

Abstract
Inhibiting the enzyme telomerase by stabilizing the G-quadruplex has potential in anticancer drug design. Diprotonated cyclo[n]pyrroles represent a set of expanded porphyrin analogues with structures similar to telomestatin, a natural product known to bind to and stabilize G-quadruplexes. As a first step towards testing whether cyclo[n]pyrroles display a similar function, a series of diprotonated cyclo[n]pyrroles (where n = 6, 7 and 8) was each added to the human telomere repeat sequence d(T2AG3)4 and examined with mass spectrometry, ion mobility and molecular dynamics calculations. Nano-ESI-MS indicated that the smaller the cyclo[n]pyrrole, the stronger it binds to the telomeric sequence. It was also found that cyclo[6]pyrrole bound to d(T2AG3)4 better than octaethylporphyrin, a finding rationalized by cyclo[6]pyrrole having a +2 charge, while octaethylporphyrin bears no charge. Ion mobility measurements were used to measure the collision cross section of each d(T2AG3)4/cyclo[n]pyrrole complex. Only one peak was observed in the arrival time distributions for all complexes and the experimental cross sections indicated that only structures with d(T2AG3)4 in an antiparallel G-quadruplex arrangement and each cyclo[n]pyrrole externally stacked below the G-quartets occur under these experimental conditions. When the cyclo[n]pyrroles were intercalated or nonspecifically bound to the quadruplex or if different conformations than antiparallel were considered for d(T2AG3)4, the theoretical cross sections did not match experiment. On this basis, it is inferred that 1) external stacking represents the dominant binding mode for the interaction of cyclo[n]pyrroles with d(T2AG3)4 and 2) the overall size and charge of the cyclo[n]pyrroles play important roles in defining the binding strength.
Introduction
The development of small molecules capable of structure-selective DNA targeting is an area of great interest due to its possible role in discovering antitumor chemotherapeutic agents.1, 2 The recent finding that there is a link between tumor immortalization and telomerase activity is providing a new focus for work in this area.3, 4 Telomerase is a ribonucleoprotein enzyme active in 85–90% of all human tumor cells 5 and undetectable in most normal somatic cells. 6 Interactions between telomerase and the specialized ends of linear chromosomes or telomeres have shown to be a key step in the immortalization of tumor cells. Human telomere DNA is comprised of 5–8 kilobases of tandem repeats of the sequence d(T2AG3)n, with a single-stranded 3′ overhang of 100–200 bases. 7, 8, 9 In normal somatic cells, telomeres are shortened by 50–200 bases after each round of cell division because of the inability of the endogenous DNA polymerase to completely replicate the lagging telomeric DNA strand.10 Once telomeres are reduced to a certain length, they become too short to form secondary structures. This precludes chromosome replication, thus causing the cells to go into senescence and eventually undergo apoptosis. However, the telomere length in cancer cells is not shortened but kept constant by telomerase, thereby allowing tumor cells to escape the senescence/apoptosis cycle and become immortal. On the basis of these observations, it has been suggested that telomerase inhibition could provide a new, useful approach to preventing tumor proliferation.11
In the context of efforts to inhibit telomerase expression, the structure of the human telomere sequence has been studied in depth. One key finding has been that the integrity of the single-strand overhang in the telomeres must be maintained for cell survival. It has been proposed that this G-rich region is protected by folding into a G-quadruplex, as observed in both the solution and the solid state structures of dAG3(T2AG3)3.12, 13 G-quadruplexes are made up of G-quartet subunits, where 4 coplanar guanines (G) are linked together by Hoogsteen hydrogen bonds as shown in Figure 1.14, 15 When the G-quartets stack on top of each other they form a G-quadruplex. There is ample evidence that quadruplex structures form in vitro for G-rich DNA sequences, but currently no direct evidence of in vivo quadruplex structures. However, indirect support for the existence and role of quadruplexes in vivo is abundant. For instance, proteins that bind to quadruplexes,16, 17 quadruplex specific nucleases,18 and some helicases that unwind quadruplexes have all been identified.19, 20 Due to the indirect nature of this evidence, a range of in vitro experiments have been carried out in an effort to elucidate possible roles of the quadruplex. Some interesting findings have emerged, including 1) the recognition that quadruplexes inhibit telomerase and 2) that certain small molecules are able to target the quadruplex structure selectively.21, 22, 23, 24 These findings have prompted efforts to develop drug candidates that bind to, and stabilize the quadruplex structure since, as implied above, it is thought that such species could inhibit telomerase and thus prevent tumor proliferation.
Figure 1.

a) The structure of a G-quartet and b) a schematic representation of a G-quadruplex.
Recently, acridine derivatives, 25 cationic porphyrin derivatives, 26 ethidium derivatives, 27 anthraquinone derivatives,28 perylene derivatives,29 and telomestatin30 have been explored for their ability to bind to quadruplexes and to inhibit telomerase activity (Figure 2). From telomeric repeat amplification protocol (TRAP) assay of those molecules, it was concluded that telomestatin, a natural product isolated from Streptomyces anulatus 3533-SV4, is the most potent inhibitor of telomerase as it displays a very impressive IC50 value (concentration required for 50% inhibition of growth in a battery of tumor and normal cells) of ~5 nM, while most of the other molecules depicted in Figure 2 displayed IC50 values in the μM range.30 Such findings have, not surprisingly, sparked interest in molecules that bear a similar structural analogy to telomestatin.
