP mimetics and provide guidelines for designing active and specific compounds for antifungal applications.
Methods
Materials
Fmoc-β-amino acids, including Fmoc-l-β-homoalanine, Fmoc-l-β-homovaline, Fmoc-l-β-homoisoleucine, Fmoc-(1S,2S)-2-aminocyclohexane carboxylic acid, Fmoc-l-β-homophenylalanine, Fmoc-O-tert-butyl-l-β-homotyrosine, Nβ-Fmoc-Nω-Boc-l-β-homolysine, and Fmoc-Nω-(2,2,5,7,8-pentamethyl-chromane-6-sulfonyl)-l-β-homoarginine were purchased from Chem-Impex International, Inc. TentaGel S RAM Fmoc, HBTU (O-(benzotriaole-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate), and HOBt·H2O (N-hydroxybenzotrizole monohydrate) were purchased from Advanced ChemTech. (S)-3-Aminopentanoic acid was purchased from Sigma-Aldrich for synthesis of Fmoc-(S)-3-aminopentanoic acid (Fmoc-β3-Et-OH).82 RPMI powder (with l-glutamine and phenol red, without HEPES and sodium bicarbonate) and 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) were purchased from Invitrogen. 3-(N-Morpholino) propanesulfonic acid (MOPS) was purchased from Fisher Scientific. Phosphate-buffered saline (PBS) liquid concentrate (10X) was purchased from OmniPur. Menadione and melittin were purchased from Sigma. Freshly expired human red blood cells were obtained from the blood bank at University of Wisconsin-Madison Hospital.
β-Peptide Synthesis
β-Peptides were synthesized using TentaGel (20–40 μmol) microwave-assisted solid phase peptide synthesis procedures similar to those reported previously.83 Briefly, Fmoc-β-amino acid, coupling reagent (HBTU, HOBt), and base (DIEA) were dissolved in DMF individually and then mixed before coupling. Microwave (CEM Discover) irradiation was used for coupling of Fmoc-β-amino acid (600 W maximum power, 70 °C, ramp 2 min, hold 12 min) and deprotection of Fmoc (600 W maximum power, 80 °C, ramp 2 min, hold 6 min). After coupling and deprotection, the resin was washed with DMF (5 times), CH2Cl2 (5 times), and DMF (5 times), and then the peptide was cleaved from the resin by TFA-containing H2O (2.5% v/v) and triisopropylsilane (2.5% v/v) for 1–2 h. The crude product was purified by preparative RP-HPLC with a gradient of 25–73% (v/v) CH3CN in water containing 0.1% (v/v) TFA.
Characterization of β-Peptide Hydrophobicity
To characterize the hydrophobicity of the 14-helical β-peptides, we measured retention times by analytical RP-HPLC using a C18 column (Waters, X-bridge). The β-peptides (dissolved to a concentration of 0.5–1 mg/mL using deionized H2O containing 20–30% ACN and 0.1% TFA(v/v)) were injected (50 μL) into the HPLC. Retention time was characterized in triplicate with a gradient of 20–80% CH3CN in water containing 0.1% TFA (v/v) over 5–35 min.
