

Abstract
Background
The microbiota of the mammalian gastrointestinal (GI) tract consists of diverse populations of commensal bacteria that interact with host physiological function. Dysregulating these populations, through exogenous means such as antibiotics or dietary changes, can have adverse consequences on the health of the host. Studies from laboratories such as ours have demonstrated that exposure to psychological stressors disrupts the population profile of intestinal microbiota. To date, such studies have primarily focused on prolonged stressors (repeated across several days) and have assessed fecal bacterial populations. It is not known whether shorter stressors can also impact the microbiota, and whether colonic mucosa-associated populations can also be affected. The mucosa-associated microbiota exist in close proximity to elements of the host immune system and the two are tightly interrelated. Therefore, alterations in these populations should be emphasized. Additionally, stressors can induce differential responses in anxiety-like behavior and corticosterone outputs in variant strains of mice. Thus, whether stressor exposure can have contrasting effects on the colonic microbiota in inbred C57BL/6 mice and outbred CD-1 mice was also examined.
Results
In the present study, we used high throughput pyrosequencing to assess the effects of a single 2-hour exposure to a social stressor, called social disruption (SDR), on colonic mucosa-associated microbial profiles of C57BL/6 mice. The data indicate that exposure to the stressor significantly changed the community profile and significantly reduced the relative proportions of two genera and one family of highly abundant intestinal bacteria, including the genus Lactobacillus. This finding was confirmed using a quantitative real-time polymerase chain reaction (qPCR) technique. The use of qPCR also identified mouse strain-specific differences in bacterial abundances. L. reuteri, an immunomodulatory species, was decreased in stressor-exposed CD-1 mice, but not C57BL/6 mice.
Conclusions
These data illustrate that stressor exposure can affect microbial populations, including the lactobacilli, that are closely associated with the colonic mucosa. Because the lactobacilli can have beneficial effects on human health, stressor-induced reductions of their population could have important health implications.
Keywords: Psychological stress, Gastrointestinal microbiota, Lactobacillus, Lactobacillus reuteri, Dysbiosis
Background
The human body is colonized by an enormous array of microbes collectively referred to as the microbiota. It is estimated that there are approximately ten times more bacterial cells than there are human cells in the human body with microbiota being found in various receptive niches such as on the skin, in the oral and respiratory tracts, reproductive tract, and most numerously, in the gastrointestinal (GI) tract [1,2]. In the GI tract, the microbiota form a community that fully interacts with one another and with the host. The GI microbiota have multiple beneficial effects on their hosts [3-5] and the structure of the GI community can impact these functions. Of the beneficial interactions between microbiota and host, those that involve the immune system are some of the most intensively studied, and several lines of experiments have demonstrated that the GI microbiota and the host immune system are deeply intertwined in each other’s development [6]. For example, germ-free mice, which have never been colonized by any type of microbe, have diminished immune responses compared to mice colonized with a healthy microbiota [7,8]. Mice deficient in Nod2, a gene that encodes for a receptor that is involved in immune recognition of bacterial muramyl dipeptide, develop a unique microbiota that can lead to colonic inflammation when transmitted to wild-type mice [9,10]. It is now well recognized that the GI microbiota have strong effects on immunoregulation, and immune system activity, in turn, helps to shape the GI microbiota [6-9].
It is estimated that over 500 different species of bacteria can colonize the GI tract [11]. Despite this enormous species-level diversity, only a small number of bacterial phyla are represented. The vast majority of bacteria in the human GI tracts belong to the phyla Firmicutes and Bacteroidetes, with bacteria in the phyla Actinobacteria and Proteobacteria also comprising a smaller portion of the overall microbiota as well [12]. The GI microbiota of the murine GI tract is similar to that of humans, with the majority of microbes belonging to the major phyla, Firmicutes and Bacteroidetes[13-15]. These populations shift slowly over time, but their general stability is important to the health of the host. Abrupt changes to the gut microbiota have been shown to potentially lead to serious negative host health outcomes, including diarrhea, opportunistic infections, and obesity [16-18]. Changes to the microbiota can be caused by factors such as antibiotic use and severe enteric infection. Data from this laboratory, as well as others, indicate that exposure to either physical or psychological stressors can also alter intestinal microbe profiles. Stressor exposure early in life has been demonstrated to alter the types and abundance of bacteria found in the intestines. Separating infant monkeys from their mothers to induce a physiological stress response resulted in a significant reduction in the number of total lactobacilli that could be cultured from the stool [19]. Reductions in lactobacilli are meaningful as certain species, including L. reuteri, are involved in immunomodulation [20,21]. In rats, separating the pups from their mothers during the first 14 days of life led to an altered GI microbiome [22]. Stressor exposure during adulthood can also impact the stability of the intestinal microbiota. For example, exposing adult mice to a prolonged restraint stressor was shown to significantly alter microbial profiles in the cecal contents [23].