Figure 2.

Telomerase inhibitors.
While not a natural product, the recently reported cyclo[8]pyrroles (Figure 3),31 a new class of aromatic expanded porphyrins, are structurally similar to that of telomestatin. In an effort to test whether cyclo[8]pyrrole and its analogues are capable of binding to quadruplexes, four diprotonated cyclo[n]pyrroles of different sizes32 (2–5, Figure 3) were allowed to interact with the intramolecular G-quadruplex d(T2AG3)4 (abbreviated as T4). Octaethylporphyrin, 1, was also studied as a control. It was chosen for this role since, under the condition of the experiment, it is neutral, whereas 2–5 are diprotonated and bear a +2 charge. In this paper, we present the results of studies between T4 and 1–5, using nano-electrospray ionization mass spectrometry (nano-ESI-MS) and ion mobility spectrometry.
Figure 3.
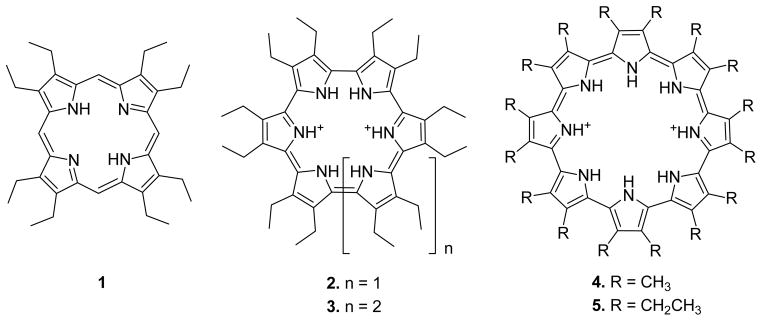
Structures of oligopyrrole macrocycles (.porphyrinoids.) used in this study: octaethylporphyrin 1, cyclo[6]pyrrole 2, cyclo[7]pyrrole 3, and cyclo[8]pyrroles 4 and 5.
ESI-MS is a very important method for the characterization of noncovalently bound biological molecules in the gas phase.33, 34, 35 Likewise, ion mobility methods have emerged as very powerful techniques for investigating the conformations of gas phase ions. 36, 37 Recently, ESI-MS in combination with ion mobility methods has served to demonstrate that DNA helices38, 39 and G-quadruplexes 40, 41 are stable when sprayed and dehydrated using nano-ESI and that they retain most of their solution phase structural characteristics. Knowing that DNA complexes remain intact in the gas phase makes ESI-MS in conjunction with ion mobility analysis an ideal method for studying the noncovalent T4/oligopyrrole macrocycle complexes. Our study goals were thus 1) to use ESI-MS to determine the relative binding strengths of each porphyrin and porphyrin analogues (collectively referred to as porphyrinoids) to T4 and then 2) to use ion mobility methods and molecular dynamics simulations to determine the specific binding mode.
Experimental
Materials
The synthesis procedures for 1–5 have previously been published.31, 32, 42 Compounds 1, 4, and 5 were prepared to a concentration of 300 μM by first adding THF and then water so that a 20% THF and 80% H2O solution was obtained. Since 1, 4 and 5 are not 100% water soluble, adding THF allows the porphyrinoids to be solublized to the point that they remain in solution once water is added. Macrocycle 2 and 3 are not quite as water soluble as 1, 4, and 5. Accordingly, a small amount of chloroform was first added to the solids, then THF, and finally H2O, such that the final solution was 2% chloroform, 18% THF, and 80% H2O.
T4 was purchased from Integrated DNA Technologies, Inc. (Coralville, IA) and used without further purification. T4 was prepared to a concentration of 300 μM in a 150 mM NH4OAc/H2O solution at pH 7. T4 was annealed by itself at 95 ºC for 10 minutes, slowly cooled to room temperature, and stored at 10 ºC. Before being combined with the porphyrinoids 1–5, T4 was diluted to 100 μM with H2O. Then, 3 uL of the annealed T4 solution and 3 μL of the porphyrinoid solution subject to study were combined to give a 1:3 DNA to macrocycle solution.