Characterization of β-Peptide Helicity
β-Peptides were dissolved at 1 mg/mL in deionized H2O, divided into desired amounts of solution using a gastight syringe (Hamilton), and then lyophilized. Peptides were then dissolved in either MeOH (0.1 mM), PBS, or a mixture of MeOH/PBS (20 to 80% (v/v)). Circular dichroism (CD) was measured using an AVIV spectrometer at 20 °C with a 1 mm path length cell and 5 s averaging times. The CD signal in 100% methanol was assumed to be 100% helical. The retained helicity in 100% PBS compared with 100% MeOH was calculated by using the following equation:
Characterization of Antifungal Minimum Inhibitory Concentration (MIC)
The antifungal activities of the β-peptides against C. albicans were assayed in 96-well plates according to the planktonic susceptibility testing guidelines provided by the Clinical and Laboratory Standards Institute (formally, National Committee for Clinical Laboratory Standards) broth microdilution assay84 modified to include a quantitative XTT assessment of cell viability. Two-fold serial dilutions (100 μL) of β-peptides in RPMI (pH adjusted to 7.2 with MOPS) were mixed with 100 μL of a C. albicans strain SC5314 cell suspension (grown for 24 h at 35 °C and concentration adjusted to (1–5) × 103 cells/mL based on absorbance at 600 nm), and the plates were incubated at 35 °C for 48 h. Wells lacking β-peptide (cell controls) and wells lacking both peptide and cells (medium sterility controls) were included in every plate that was assayed. After 48 h, 100 μL of XTT solution (0.5 g L–1 in PBS, pH 7.4, containing 3 μM menadione in acetone) was added to all wells, and plates were incubated at 37 °C in the dark for 1.5 h. The supernatants (75 μL) from all wells were transferred to a fresh plate, and absorbance measurements at 490 nm were recorded using a plate reader (EL800 Universal Microplate Reader, Bio-Tek instruments, Inc.). The cell viability was plotted as a function of β-peptide concentration. Percent cell viability was calculated as
where A490, A490cell control, and A490 are the average absorbance values at 490 nm of the supernatant from wells containing a specific concentration of β-peptide, cell control wells lacking β-peptide, and medium sterility control wells, respectively. Experiments were performed in triplicate and repeated on at least three different days. The lowest assayed concentration of β-peptide that resulted in a decrease in absorbance of at least 90% of the mean was taken as the minimum inhibitory concentration (MIC) of that peptide.
Hemolysis Assays
Hemolysis assays were performed as previously described.57 Red blood cells (RBCs) were washed multiple times with Tris-buffered saline (TBS, 10 mM Tris-HCl, 100 mM NaCl, pH 7.5), until a clear supernatant was obtained. In a 96-well plate, 80 μL of RBCs was mixed with 20 μL of 2-fold serial dilutions of β-peptides in TBS and incubated at 37 °C for 1 h. Melittin served as a positive lysis control, and TBS was used as a negative lysis control. Plates were then centrifuged at 3000 rpm for 10 min, 50 μL of the supernatant was diluted with 50 μL of water in a fresh plate, and absorbance was measured at 405 nm using a plate reader. β-Peptide hemolysis was plotted as percent hemolysis as a function of β-peptide concentration. The percent hemolysis was calculated as
where A405, A405negative control, and A405 are the average absorbance values at 405 nm of the supernatant of RBCs treated with peptides, RBCs in TBS, and RBCs in melittin, respectively. Experiments were performed in duplicate and repeated on at least three different days.
Acknowledgments
This work was supported by NIH grant 1R01 AI092225. The peptide quantum calculations in Figure 1 were supported in part by National Science Foundation Grant CHE-0840494.
Supporting Information Available
Detailed synthetic procedures of Fmoc-β3-Et-OH, HPLC profiles, MIC analyses, hemolysis data, MALDI-TOF data, circular dichroism titration curves, and the plots illustrating relationships between hydrophobicity, helicity, and activity. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Mavor A. L.; Thewes S.; Hube B. (2005) Systemic fungal infections caused by Candida species: Epidemiology, infection process and virulence attributes. Curr. Drug Targets 6, 863–874. [DOI] [PubMed] [Google Scholar]