Similar findings have been associated with a social stressor called social disruption (SDR) [15] that involves repeated social defeat as a result of inter-male aggression over a 2 hr period. When repeated over 6 consecutive nights, this stressor induces a physiological stress response marked by the activation of the hypothalamic pituitary adrenal (HPA) axis and the sympathetic nervous system [24]. The study by Bailey et al. demonstrated that this week-long exposure to the stressor can alter the cecal luminal microbiome in outbred CD-1 mice [15]. However, it is not known whether a single 2 hr exposure to the stressor could induce similar alterations, or whether other mouse strains, such as inbred C57BL/6 mice that are widely used in infectious disease research, are affected by stressor exposure. Knowing whether short-lasting stressors can impact the microbiota has translational importance particularly for the inflammatory bowel diseases (IBD) and irritable bowel syndrome (IBS). Patients with IBD often report stressful periods preceding symptom flares [25]. It is not clear whether acute stress can exacerbate IBD symptomatology, but it is recognized that acute stressor exposure is associated with increased visceral sensitivity in IBS [26]. Because alterations in gut microbiota community structure are evident in IBD and IBS [27,28], and because these alterations are thought to possibly contribute to these diseases [29,30], we determined whether a short-lasting stressor was sufficient to impact gut microbiota community structure.
Previous studies assessing the effects of stress on the microbiota have relied on assessment of intestinal lumen or fecal populations [15,23,31]. Studies in healthy individuals as well as patients with IBS, IBD, or hepatic encephalopathy demonstrate that luminal/fecal microbiota are significantly different than mucosa-associated microbiota [32,33]. Because shifts in microbial populations that are in close proximity to the intestinal epithelium are thought to have the strongest effects on host immunity [34], an additional aspect of this study was to determine whether stressor exposure could impact tissue-associated microbial community profiles.
Stressor-induced changes in lactobacilli have reliably been found in laboratory animals as well as in humans [19,35,36]. However, stressor-induced changes in other taxa have not been widely reported. Thus, 454 pyrosequencing was performed on the colonic tissue of inbred C57BL/6 mice to determine whether a single 2 hr stressor exposure could change colonic tissue-associated microbial community profiles. Quantitative PCR (qPCR) was then used to determine whether stressor-induced alterations in relative abundances evident with the 454 pyrosequencing were also evident as alterations in absolute abundance in both inbred and outbred mice.
Results
Social stress affects the community structure of the colonic mucosa-associated microbiota
We analyzed the effect of a short term SDR stressor on the colonic mucosa-associated microbiota using 16S rRNA gene pyrosequencing of samples from both SDR C57BL/6 stressor-exposed mice and non-stressed HCC control mice. After normalization of the datasets by rarefaction, we observed no statistical difference in the total number of operational taxonomic units (OTU) between the SDR and control mice (data not shown). Additional analysis of the Shannon, equitability and Chao1 alpha diversity metrics showed no statistical difference between the two groups (Figure 1A-C). Thus, a single two-hour cycle of exposure to the social stressor did not affect the alpha diversity of the mucosa-associated microbiota.While we did not observe any statistically significant differences in the alpha diversity of the microbiota between the SDR and control group, beta diversity analyses did reveal differences in composition. A principal coordinates analysis (PCoA) of the UniFrac distances between samples showed that they clustered into separate groups according to treatment (Figure 2A). An unweighted pair group with arithmetic mean (UPGMA) hierarchical clustering dendrogram confirmed this clustering (Figure 2B). Statistical analysis of the UniFrac distances using the analysis of similarity statistic (ANOSIM) indicated that colonic mucosal microbiota from SDR stressor-exposed mice were significantly different from those of the home cage control (HCC) mice (p < 0.05).