Mass Spectra and Ion Mobility Experiments
Details concerning the experimental setup for the mass spectra and ion mobility measurements have been published previously.43 Accordingly, only a brief description will be given here. Approximately 6 μL of one of the solutions prepared as described above was placed in a metalized glass needle (spray tip). Ions were formed by nano-ESI and injected into a specially designed ion funnel. The ions were then carefully injected into a 4.5 cm long drift cell filled with ~5 torr of helium gas and gently pulled through the He gas at a constant drift velocity by a weak electric field. After exiting the drift cell, the ions were mass analyzed by a quadrupole mass filter and detected. The quadrupole mass filter can either be set to select a mass range of interest for the acquisition of a mass spectrum or in a pulsed experiment it can be set to detect one specific m/z as a function of time, yielding an arrival time distribution or ATD. The reduced mobility, Ko, of a specific ion is accurately determined using Equation 144
| (1) |
where l is the length of the cell, T is the temperature in Kelvin, p is the pressure of the He gas (in torr), V is the voltage applied to the drift cell, tA is the arrival time of the ions taken from the center of the ATD peak, and to is the amount of time the ion spends outside the drift cell before reaching the detector. A series of arrival times is measured by changing the voltage applied to the drift cell. A plot of tA vs. p/V yields a straight line with a slope inversely proportional to Ko and an intercept of to. Once Ko is determined in this way, the collisional cross section of the ion, σ, could be calculated using Equation 2,
| (2) |
where e is the charge of the ion, No is the number density of He at STP, T is temperature, kb is Boltzmann’s constant, and μ is the ion-He reduced mass.44
Theoretical Calculations
Structural information relevant to the ion mobility experiments was obtained by comparing the experimental cross sections determined from the ATDs to the cross sections calculated for various theoretical structures. Starting structures for T4 were created using the antiparallel NMR structure 143D,12, 45 the parallel X-ray structure 1KF113, 45 and the mixed parallel/ antiparallel NMR structure 186D.45, 46 HyperChem47 was used to add 2 Ts to the start of both the NMR and X-ray structures since they were for the sequence dAG3(T2AG3)3 and to edit the mixed parallel/antiparallel sequence to reflect T4. HyperChem was also used to add the porphyrin and cyclo[n]pyrroles to various locations around the initial quadruplex structures. 300 K molecular dynamics simulations were run on each complex for 2 ns using the AMBER 748 set of programs and every 5 ps a structure was saved. Each structure was then energy minimized and its cross section calculated. For ions with more than 200 atoms, collisional cross sections were calculated using hard-sphere scattering and trajectory models developed by the Jarrold group.49, 50 From the calculations of each of the T4/porphyrinoid complexes, it was found that the various starting structures eventually converge to give one steady state structure where the cross section remains relatively constant. The average cross sections of the final 100 steady state structures were used for comparison with the experimental values.
The overall charge state of the complexes observed in the experiments can be readily identified from the mass spectra, but the exact locations of the deprotonation sites needed for the modeling are not known. Cyclo[n]pyrroles 2–5 carry a charge of +2, whereas porphyrin 1 is neutral, as inferred from mass spectra (not shown). On the basis of such analysis, it was deduced that T4 can carry a charge of −5, −6 or −7 in the different complexes observed. Consequently, the same structure for T4 was modeled with different deprotonation sites, but no changes in cross section or conformation were observed.
Results and Discussion
Mass Spectra
The nano-ESI mass spectra of T4 in a 1 to 3 ratio with 1–5 are shown in Figure 4. For all of the spectra, the most intense peaks are for T46- and T45- with NH4+ adducts broadening the peaks on the high mass side. Normally quadruplexes are stabilized by having K+, Na+ or NH4 + between each G-quartet layer.51, 52, 53 Thus, in the absence of alkali cation stabilization, it was expected that (T4 + 2NH4 +) at multiple charges states would be the dominant peaks in the mass spectra, rather than T4. However, it has been reported that the dominant peaks in the ESI-MS spectra of T4 and dAG3(T2AG3)3 occur in the absence of any cation stabilization.54 Ion mobility studies were carried out in an effort to determine whether these solvent-free complexes without metal ion stabilization are really quadruplexes or just globular forms.41 From the ion mobility measurements,41 it was found that the experimental cross section of T4 without NH4 + matches a stable antiparallel basket conformation (the conformation observed by NMR), an antiparallel chair conformation, or both as shown in Figure 5. Distinction could not be made between the basket and chair conformers because they both have the same cross section. However, model cross sections of globular forms of T4 were too small to match the experimental cross section and the model cross sections of the parallel propeller conformation observed in X-ray crystallography and the mixed parallel/antiparallel structure were too large (Figure 5). On this basis, it was concluded that specific quadruplex conformations are present for T4 without NH4 + stabilization and that the conformations are similar to those seen in solution, as inferred from NMR structural studies.
Figure 4.

Nano-ESI mass spectra of T4 in a 1 to 3 ratio with a) 1, b) 2, c) 3, d) 4 and e) 5. Except for 5, −5 and −6 T4/porphyrinoid complexes were observed for all macrocycles.
Figure 5.

a) Four different strand orientations for T4. b) Possible binding sites for the porphyrinoid 1–5 as illustrated using the antiparallel basket conformation of T4.
T4/porphyrinoid complexes with charge states of −5 and −6 are also present in all of the mass spectra except when T4 and 5 are combined, as can be seen in Figure 4. Similar to the T4 peaks, the dominant T4/porphyrinoid peaks in the mass spectra are not stabilized by NH4 + adducts. In an effort to increase the abundance of the T4/porphyrinoid complexes, various ratios of T4 to 1–5 were tried. Unfortunately, at higher concentration of 1–5, π-stacking between pairs of macrocycles is apparently favored and even less evidence of complexation with T4 is observed. A T4 to porphyrinoid ratio of 1 to 3 seemed to give rise to the greatest level of complexation.