- Brown G. D.; Denning D. W.; Levitz S. M. (2012) Tackling human fungal infections. Science 336, 647–647. [DOI] [PubMed] [Google Scholar]
- Fishman J. A. (2007) Infection in solid-organ transplant recipients. N. Engl. J. Med. 357, 2601–2614. [DOI] [PubMed] [Google Scholar]
- Fridkin S. K.; Kaufman D.; Edwards J. R.; Shetty S.; Horan T. (2006) Changing incidence of Candida bloodstream infections among NICU patients in the United States: 1995–2004. Pediatrics 117, 1680–1687. [DOI] [PubMed] [Google Scholar]
- Kontoyiannis D. P.; Marr K. A.; Park B. J.; Alexander B. D.; Anaissie E. J.; Walsh T. J.; Ito J.; Andes D. R.; Baddley J. W.; Brown J. M.; Brumble L. M.; Freifeld A. G.; Hadley S.; Herwaldt L. A.; Kauffman C. A.; Knapp K.; Lyon G. M.; Morrison V. A.; Papanicolaou G.; Patterson T. F.; Perl T. M.; Schuster M. G.; Walker R.; Wannemuehler K. A.; Wingard J. R.; Chiller T. M.; Pappas P. G. (2010) Prospective surveillance for invasive fungal infections in hematopoietic stem cell transplant recipients, 2001–2006: Overview of the transplant-associated infection surveillance network (TRANSNET) database. Clin. Infect. Dis. 50, 1091–1100. [DOI] [PubMed] [Google Scholar]
- Neofytos D.; Fishman J. A.; Horn D.; Anaissie E.; Chang C. H.; Olyaei A.; Pfaller M.; Steinbach W. J.; Webster K. M.; Marr K. A. (2010) Epidemiology and outcome of invasive fungal infections in solid organ transplant recipients. Transpl. Infect. Dis. 12, 220–229. [DOI] [PubMed] [Google Scholar]
- Neofytos D.; Horn D.; Anaissie E.; Steinbach W.; Olyaei A.; Fishman J.; Pfaller M.; Chang C.; Webster K.; Marr K. (2009) Epidemiology and outcome of invasive fungal infection in adult hematopoietic stem cell transplant recipients: Analysis of multicenter prospective antifungal therapy (PATH) alliance registry. Clin. Infect. Dis. 48, 265–273. [DOI] [PubMed] [Google Scholar]
- Singh N. (2003) Fungal infections in the recipients of solid organ transplantation. Infect. Dis. Clin. North Am. 17, 113–134. [DOI] [PubMed] [Google Scholar]
- Jabra-Rizk M. A.; Johnson J. K.; Forrest G.; Mankes K.; Meiller T. F.; Venezia R. A. (2005) Prevalence of Candida dubliniensis fungemia at a large teaching hospital. Clin. Infect. Dis. 41, 1064–1067. [DOI] [PubMed] [Google Scholar]
- Walsh T. J.; Groll A. H. (1999) Emerging fungal pathogens: Evolving challenges to immunocompromised patients for the twenty-first century. Transpl. Infect. Dis. 1, 247–261. [DOI] [PubMed] [Google Scholar]
- Pappas P. G.; Rex J. H.; Lee J.; Hamill R. J.; Larsen R. A.; Powderly W.; Kauffman C. A.; Hyslop N.; Mangino J. E.; Chapman S.; Horowitz H. W.; Edwards J. E.; Dismukes W. E. (2003) NAIAD Mycoses Study Group, A prospective observational study of candidemia: Epidemiology, therapy, and influences on mortality in hospitalized adult and pediatric patients. Clin. Infect. Dis. 37, 634–643. [DOI] [PubMed] [Google Scholar]
- Mukherjee P. K.; Zhou G. Y.; Munyon R.; Ghannoum M. A. (2005) Candida biofilm: A well-designed protected environment. Med. Mycol. 43, 191–208. [DOI] [PubMed] [Google Scholar]
- Kojic E. M.; Darouiche R. O. (2004) Candida infections of medical devices. Clin. Microbiol. Rev. 17, 255–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawser S. P.; Douglas L. J. (1995) Resistance of Candida-albicans biofilms to antifungal agents in-vitro. Antimicrob. Agents Chemother. 39, 2128–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz T. (2003) Defensins: Antimicrobial peptides of innate immunity. Nat. Rev. Immunol. 3, 710–720. [DOI] [PubMed] [Google Scholar]
- Gallo R. L.; Nizet V. (2003) Endogenous production of antimicrobial peptides in innate immunity and human disease. Curr. Allergy Asthma Rep. 3, 402–409. [DOI] [PubMed] [Google Scholar]
- Beutler B. (2004) Innate immunity: an overview. Mol. Immunol. 40, 845–859. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W.; Lehrer R. (1998) Cationic peptides: a new source of antibiotics. Trends Biotechnol. 16, 82–88. [DOI] [PubMed] [Google Scholar]
- Zasloff M. (2002) Antimicrobial peptides of multicellular organisms. Nature 415, 389–395. [DOI] [PubMed] [Google Scholar]
- Brogden K. A.; Ackermann M.; McCray P. B.; Tack B. F. (2003) Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob. Agents 22, 465–478. [DOI] [PubMed] [Google Scholar]
- Antimicrobial Peptide Database (http://aps.unmc.edu/AP/main.php).