Figure 1.
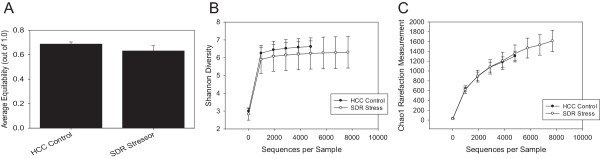
Exposure to the SDR stressor did not impact alpha-diversity. Mice were exposed to the SDR stressor, or were left undisturbed as non-stressed Home Cage controls (HCC control). After 454 pyrosequencing, three measures of community alpha-diversity were calculated using QIIME. A. Equitability Index, B. Shannon Diversity Index, and C. Chao1 Rarefaction Measurement were unaffected by exposure to the SDR stressor. Data are from n = 5 mice per group.
Figure 2.
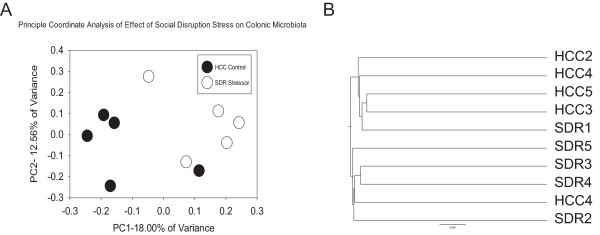
Exposure to the SDR stressor significantly changes beta-diversity. After 454 pyrosequencing, UniFrac distances were calculated using QIIME and plotted on a 2-D principal coordinates graph (A). Exposure to the SDR stressor shifted the colonic microbiota, with 4 of 5 SDR stressor samples clustering apart from HCC controls. UniFrac distances were also used for construction of an unweighted pair group method by arithmetic mean (UPGMA) dendrogram, in which 4 of 5 SDR stressor samples clustered together (B). The clustering in Figure 2A and 2B was statistically significant (p < 0.05) after performing the ANOSIM test to measure differences between distance matrices of SDR stressor and HCC control groups. Data are from n = 5 mice per group.
Populations of the genus Lactobacillus and L. reuteri are reduced in mice exposed to a social stressor
The taxonomic profile of the microbiota at the phylum level showed no significant differences between the two treatment groups (Figure 3). Analysis of relative abundances at lower taxonomic levels showed a reduction in the family Porphyromonadaceae in SDR stressor-exposed mice compared to non-stressed HCC mice (p < 0.01) (Table 1). Exposure to the SDR stressor also reduced the relative abundance of bacteria in the family Lactobacillaceae (p < .05) (Table 1). This reduction in the Lactobacillaceae was reflected by a reduction in the relative abundance of bacteria in the genus Lactobacillus (p < .05) (Table 2). In addition to the reduction in the abundance of lactobacilli, it was evident that exposure to the SDR stressor significantly reduced the relative abundance of the genus Parabacteroides (p < .01), as well as an unclassified group within the phylum Firmicutes (p < .05) and an unclassified group within the class Bacilli (p < .05) (Table 2).
Figure 3.
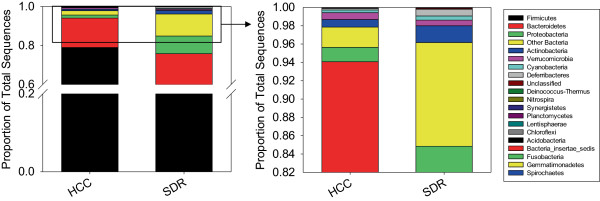
The relative abundances of mucosal-associated bacterial phyla were unaffected by exposure to the SDR stressor. Data are the mean+/-S.E. of the relative abundances calculated from 454 pyrosequencing data and are from n = 5 mice per group.
Table 1.