The relative intensities of the various porphyrinoids in the mass spectra can be related to the relative stabilities of the complexes. A quantitative relationship requires that the relative binding probabilities occurring in solution are maintained in the ESI mass spectra and there are two main reasons to believe this is so. First, identical solution conditions were maintained for each sample and second, the energy with which the ions were transferred from the ion funnel into the drift cell was varied over a wide range with essentially no change in the spectra observed. A certain minimal amount of energy (20 eV) is required to go from the low pressure ion funnel .upstream. into the much higher pressure drift cell. This injection energy initially imparts a collisionally induced transient internal energy spike into the complex that is subsequently removed by collisions in the cell. Increasing the injection energy from 20 to 100 eV had little effect on the spectra suggesting all systems are strongly bound in the gas phase and the relative solution affinities are the reasons for the variations in the intensities of the complexes of 1–5 with T4. This data yields a binding strength order of 2 > 1 > 3 > 4 ≫ 5.
To begin evaluating why the porphyrinoids of this study bind in this order, the sizes of 1–5 were considered. As can be inferred from Figure 3, the size of the porphyrinoid increases in the order 1 < 2 < 3 < 4 < 5, so consequently, we can conclude that smaller porphyrinoid compounds bind more strongly to T4 than their larger congeners. This result can be rationalized using Figure 6, where a footprint is shown of each porphyrinoid above the 3 stacked quartets (the sugars and phosphate backbone are omitted for clarity). Porphyrinoids 1 and 2 provide a good size match for the quartet, whereas 3 and 4 are slightly larger and 5 is much larger. The footprint of 5 clearly illustrates that in order for it to bind to a quartet it would have to interact with the sugar-phosphate backbone of the DNA strand. These interactions are almost certainly repulsive and would make it very difficult for 5 to bind to the G-quartets, a conclusion that is illustrated by the lack of (T4 + 5) complexes formed in the mass spectra in Figure 4. In a similar vein, porphyrinoids 3 and 4 are also larger than the quartets being targeted and would interact slightly with the sugars upon binding, consistent with the fact that these two species do not bind as well as 1 and 2.
Figure 6.
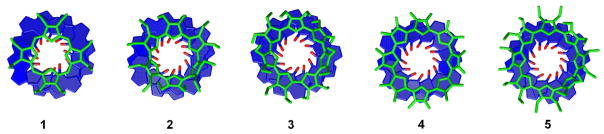
The footprint of each porphyrinoid above 12 guanines in a G-quadruplex arrangement. This figure is designed to show the size difference between the porphyrinoid and stacked G-quartets. 1 is octaethylporphyrin and 2 is the smallest macrocycle whereas 5 is the largest. The sugars and phosphate backbones are omitted for clarity.
One exception to this size dependent binding trend is provided by 1. Although 1 is the smallest porphyrinoid, being slightly smaller than 2, it does not bind as well as 2. However, unlike 2 and its larger congeners 3–5, porphyrin 1 was the only neutral porphyrinoid studied (2–5 have a charge of +2). It thus serves as a control to determine if charge plays a significant role in modulating the binding strength of porphyrinoids. The most important interaction between G-quartets and porphyrinoids in solution is thought to be π-stacking,55 but since quartets have a negative charge it has also been suggested that positively charged porphyrins should bind more strongly than neutral porphyrins.56 The results of this study are consistent with this suggestion since 2 is bound more strongly than 1 even though this latter system is smaller, and based on considerations of size alone it was expected to bind better than 2. We thus conclude that an optimized combination of size and positive charge are necessary to achieve the highest level of T4/porphyrinoid binding in solution.
The solutions used in these experiments contained approximately 90% water and either 10% THF or 9% THF and 1% chloroform along with some ammonium acetate buffer. Hence, while the pH was near physiological the solutions themselves were not. The 10% THF could have some effect on the relative binding strengths of the complexes, but if so this is probably only a minor effect. The T4 does form a stable quadruplex in these solutions and there is no indication the binding of the porphyrinoids to T4 is solvent mediated. However, this point will be explored in the future when water soluble porphyrinoids become available. Research is underway on the synthesis of these species.
Ion Mobility
In order to examine the conformational properties of the T4/porphyrinoid complexes and determine where 1–4 bind on T4, ion mobility experiments were performed. In each case, the appropriate ion for each complex was gently injected into the drift cell and its ATD collected.
A typical ATD obtained for all of the T4/porphyrinoid complexes is shown in Figure 7. Only one peak appears in the ATDs for all of the −5 and −6 complexes, and a single symmetric peak indicates that either only one family of conformers is present in the distribution or all of the families of conformers present have essentially the same cross section. Experimental cross sections were extracted from the ATDs for each of the various complexes and are listed in Table 1.
Figure 7.

A single peak is observed in the ATD for all of the T4/porphyrinoid complexes analyzed in this study. This supports the notion that each complex consists of only one conformation. Here the ATD for (T4+1)5- is illustrated.
Table 1.