- Chu H. L.; Yu H. Y.; Yip B. S.; Chih Y. H.; Liang C. W.; Cheng H. T.; Cheng J. W. (2013) Boosting salt resistance of short antimicrobial peptides. Antimicrob. Agents Chemother. 57, 4050–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J. J.; Travis S. M.; Greenberg E. P.; Welsh M. J. (1996) Cystic fibrosis airway epithelia fail to kill bacteria because of abnormal airway surface fluid. Cell 85, 229–236. [DOI] [PubMed] [Google Scholar]
- Bowdish D. M. E.; Davidson D. J.; Lau Y. E.; Lee K.; Scott M. G.; Hancock R. E. W. (2005) Impact of LL-37 on anti-infective immunity. J. Leukocyte Biol. 77, 451–459. [DOI] [PubMed] [Google Scholar]
- Goldman M. J.; Anderson G. M.; Stolzenberg E. D.; Kari U. P.; Zasloff M.; Wilson J. M. (1997) Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell 88, 553–560. [DOI] [PubMed] [Google Scholar]
- Huang J. F.; Hao D. M.; Chen Y.; Xu Y. M.; Tan J. J.; Huang Y. B.; Li F.; Chen Y. X. (2011) Inhibitory effects and mechanisms of physiological conditions on the activity of enantiomeric forms of an alpha-helical antibacterial peptide against bacteria. Peptides 32, 1488–1495. [DOI] [PubMed] [Google Scholar]
- Yeaman M. R.; Yount N. Y. (2003) Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev. 55, 27–55. [DOI] [PubMed] [Google Scholar]
- Shai Y. (1999) Mechanism of the binding, insertion and destabilization of phospholipid bilayer membranes by alpha-helical antimicrobial and cell non-selective membrane-lytic peptides. Biochim. Biophys. Acta, Biomembr. 1462, 55–70. [DOI] [PubMed] [Google Scholar]
- Huang H. W. (2000) Action of antimicrobial peptides: Two-state model. Biochemistry 39, 8347–8352. [DOI] [PubMed] [Google Scholar]
- Nizet V. (2006) Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr. Issues Mol. Biol. 8, 11–26. [PubMed] [Google Scholar]
- Schmidtchen A.; Frick I. M.; Andersson E.; Tapper H.; Bjorck L. (2002) Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol. 46, 157–168. [DOI] [PubMed] [Google Scholar]
- Andersson D. I.; Hughes D. (2010) Antibiotic resistance and its cost: is it possible to reverse resistance?. Nat. Rev. Microbiol. 8, 260–271. [DOI] [PubMed] [Google Scholar]
- Brogden K. A. (2005) Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria?. Nat. Rev. Microbiol. 3, 238–250. [DOI] [PubMed] [Google Scholar]
- Hilchie A. L.; Wuerth K.; Hancock R. E. W. (2013) Immune modulation by multifaceted cationic host defense (antimicrobial) peptides. Nat. Chem. Biol. 9, 761–768. [DOI] [PubMed] [Google Scholar]
- Padhee S.; Hu Y. G.; Niu Y. H.; Bai G.; Wu H. F.; Costanza F.; West L.; Harrington L.; Shaw L. N.; Cao C. H.; Cai J. F. (2011) Non-hemolytic alpha-AApeptides as antimicrobial peptidomimetics. Chem. Commun. 47, 9729–9731. [DOI] [PubMed] [Google Scholar]
- Chongsiriwatana N. P.; Miller T. M.; Wetzler M.; Vakulenko S.; Karlsson A. J.; Palecek S. P.; Mobashery S.; Barron A. E. (2011) Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrob. Agents Chemother. 55, 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chongsiriwatana N. P.; Patch J. A.; Czyzewski A. M.; Dohm M. T.; Ivankin A.; Gidalevitz D.; Zuckermann R. N.; Barron A. E. (2008) Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc. Natl. Acad. Sci. U.S.A. 105, 2794–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryge T. S.; Hansen P. R. (2005) Novel lysine-peptoid hybrids with antibacterial properties. J. Pept. Sci. 11, 727–734. [DOI] [PubMed] [Google Scholar]