Top 10 most abundant colonic tissue-associated bacterial families
| HCC | SDR | |
|---|---|---|
|
Lachnospiraceae |
38.71 ± 5.22 |
54.10 ± 5.81 |
|
Clostridiales; Other |
9.01 ± 0.56 |
10.14 ± 0.96 |
|
Bacteroidaceae |
10.77 ± 1.49 |
6.67 ± 2.11 |
|
Lactobacillaceae |
13.66 ± 4.81 |
3.13 ± 100* |
|
Ruminococcaceae |
5.22 ± 0.36 |
4.35 ± 0.62 |
|
Acetobacteraceae |
0.001 ± 0.004 |
8.86 ± 8.83 |
|
Clostridiaceae |
6.34 ± 3.62 |
0.92 ± 0.78 |
|
Porphyromonadaceae |
2.67 ± 0.20 |
1.59 ± 0.25** |
|
Bacteria; Other |
2.21 ± 0.25 |
1.84 ± 0.46 |
| Peptostreptococcaceae | 1.81 ± 0.35 | 0.94 ± 0.40 |
Data are the mean ± standard error. *p < .05 vs. HCC; **p < .01 vs. HCC.
Table 2.
Top 40 most abundant tissue-associated bacterial genera
| HCC | SDR | |
|---|---|---|
|
Lachnospiraceae; Other |
37.61 ± 5.23 |
53.02 ± 5.74 |
| Clostridiales; Other; Other |
9.01 ± 0.56 |
10.14 ± 0.96 |
|
Bacteroides spp. |
10.77 ± 0.56 |
6.67 ± 2.11 |
|
Lactobacillus spp |
13.53 ± 4.76 |
3.11 ± 1.00* |
|
Rosemonas spp |
0.00 ± 0.00 |
8.83 ± 8.83 |
|
Ruminococcaceae; Other |
3.81 ± 0.44 |
2.95 ± 0.45 |
|
Clostridium spp. |
5.86 ± 3.37 |
0.82 ± 0.70 |
| Unclassified Bacteria |
2.21 ± 0.25 |
1.84 ± 0.46 |
|
Parabacteroides spp. |
2.25 ± 0.12 |
1.35 ± 0.20** |
|
Peptostreptococcaceae; Other |
1.80 ± 0.35 |
0.93 ± 0.40 |
| Unclassified Firmicutes |
1.25 ± 0.20 |
0.69 ± 0.13* |
|
Marvinbryantia spp. |
0.79 ± 0.11 |
0.81 ± 0.24 |
|
Turicibacter spp. |
0.94 ± 0.30 |
0.52 ± 0.20 |
|
Oscillibacter spp. |
0.62 ± 0.29 |
0.65 ± 0.28 |
|
Asaccharobacter spp. |
0.81 ± 0.25 |
0.39 ± 0.12 |
|
Akkermansia spp. |
0.73 ± 0.32 |
0.39 ± 0.22 |
| Unclassified Bacteroidetes |
0.60 ± 0.05 |
0.33 ± 0.16 |
|
Butyricicoccus spp. |
0.40 ± 0.03 |
0.52 ± 0.19 |
| Unclassified Alphaproteobacteria |
0.62 ± 0.40 |
0.14 ± 0.11 |
|
Erysipelotrichaceae; Other |
0.47 ± 0.14 |
0.24 ± 0.07 |
|
Butyricimonas spp. |
0.43 ± 0.09 |
0.22 ± 0.07 |
|
Hyphomonadaceae; Other |
0.01 ± 0.00 |
0.58 ± 0.55 |
|
Clostridiaceae; Other |
0.48 ± 0.26 |
0.10 ± 0.08 |
|
Alistepes spp. |
0.38 ± 0.09 |
0.18 ± 0.05 |
| Unclassified Cyanobacteria |
0.01 ± 0.00 |
0.45 ± 0.42 |
|
Chitinophagaceae; Other |
0.39 ± 0.35 |
0.00 ± 0.00 |
|
Bacillariophyta; Other |
0.06 ± 0.05 |
0.28 ± 0.17 |
|
Anaerotruncus spp. |
0.15 ± 0.04 |
0.17 ± 0.02 |
|
Coprobacillus spp. |
0.15 ± 0.05 |
0.14 ± 0.06 |
|
Mucispirillium spp. |
0.17 ± 0.10 |
0.11 ± 0.05 |
|
Ruminococcus spp. |
0.22 ± 0.08 |
0.05 ± 0.03 |
|
Lactobacillales; Other; Other |
0.21 ± 0.07 |
0.06 ± 0.02 |
|
Blautia spp. |
0.19 ± 0.05 |
0.07 ± 0.02 |
| Unclassified Bacilli |
0.18 ± 0.04 |
0.05 ± 0.02* |
|
Anaerostipes spp. |
0.16 ± 0.05 |
0.07 ± 0.01 |
|
Ponticaulis spp. |
0.00 ± 0.00 |
0.22 ± 0.21** |
|
Anaerovorax spp. |
0.14 ± 0.05 |
0.08 ± 0.02 |
|
Holdemania spp. |
0.16 ± 0.06 |
0.04 ± 0.01 |
|
Bacillus spp. |
0.19 ± 0.19 |
0.01 ± 0.00 |
| Roseburia spp. | 0.09 ± 0.03 | 0.10 ± 0.04 |
Data are the mean relative abundance ± standard error. *p < .05 vs. HCC; **p < .01 vs. HCC.