Experimental and Theoretical Cross Sections (Å2) for T4/Porphyrinoid Complexes
| Expta | Theoryb | ||
|---|---|---|---|
| Complex | Basketc Externally Stackedd |
Chairc Externally Stackedd |
|
| (T4+1)5- | 854 | 858 | 860 |
| (T4+1)6- | 867 | 872 | 874 |
|
| |||
| (T4+2)5- | 864 | 868 | 872 |
| (T4+2)6- | 872 | 877 | 876 |
|
| |||
| (T4+3)5- | 862 | 869 | 870 |
| (T4+3)6- | 874 | 879 | 881 |
|
| |||
| (T4+4)5- | 859 | 866 | 869 |
| (T4+4)6 | 870 | 881 | 880 |
1% reproducibility error,
≤ 2% standard deviation,
the conformation of T4,
the location of the porphyrinoids
In order to obtain structures for the conformers observed in the ATDs, 300 K molecular dynamics calculations were carried out on the multiple complex geometries possible for the T4/porphyrinoid complexes (Figure 5). Seenisamy et al. have indicated that when a ligand binds it can possibly change the conformation of the quadruplex, 57, 58, 59 so in addition to the basket and chair conformation observed for T4 in the ion mobility studies,41 a parallel propeller, mixed parallel/antiparallel and globular conformations were also used as starting geometries. Porphyrinoids can potentially bind to a quadruplex by externally stacking below the quartets, intercalating between the quartets or nonspecifically binding to some random location on the DNA strand. Each of these binding locations was modeled with the basket, chair, parallel/antiparallel mix and propeller T4 conformations. The globular conformation was modeled using only nonspecifically bound porphyrinoids because of its lack of quartets.
300 K dynamics simulations were performed on each different starting structure and a typical dynamics plot of cross section versus time is shown in Figure 8. Only one steady state was observed during the dynamics runs on each of the different possible complexes. However, different cross sections were observed for each complex as illustrated for the −5 complexes in Table 2. Similar cross section values to those given in Table 2 were obtained for the −6 complexes. The cross sections for the basket and chair form of T4 with the porphyrinoids externally stacked agree very well with the appropriate experimental cross section (Table 1). Papers by the Hurley group57–59 have suggested the main form of binding for porphyrins and porphyrin-like compounds of about the same size as those considered here to G-quadruplexes is external stacking, consistent with our results.
Figure 8.

The plot of cross section versus dynamics time for (T4+2)5-. Dynamics simulations were run at 300 K for 2 ns and every 5 ps a structure was saved and its cross section calculated. Only one steady state is observed in the dynamics plot for each of the complexes studied.
Table 2.
Theoretical Cross Sections (Å2) for Various (T4+Porphyrinoid)5- Conformationsa
| T4 / Porphyrinoidb Conformation | Cross Sections
|
|||
|---|---|---|---|---|
| (T4+1)5- | (T4+2)5- | (T4+3)5- | (T4+4)5- | |
|
| ||||
| Basket / ES | 858 | 868 | 869 | 866 |
| Basket / I | 921 | 923 | 922 | 920 |
| Basket / N | 929 | 930 | 926 | 925 |
|
| ||||
| Chair / ES | 860 | 872 | 870 | 869 |
| Chair / I | 918 | 920 | 925 | 922 |
| Chair / N | 932 | 929 | 927 | 924 |
|
| ||||
| Mixed / ES | 887 | 890 | 888 | 891 |
| Mixed / I | 933 | 931 | 936 | 932 |
| Mixed / N | 940 | 937 | 940 | 938 |
|
| ||||
| Propeller / ES | 908 | 912 | 911 | 914 |
| Propeller / I | 956 | 961 | 958 | 960 |
| Propeller / N | 964 | 967 | 970 | 968 |
|
| ||||
| Globular / N | 813 | 817 | 819 | 823 |
≤ 2% standard deviation,
ES = externally stacked, I = intercalated, N = nonspecific
When the porphyrinoids were intercalated or nonspecifically bound to the antiparallel basket or chair conformation, the calculated cross sections were much too large to match the experimental cross sections. The theoretical cross sections for T4 in either the parallel/antiparallel mix or propeller conformation also proved too large to match the experimental numbers, no matter how the porphyrinoids were bound. Likewise, T4 in a globular conformation with nonspecifically bound porphyrinoids gave values that were too small to match the experimental findings. From these results, it can be concluded that porphyrinoids 1–4 bind to T4 via external stacking to the antiparallel basket or chair T4 conformation. Alternative modes, such as intercalation or nonspecific binding are ruled out.
Representative structures of (T4+1)5-, (T4+2)5-, (T4+3)5- and (T4+4)5- with the porphyrinoids externally stacked below the quartets and T4 in an antiparallel basket conformation are shown in Figure 9. Complexes where T4 is in a chair conformation look very similar to those for the basket conformation. The quadruplex structure of T4 is retained in all of the complexes, with the exception of a slight disruption observed in the quartet directly above 2, 3 and 4. This disruption is probably due to the positive charge of the porphyrinoid, which appears to attract the oxygen atoms from the guanine residues above it. However, all of the other quartets are stable and the quadruplex for T4 is intact even without NH4 + cation stabilization.
Figure 9.

Theoretical structures of each externally stacked T4/porphyrinoid complex generated after 2 ns of 300 K molecular dynamics. The externally stacked complexes are the only ones that match the experimental cross sections. Gs are illustrated in dark blue, T and A are gray, and each porphyrinoid is green.