- Ivankin A.; Livne L.; Mor A.; Caputo G. A.; DeGrado W. F.; Meron M.; Lin B.; Gidalevitz D. (2010) Role of the conformational rigidity in the design of biomimetic antimicrobial compounds. Angew. Chem., Int. Ed. 49, 8462–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R. W.; DeGrado W. F.; Tew G. N. (2008) De novo designed synthetic mimics of antimicrobial peptides. Curr. Opin. Biotechnol. 19, 620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudon P.; Violette A.; Lamour K.; Decossas M.; Fournel S.; Heurtault B.; Godet J.; Mely Y.; Jamart-Gregoire B.; Averlant-Petit M. C.; Briand J. P.; Duportail G.; Monteil H.; Guichard G. (2010) Consequences of isostructural main-chain modifications for the design of antimicrobial foldamers: Helical mimics of host-defense peptides based on a heterogeneous amide/urea backbone. Angew. Chem., Int. Ed. 49, 333–336. [DOI] [PubMed] [Google Scholar]
- Liu R. H.; Chen X. Y.; Falk S. P.; Mowery B. P.; Karlsson A. J.; Weisblum B.; Palecek S. P.; Masters K. S.; Gellman S. H. (2014) Structure-activity relationships among antifungal nylon-3 polymers: Identification of materials active against drug-resistant strains of Candida albicans. J. Am. Chem. Soc. 136, 4333–4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H.; Palermo E. F.; Yasuhara K.; Caputo G. A.; Kuroda K. (2013) Molecular design, structures, and activity of antimicrobial peptide-mimetic polymers. Macromol. Biosci. 13, 1285–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uppu D.; Akkapeddi P.; Manjunath G. B.; Yarlagadda V.; Hoque J.; Haldar J. (2013) Polymers with tunable side-chain amphiphilicity as non-hemolytic antibacterial agents. Chem. Commun. 49, 9389–9391. [DOI] [PubMed] [Google Scholar]
- Oda Y.; Kanaoka S.; Sato T.; Aoshima S.; Kuroda K. (2011) Block versus random amphiphilic copolymers as antibacterial agents. Biomacromolecules 12, 3581–3591. [DOI] [PubMed] [Google Scholar]
- Kuroda K.; DeGrado W. F. (2005) Amphiphilic polymethacrylate derivatives as antimicrobial agents. J. Am. Chem. Soc. 127, 4128–4129. [DOI] [PubMed] [Google Scholar]
- Tew G. N.; Liu D. H.; Chen B.; Doerksen R. J.; Kaplan J.; Carroll P. J.; Klein M. L.; DeGrado W. F. (2002) De novo design of biomimetic antimicrobial polymers. Proc. Natl. Acad. Sci. U.S.A. 99, 5110–5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epand R. F.; Schmitt M. A.; Gellman S. H.; Epand R. M. (2006) Role. of membrane lipids in the mechanism of bacterial species selective toxicity by two alpha/beta-antimicrobial peptides. Biochim. Biophys. Acta, Biomembr. 1758, 1343–1350. [DOI] [PubMed] [Google Scholar]
- Epand R. F.; Schmitt M. A.; Gellman S. H.; Sen A.; Auger M.; Hughes D. W.; Epand R. M. (2005) Bacterial species selective toxicity of two isomeric alpha/beta-peptides: Role of membrane lipids. Mol. Membr. Biol. 22, 457–469. [DOI] [PubMed] [Google Scholar]
- Seebach D.; Matthews J. L. (1997) Beta-peptides: a surprise at every turn. Chem. Commun. 2015–2022. [Google Scholar]
- Cheng R. P.; Gellman S. H.; DeGrado W. F. (2001) Beta-peptides: From structure to function. Chem. Rev. 101, 3219–3232. [DOI] [PubMed] [Google Scholar]
- Hintermann T.; Seebach D. (1997) The biological stability of beta-peptides: No interactions between alpha- and beta-peptidic structures. Chimia 51, 244–247. [Google Scholar]
- Raguse T. L.; Porter E. A.; Weisblum B.; Gellman S. H. (2002) Structure-activity studies of 14-helical antimicrobial beta-peptides: Probing the relationship between conformational stability and antimicrobial potency. J. Am. Chem. Soc. 124, 12774–12785. [DOI] [PubMed] [Google Scholar]