Many bacteria in the genus Lactobacillus have been shown to have immunomodulatory functions in the colon, and can be used as a probiotic to treat inflammation. Thus, we used qPCR to quantify the effect of the SDR stressor on the abundance of this group of bacteria [20,21]. When the a priori hypothesis that repeated administration of the SDR stressor would reduce the abundance of lactobacilli was tested, it was determined that the absolute abundance of Lactobacillus spp. was significantly lower (p < .05) after 6 cycles of the stressor in comparison to baseline levels in C57BL/6 mice (Figure 4A). This reduction in lactobacilli abundance was also observed in outbred CD-1 mice (p < .05), with the largest reduction in mean levels of lactobacilli also occurring after 6 days of the SDR stressor (Figure 4B). In addition, qPCR analyses also revealed that exposure to the SDR stressor significantly reduced the abundance of the immunomodulatory species L. reuteri over the course of six cycles of SDR (p < .05). This difference was only evident in outbred CD-1 mice (Figure 4C). L. reuteri levels were below the qPCR limit of detection of 4.5 copies/gram of wet tissue (log10) in inbred C57BL/6 mice.
Figure 4.
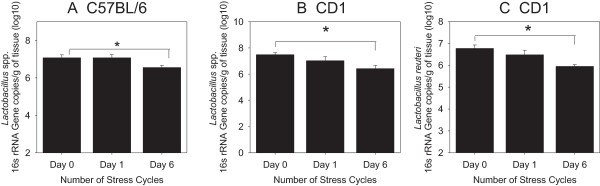
The absolute abundance of bacteria in the genus Lactobacillus is reduced by exposure to the SDR stressor. Mice were exposed to either 0, 1, or 6 consecutive days of the SDR stressor and qPCR used to quantify Lactobacillus group bacteria (A and B) or L. reuteri (C) in the colon. *indicates p < .05 vs. 0 cycles of SDR (0 cycles is equivalent to HCC controls). Inbred C57Bl/6- n = 16 at Day 0, n = 7 on Day 1, and n = 8 on Day 6. Outbred CD-1- n = 21 at Day 0, n = 15 at Day 1, and n = 6 on Day 6.
Relative bacterial levels differ by mouse strain
In order to confirm that stressor exposure was reducing the absolute abundance of other bacterial groups that were reduced in relative abundance in the pyrosequencing analysis, qPCR was performed on colonic tissues from both inbred C57BL/6 mice and outbred CD1 mice. Primers targeting the 16 s ribosomal RNA genes of Parabacteroides distasonis, a member of the Parabacteroides genus, and Bacteroides-Prevotella-Porphyromonas were used. Stressor exposure did not significantly affect the absolute abundance of either P. distasonis or Bacteroides-Prevotella-Porphyromonas genera (Table 3). In addition, levels of Bacteroides-Prevotella-Porphyromonas group and P. distasonis were similar in the two strains of mice.
Table 3.