The structures of T4 in a mixed parallel/antiparallel arrangement, propeller conformation and those of the intercalated and nonspecifically bound complexes were analyzed to understand why the cross sections were so much larger than what was observed for the basket and chair externally stacked structures. It has been shown by ion mobility studies that the loop arrangements on the propeller and parallel/antiparallel mix structures for T4 cause it to have a larger cross section than the antiparallel basket and chair conformations.41 Hence, attachment of porphyrinoids to these T4 structures with parallel loops would result in larger cross sections for the complexes. Since these are not observed by experiment, it is inferred that T4 does not have a propeller or parallel/antiparallel mix conformation in the complexes studied. When the porphyrinoids were placed between two quartets (intercalated complexes), the thermally induced movement of the methyl or ethyl groups serves to break up most of the quartets, as shown in Figure 10. As a consequence, the guanine residues reposition themselves in a way that stabilizes a new, more globular structure. This structure also has a larger theoretical cross section than that observed by experiment. In contrast, porphyrinoids in nonspecifically bound complexes simply attach themselves to the sugar-phosphate backbone on the DNA strand presumably via electrostatic means. While no breakage of the quartets occurs (Figure 10), the resulting structures remain larger than the corresponding externally stacked structures and larger than the experimental cross sections. Such considerations lead us to propose that nonspecific binding is not an important binding mode and that the porphyrinoids are structure-selective for the G-quartets.
Figure 10.
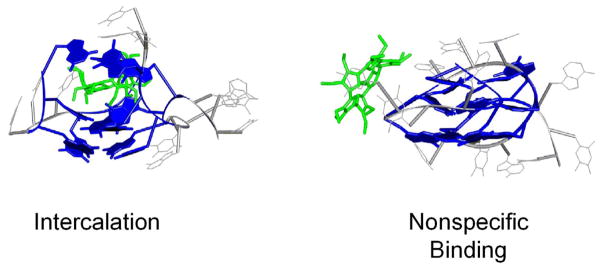
Theoretical structures for (T4+2)5- when 2 is intercalated and nonspecifically bound. When 2 is intercalated, its ethyl groups serve to break up the G-quartets leading to the formation of a complex that is larger than the experimental cross section. When 2 is bound nonspecifically it is also too large to match the experimental cross section. Similar structures are observed for all of the T4/porphyrinoid complexes. Guanine residues are illustrated in dark blue, T and A are gray, and the porphyrinoid 2 is green.
Supplementary Material
Summary.
The mass spectrometry, ion mobility, and molecular dynamics results presented in this paper provide insights into the interactions between cyclo[n]pyrroles and the human telomere G-quadruplex sequence T4. In particular, we conclude that:
Porphyrinoids 1–4 bind to T4, but 5 does not. From the intensities of the T4/porphyrinoid complexes in the mass spectra, the order of solution binding affinity is 2 > 1 > 3 > 4 ≫ 5.
An optimized combination of size and positive charge results in the highest level of binding affinity. Smaller porphyrinoid systems tend to bind more strongly as long as the positive charge on the macrocycle is retained.
Ion mobility studies illustrate that the binding of 1–4 to T4 takes place via external stacking with T4 in either an antiparallel basket conformation, an antiparallel chair conformation, or both. No evidence of intercalation or nonspecific binding was seen for any of the systems included in this study.
Given the above, we propose that modulations in macrocycle structure can influence how expanded porphyrins, such as 2–4, interact with G-quadruplexes and that, more specifically, soluble forms of these systems or their analogues may have a role to play as telomerase inhibitors.
Acknowledgments
The support of the NIH under grant CA 68682 to J.L.S and the National Science Foundation under grant CHE-0503728 to M.T.B. is gratefully acknowledged.
Footnotes
Supporting Information Available
The full citation for reference 48 and coordinates for the structures in Figure 9. This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Jonathan L. Sessler, Email: sessler@mail.utexas.edu.
Michael T. Bowers, Email: bowers@chem.ucsb.edu.
References
- 1.Thurston DE. Brit J Cancer. 1999;80(Supp I):65. [PubMed] [Google Scholar]
- 2.Jenkins TC. Curr Med Chem. 2000;7:99. doi: 10.2174/0929867003375551. [DOI] [PubMed] [Google Scholar]