- Liu D. H.; DeGrado W. F. (2001) De novo design, synthesis, and characterization of antimicrobial beta-peptides. J. Am. Chem. Soc. 123, 7553–7559. [DOI] [PubMed] [Google Scholar]
- Hamuro Y.; Schneider J. P.; DeGrado W. F. (1999) De novo design of antibacterial beta-peptides. J. Am. Chem. Soc. 121, 12200–12201. [Google Scholar]
- Arvidsson P. I.; Frackenpohl J.; Ryder N. S.; Liechty B.; Petersen F.; Zimmermann H.; Camenisch G. P.; Woessner R.; Seebach D. (2001) On the antimicrobial and hemolytic activities of amphiphilic beta-peptides. ChemBioChem 2, 771–773. [DOI] [PubMed] [Google Scholar]
- Karlsson A. J.; Pomerantz W. C.; Neilsen K. J.; Gellman S. H.; Palecek S. P. (2009) Effect of sequence and structural properties on 14-helical beta-peptide activity against Candida albicans planktonic cells and biofilms. ACS Chem. Biol. 4, 567–579. [DOI] [PubMed] [Google Scholar]
- Karlsson A. J.; Pomerantz W. C.; Weisblum B.; Gellman S. H.; Palecek S. P. (2006) Antifungal activity from 14-helical beta-peptides. J. Am. Chem. Soc. 128, 12630–12631. [DOI] [PubMed] [Google Scholar]
- Epand R. F.; Umezawa N.; Porter E. A.; Gellman S. H.; Epand R. M. (2003) Interactions of the antimicrobial beta-peptide beta-17 with phospholipid vesicles differ from membrane interactions of magainins. Eur. J. Biochem. 270, 1240–1248. [DOI] [PubMed] [Google Scholar]
- Porter E. A.; Weisblum B.; Gellman S. H. (2002) Mimicry of host-defense peptides by unnatural oligomers: Antimicrobial beta-peptides. J. Am. Chem. Soc. 124, 7324–7330. [DOI] [PubMed] [Google Scholar]
- Porter E. A.; Wang X. F.; Lee H. S.; Weisblum B.; Gellman S. H. (2000) Antibiotics - Non-haemolytic beta-amino-acid oligomers. Nature 404, 565–565. [DOI] [PubMed] [Google Scholar]
- Arvidsson P. I.; Ryder N. S.; Weiss H. M.; Gross G.; Kretz O.; Woessner R.; Seebach D. (2003) Antibiotic and hemolytic activity of a beta(2)/beta(3) peptide capable of folding into a 12/10-helical secondary structure. ChemBioChem 4, 1345–1347. [DOI] [PubMed] [Google Scholar]
- Parker J. M. R.; Guo D.; Hodges R. S. (1986) New hydrophiliity scale derived from high-performance liquid-chromatography peptide retention data - correlation of predicted surface residues with antigenicity and x-ray derived accessible sites. Biochemistry 25, 5425–5432. [DOI] [PubMed] [Google Scholar]
- Browne C. A.; Bennett H. P. J.; Solomon S. (1982) The isolation of peptides by high-performance liquid-chromatography using predicted elution positions. Anal. Biochem. 124, 201–208. [DOI] [PubMed] [Google Scholar]
- Wilson K. J.; Honegger A.; Stotzel R. P.; Hughes G. J. (1981) The behavior of peptides on reverse-phase supports during high-pressure liquid-chromatorraphy. Biochem. J. 199, 31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondejewski L. H.; Jelokhani-Niaraki M.; Farmer S. W.; Lix B.; Kay C. M.; Sykes B. D.; Hancock R. E. W.; Hodges R. S. (1999) Dissociation of antimicrobial and hemolytic activities in cyclic peptide diastereomers by systematic alterations in amphipathicity. J. Biol. Chem. 274, 13181–13192. [DOI] [PubMed] [Google Scholar]
- Kim S.; Kim S. S.; Lee B. J. (2005) Correlation between the activities of alpha-helical antimicrobial peptides and hydrophobicities represented as RP HPLC retention times. Peptides 26, 2050–2056. [DOI] [PubMed] [Google Scholar]