Real-time PCR assessment of bacterial group abundances
| C57BL6 | CD-1 | ||
|---|---|---|---|
|
Parabacteroides distasonis |
Day 0 |
6.27 (6.02 – 7.42) |
6.65 (6.53 – 6.78) |
| Day 1 |
6.94 (6.66 – 7.23) |
7.54 (7.30 – 7.77) |
|
| Day 6 |
6.52 (6.10 – 6.94) |
7.00 (6.26 – 7.74) |
|
| Group Porphyromonas-Bacteroides - Prevotella |
Day 0 |
7.61 (7.39 – 7.85) |
7.20 (7.02 – 7.39) |
| Day 1 |
8.10 (7.82 – 8.37) |
7.37 (6.93 – 7.82) |
|
| Day 6 | 7.46(7.17 – 7.75) | 6.97 (6.44 – 7.50) |
Data are the mean (standard error) of the copy number (log10) of target 16 s rRNA per gram of tissue.
Colonic inflammatory cytokine mRNA levels are not affected by social stressor exposure
The colonic mRNA levels for IL-1β, TNF-α, and iNOS were unaffected by exposure to the SDR stressor, suggesting that exposing C57BL/6 and CD-1 mice to the SDR stressor did not result in detectable increases in colonic cytokines or inflammatory mediators (Table 4).
Table 4.
Real-time PCR assessment of colonic inflammation
| |
|
C57BL6 |
CD-1 |
||
|---|---|---|---|---|---|
| DAY 1 | DAY 6 | DAY 1 | DAY 6 | ||
| IL1β |
HCC |
1.00 (0.57 – 1.75) |
1.00 (0.55 – 1.82) |
1.00 (0.37 – 2.68) |
1.00 (0.64 – 1.57) |
| SDR |
0.95 (0.49 – 1.84) |
1.37 (1.00 – 1.37) |
3.18 (2.92 – 3.47) |
1.11 (0.59 – 2.11) |
|
| iNOS |
HCC |
1.00 (0.56 – 1.78) |
1.00 (0.50 – 2.01) |
1.00 (0.56 – 1.78) |
1.00 (0.69 – 1.46) |
| SDR |
0.62 (0.46 – 0.83) |
2.50 (1.92 – 3.25) |
0.32 (0.24 – 0.43) |
0.23 (0.14 – 0.37) |
|
| TNFα |
HCC |
1.00 (0.74 – 1.35) |
1.00 (0.51 – 1.98) |
1.00 (0.76 – 1.32) |
1.00 (0.61 – 1.65) |
| SDR | 0.50 (0.35 – 0.73) | 1.03 (0.71 – 1.49) | 0.92 (0.69 – 1.22) | 0.92 (0.63 – 1.33) | |
Data are the mean (standard error) of the fold change in cytokine gene expression over non-stressed HCC controls.
Discussion
Exposure to physical and psychological stressors has been shown to impact the gut microbiota of both laboratory animals and humans [19,22,35]. However, the majority of the previous studies have utilized repeated and prolonged stressors, and have assessed microbial populations in the lumen of the intestines or present in the fecal matter. Whether stressor exposure has different effects on the microbiota of different strains of mice is also poorly understood. Because gut microbes that adhere to the colonic mucosa can have different effects on the host [34], this study assessed whether stressor exposure could alter the community structure of mucosa-associated microbes. This study demonstrated that as little as 2 hrs of stressor exposure is enough to significantly change the structure of the microbiota associated with the colonic mucosa. This effect was not manifest as alterations in alpha diversity, but rather as alterations in beta diversity. The ANOSIM distance matrix analysis and cluster analysis based on unweighted UniFrac demonstrated that microbiota within the samples from stressor-exposed mice were significantly different than those from the non-stressed HCC control mice. In addition to significantly changing beta diversity, exposure to the stressor significantly changed the relative abundance of 2 bacterial genera, namely Parabacteroides and Lactobacillus, and one bacterial family, Porphyromonadaceae.
This study targeted mucosa-associated populations, but true stratification of luminal and mucosal populations does not exist. There is much crossover between microbes often associated with the lumen and those that can adhere to the mucus layer of the GI tract, as the former becomes trapped in the mucus layer and the latter sheds into the lumen. Steps were taken to remove the majority of the fecal matter from tissue samples, wherein lie the majority of the luminal portion of the GI microbiota. This study extends previous studies that indicated that luminal and fecal microbiota can change as a consequence of stressor exposure to now include mucosa-associated populations.