- 3.Weitzman JB, Yaniv M. Nature. 1999;400:401. doi: 10.1038/22637. [DOI] [PubMed] [Google Scholar]
- 4.Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, Weinberg RA. Nature. 1999;400:464. doi: 10.1038/22780. [DOI] [PubMed] [Google Scholar]
- 5.Greider CW, Blackburn EH. Cell. 1985;43:405. doi: 10.1016/0092-8674(85)90170-9. [DOI] [PubMed] [Google Scholar]
- 6.Cech T. Cell. 2004;116:273. doi: 10.1016/s0092-8674(04)00038-8. [DOI] [PubMed] [Google Scholar]
- 7.McElligott R, Wellinger RJ. EMBO J. 1997;16:3705. doi: 10.1093/emboj/16.12.3705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huffman KE, Levene SD, Tesmer VM, Shay JW, Wright WE. J Biol Chem. 2000;275:19719. doi: 10.1074/jbc.M002843200. [DOI] [PubMed] [Google Scholar]
- 9.Wright WE, Tesmer VM, Huffman KE, Levene SD, Shay JW. Genes Dev. 1997;11:2801. doi: 10.1101/gad.11.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neidle S, Parkinson G. Nature Reviews Drug Discovery. 2002;1:383. doi: 10.1038/nrd793. [DOI] [PubMed] [Google Scholar]
- 11.Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PLC, Coviello GM, Wright WE, Weinrich SL, Shay JW. Science. 1994;266:2011. doi: 10.1126/science.7605428. [DOI] [PubMed] [Google Scholar]
- 12.Wang Y, Patel DJ. Structure. 1993;1:263. doi: 10.1016/0969-2126(93)90015-9. [DOI] [PubMed] [Google Scholar]
- 13.Parkinson GN, Lee MP, Neidle S. Nature. 2002;417:876. doi: 10.1038/nature755. [DOI] [PubMed] [Google Scholar]
- 14.Neidle S, Read MA. Biopolymers. 2001;56:195. doi: 10.1002/1097-0282(2000)56:3<195::AID-BIP10009>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 15.Blackburn EH. Cell. 1994;77:621. doi: 10.1016/0092-8674(94)90046-9. [DOI] [PubMed] [Google Scholar]
- 16.Erlitzki R, Fry M. J Biol Chem. 1997;272:15881. doi: 10.1074/jbc.272.25.15881. [DOI] [PubMed] [Google Scholar]
- 17.Fang G, Cech TR. Cell. 1993;74:875. doi: 10.1016/0092-8674(93)90467-5. [DOI] [PubMed] [Google Scholar]
- 18.Liu Z, Frantz JD, Gilbert W, Tye BK. Proc Natl Acad Sci USA. 1993;90:3157. doi: 10.1073/pnas.90.8.3157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baran N, Pucshansky L, Marco Y, Benjamin S, Manor H. Nucleic Acids Res. 1997;25:297. doi: 10.1093/nar/25.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun H, Karrow JK, Hickson ID, Maizels N. J Biol Chem. 1998;273:27587. doi: 10.1074/jbc.273.42.27587. [DOI] [PubMed] [Google Scholar]
- 21.Han H, Hurley LH. Trends Pharm Sci. 2000;21:136. doi: 10.1016/s0165-6147(00)01457-7. [DOI] [PubMed] [Google Scholar]
- 22.Arthanari H, Basu S, Kawano TL, Bolton PH. Nucleic Acids Res. 1998;26:3274. doi: 10.1093/nar/26.16.3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mergny J-L, Hélèlne C. Nature Med. 1998;4:1366. doi: 10.1038/3949. [DOI] [PubMed] [Google Scholar]
- 24.Arthanari H, Bolton PH. Anti-Cancer Drug Des. 1999;14:317. [PubMed] [Google Scholar]
- 25.(a) Gowan SM, Heald R, Stevens MFG, Kelland LR. Mol Pharmacol. 2001;60:981. doi: 10.1124/mol.60.5.981. [DOI] [PubMed] [Google Scholar]; (b) Read MA, Harrison JR, Romagnoli B, Tanious FA, Gowan SH, Reszka AP, Wilson WD, Kelland LR, Neidle S. Proc Natl Acad Sci USA. 2001;98:4844. doi: 10.1073/pnas.081560598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Dixon IM, Lopez F, Esteve JP, Tejera AM, Blasco MA, Pratviel G, Meunier B. Chem Bio Chem. 2005;6:123. doi: 10.1002/cbic.200400113. [DOI] [PubMed] [Google Scholar]; (b) Keating LR, Szalai V. Biochemistry. 2004;43:15891. doi: 10.1021/bi0483209. [DOI] [PubMed] [Google Scholar]; (c) Wheelhouse RT, Sun D, Han H, Han FX, Hurley LH. J Am Chem Soc. 1998;120:3261. [Google Scholar]
- 27.Koeppel F, Riou JF, Laoui A, Mailliet P, Arimondo PB, Labit D, Petitgenet O, Helene C, Mergny JL. Nucleic Acids Res. 2001;29:1087. doi: 10.1093/nar/29.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(a) Clark GR, Pytel PD, Squire CJ, Neidle S. J Am Chem Soc. 2003;125:4066. doi: 10.1021/ja0297988. [DOI] [PubMed] [Google Scholar]; (b) Sun D, Thompson B, Cathers BE, Salazar M, Kerwin SM, Trent JO, Jenkins TC, Neidle S, Hurley LH. J Med Chem. 1997;40:2113. doi: 10.1021/jm970199z. [DOI] [PubMed] [Google Scholar]