- Jiang Z. Q.; Kullberg B. J.; van der Lee H.; Vasil A. I.; Hale J. D.; Mant C. T.; Hancock R. E. W.; Vasil M. L.; Netea M. G.; Hodges R. S. (2008) Effects of hydrophobicity on the antifungal activity of alpha-helical antimicrobial peptides. Chem. Biol. Drug Des. 72, 483–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. (1987) Magainins, a class of antimicrobial peptides from xenopus skin - isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. U.S.A. 84, 5449–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosetto M.; Manetti A. G. O.; Marchini D.; Dallai R.; Telford J. L.; Baldari C. T. (1993) Sequences of two cDNA clones from the medfly ceratitis-capitata encoding antibacterial peptides of the ceropin family. Gene 134, 241–243. [DOI] [PubMed] [Google Scholar]
- Mor A.; Nicolas P. (1994) Isolation and structure of novel defensive pepptides from frog-skin. Eur. J. Biochem. 219, 145–154. [DOI] [PubMed] [Google Scholar]
- Kreil G. (1973) Structure of melittin isolated from two species of honey bees. FEBS Lett. 33, 241–244. [Google Scholar]
- Bagella L.; Scocchi M.; Zanetti M. (1995) cDNA sequences of three sheep myeloid cathelicidins. FEBS Lett. 376, 225–228. [DOI] [PubMed] [Google Scholar]
- Duvick J. P.; Rood T.; Rao A. G.; Marshak D. R. (1992) Purification and characterization of a novel antimicrobial peptide from maize(Zea-mays L.) kernels. J. Biol. Chem. 267, 18814–18820. [PubMed] [Google Scholar]
- Rasul R.; Cole N.; Balasubramanian D.; Chen R.; Kumar N.; Willcox M. D. P. (2010) Interaction of the antimicrobial peptide melimine with bacterial membranes. Int. J. Antimicrob. Agents 35, 566–572. [DOI] [PubMed] [Google Scholar]
- Wieprecht T.; Dathe M.; Beyermann M.; Krause E.; Maloy W. L.; MacDonald D. L.; Bienert M. (1997) Peptide hydrophobicity controls the activity and selectivity of magainin 2 amide in interaction with membranes. Biochemistry 36, 6124–6132. [DOI] [PubMed] [Google Scholar]
- Chen Y. X.; Vasil A. I.; Rehaume L.; Mant C. T.; Burns J. L.; Vasil M. L.; Hancock R. E. W.; Hodges R. S. (2006) Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des. 67, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y. X.; Mant C. T.; Farmer S. W.; Hancock R. E. W.; Vasil M. L.; Hodges R. S. (2005) Rational design of alpha-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 280, 12316–12329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H. C.; Brown J. H.; Morell J. L.; Huang C. M. (1988) Synthetic magainin analogs with improved antimicrobial activity. FEBS Lett. 236, 462–466. [DOI] [PubMed] [Google Scholar]
- Zhang L. J.; Benz R.; Hancock R. E. W. (1999) Influence of proline residues on the antibacterial and synergistic activities of alpha-helical peptides. Biochemistry 38, 8102–8111. [DOI] [PubMed] [Google Scholar]
- Grieco P.; Carotenuto A.; Auriemma L.; Saviello M. R.; Campiglia P.; Gomez-Monterrey I. M.; Marcellini L.; Luca V.; Barra D.; Novellino E.; Mangoni M. L. (2013) The effect of D-amino acid substitution on the selectivity of temporin L towards target cells: Identification of a potent anti-Candida peptide. Biochim. Biophys. Acta, Biomembr. 1828, 652–660. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for detailed synthetic procedure (Scheme S1).
- Murray J. K.; Gellman S. H. (2005) Application of microwave irradiation to the synthesis of 14-helical beta-peptides. Org. Lett. 7, 1517–1520. [DOI] [PubMed] [Google Scholar]
- National Committee for Clinical Laboratory Standards (2002) Reference method for broth dilution antifungal susceptibility testing of yeasts: Approved standard, 2nd ed., NCCL document M27-A2, NCCLS, Wayne, PA. [Google Scholar]
- Daniels D. S.; Petersson E. J.; Qiu J. X.; Schepartz A. (2007) High-resolution structure of a beta-peptide bundle. J. Am. Chem. Soc. 129, 1532–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.