- 29.(a) Tuntiwechapikul W, Lee JT, Salazar M. J Am Chem Soc. 2001;123:5606. doi: 10.1021/ja0156439. [DOI] [PubMed] [Google Scholar]; (b) Fedoroff OY, Salazar M, Han H, Chemeris VV, Kerwin SM, Hurley LH. Biochemistry. 1998;37:12367. doi: 10.1021/bi981330n. [DOI] [PubMed] [Google Scholar]
- 30.(a) Kim M-Y, Vankayalapati H, Shin-ya K, Wierzba K, Hurley LH. J Am Chem Soc. 2002;124:2098. doi: 10.1021/ja017308q. [DOI] [PubMed] [Google Scholar]; (b) Shin-ya K, Wierzba K, Matsuo K-L, Ohtani T, Yamada Y, Furihata K, Hayakawa Y, Seto H. J Am Chem Soc. 2001;123:1262. doi: 10.1021/ja005780q. [DOI] [PubMed] [Google Scholar]
- 31.Seidel D, Lynch V, Sessler JL. Angew Chem Int Ed. 2002;41:1422. doi: 10.1002/1521-3773(20020415)41:8<1422::aid-anie1422>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 32.Köhler T, Seidel D, Lynch V, Arp FO, Ou Z, Kadish KM, Sessler JL. J Am Chem Soc. 2003;125:6872. doi: 10.1021/ja035089y. [DOI] [PubMed] [Google Scholar]
- 33.Fenn JB, Mann M, Meng CK, Wong SF, Whitehouse CM. Science. 1989;246:64. doi: 10.1126/science.2675315. [DOI] [PubMed] [Google Scholar]
- 34.Smith RD, Loo JA, Edmonds CG, Barinaga HR, Udseth HR. Anal Chem. 1990;62:882. doi: 10.1021/ac00208a002. [DOI] [PubMed] [Google Scholar]
- 35.Chait B, Kent SBH. Science. 1992;257:1885. doi: 10.1126/science.1411504. [DOI] [PubMed] [Google Scholar]
- 36.Bowers MT, Kemper PR, von Helden G, van Koppen PAM. Science. 1993;260:1446. doi: 10.1126/science.260.5113.1446. [DOI] [PubMed] [Google Scholar]
- 37.Clemmer DE, Jarrold MF. J Mass Spectrom. 1997;32:577. [Google Scholar]
- 38.Gidden J, Ferzoco A, Baker ES, Bowers MT. J Am Chem Soc. 2004;126:15132. doi: 10.1021/ja046433+. [DOI] [PubMed] [Google Scholar]
- 39.Gidden J, Baker ES, Ferzoco A, Bowers MT. Int J Mass Spectrom. 2005;240:183. [Google Scholar]
- 40.Baker ES, Bernstein SL, Bowers MT. J Am Soc Mass Spectrom. 2005;16:989. doi: 10.1016/j.jasms.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Baker ES, Bernstein SL, Gabelica V, De Pauw E, Bowers MT. submitted to J. Am. Chem. Soc. [Google Scholar]
- 42.Sessler JL, Mozaffari A, Johnson MR. Org Synth. 1992;70:68. [Google Scholar]
- 43.Wyttenbach T, Kemper PR, Bowers MT. Int J Mass Spectrom. 2001;212:13. [Google Scholar]
- 44.Mason EA, McDaniel EW. Transport Properties of Ions in Gases. Wiley; New York: 1988. [Google Scholar]
- 45.HyperChem 7.0. Hypercube Inc; 2002. [Google Scholar]
- 46.Wang Y, Patel DJ. Structure. 1994;2:1141. doi: 10.1016/s0969-2126(94)00117-0. [DOI] [PubMed] [Google Scholar]
- 47.Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. Nucleic Acids Res. 2000;28:235. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Case DA, et al. AMBER. Vol. 7. University of California; San Francisco: 2002. [Google Scholar]
- 49.Mesleh MF, Hunter JM, Shvartsburg AA, Schwartz GC, Jarrold MF. J Phys Chem. 1996;100:16082. [Google Scholar]
- 50.Shvartsburg AA, Jarrold MF. Chem Phys Lett. 1996;261:86. [Google Scholar]
- 51.Nagesh N, Chatterji D. J Biochem Biophys Methods. 1995;30:1. doi: 10.1016/0165-022x(94)00057-k. [DOI] [PubMed] [Google Scholar]
- 52.Guschlbauer W, Chantot JF, Thiele D. J Biomol Struct Dyn. 1990;8:491. doi: 10.1080/07391102.1990.10507825. [DOI] [PubMed] [Google Scholar]
- 53.Rovnyak D, Baldus M, Wu G, Hud NV, Feigon J, Griffin RG. J Am Chem Soc. 2000;122:11423. [Google Scholar]
- 54.Rosu F, Gabelica B, Houssier C, Colson P, De Pauw E. Rapid Commun Mass Spectrom. 2002;16:1729. doi: 10.1002/rcm.778. [DOI] [PubMed] [Google Scholar]
- 55.Kerwin SM. Current Pharmaceutical Design. 2000;6:441. doi: 10.2174/1381612003400849. [DOI] [PubMed] [Google Scholar]
- 56.Read MA, Neidle S. Biochemistry. 2000;39:13422. doi: 10.1021/bi001584k. [DOI] [PubMed] [Google Scholar]
- 57.Seenisamy J, Rezler EM, Powell TJ, Tye D, Gokhale V, Joshi CS, Siddiqui-Jain A, Hurley LH. J Am Chem Soc. 2004;126:8702. doi: 10.1021/ja040022b. [DOI] [PubMed] [Google Scholar]
- 58.Seenisamy J, Bashyam S, Gokhale V, Vankayalapati H, Sun D, Siddiqui-Jain A, Streiner N, Sin-ya K, White E, Wilson WD, Hurley LH. J Am Chem Soc. 2005;127:2944. doi: 10.1021/ja0444482. [DOI] [PubMed] [Google Scholar]
- 59.Rezler EM, Seenisamy J, Bashyam S, Kim MY, White E, Wilson WD, Hurley LH. J Am Chem Soc. 2005;127:9439. doi: 10.1021/ja0505088. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.