To determine whether the stressor effects only encompassed changes to bacterial relative abundance or whether changes in absolute abundance (as assessed by determining copies of 16 s rRNA gene per gram of sample) may also result from stressor exposure, qPCR was used to further investigate the effects of the stressor on bacterial abundance. This was performed in both an inbred (i.e., C57BL/6) and an outbred (i.e., CD-1) mouse strain, because studies consistently show that microbiota composition is associated with mouse strain [37,38]. In addition, different mouse strains can also have different physiological and behavioral responses to stressor exposure, including changes to anxiety-like behavior, as well as diarrhea output and colonic serotonin concentration [39,40]. Thus, it was important to determine whether stressor effects were conserved across mouse strains. While 2 hrs of stressor exposure was enough to reduce the relative abundance of the genus Lactobacillus, a reduction in the absolute abundance was only evident after repeated exposure to the stressor. This indicates that some of the effects that stressor exposure has upon the microbiota are additive. Both pre- and post-stressor lactobacilli levels were similar in outbred CD-1 mice and inbred C57BL/6 mouse strains, demonstrating that the effects of the stressor are consistent across mouse strains. This finding was not surprising given that stressor-induced reduction in lactobacilli levels have been documented in other host species, including human and non-human primates.
The finding that stressor exposure reduced the relative and absolute abundance of lactobacilli is consistent with previous studies demonstrating that stressor exposure can reduce the lactobacilli [19,31,35,36]. To date, however, the Lactobacillus species that can be reduced by stressor exposure has not been addressed. Many species of bacteria in the genus Lactobacillus are known to have health promoting effects, and studies from this laboratory, as well as others, indicate that administering probiotic L. reuteri to mice can reduce colonic inflammatory responses [20,21,41]. Because stressor exposure can exacerbate colonic inflammation [42], we focused on whether indigenous L. reuteri was reduced in stressor-exposed animals. As predicted, L. reuteri was significantly reduced in colonic mucosa from outbred CD-1 mice exposed to the SDR stressor. Interestingly, however, L. reuteri was not consistently detectable in inbred C57BL/6 mice. Thus, it is apparent that L. reuteri levels on the mucosa of C57BL/6 mice are lower than levels observed in outbred CD-1 mice and below the detection limit (4.5 log copies/gram of wet tissue).
Although it is difficult to compare deep sequencing results across different experiments that assess different body niches and encompass different stressors, the use of 454 pyrosequencing demonstrated that the effects of stressor exposure are not confined to effects on the lactobacilli. The relative abundance of bacteria in the family Porphyromonadaceae was found to be significantly reduced by exposure to the SDR stressor. This finding is consistent with a previous study demonstrating that exposure to a prolonged restraint stressor in oubred CD-1 mice was sufficient to reduce the relative abundance of cecal Porphyromonadaceae[23]. However, it was not previously known whether stressor exposure could reduce the absolute abundance of the Porphyromonadaceae family. Thus, qPCR was used to determine whether this stressor also affected the absolute abundance of Porphyromonadaceae by assessing levels of bacteria in the Porphyromonas-Bacteroides-Prevotella group as a surrogate marker. This was performed because of the problems designing qPCR primers specific for the Porphyromonadaceae family. The absolute abundance of the Porphyromonas-Bacteorides-Prevotella group was similar in CD-1 mice and inbred C57BL/6 mice, and stressor exposure had no effect on the absolute abundance in either mouse strain. Thus, although this study and others [23] have found stressor-induced reductions in the relative abundance of Porphyromonadaceae, these results indicate that absolute abundance of the Porphyromonadaceae is unaffected by stressor exposure. Stressor exposure also did not affect the absolute abundance of P. diastonosis in either strain of mice, even though the relative abundance of Parabacteroides was reduced as detected by 454 pyrosequencing.
Colonic inflammation is well known to impact the gut microbiota, and studies have found that intestinal inflammation can also reduce the abundance of the family Lactobacillaceae[43,44] and Porphyromonadaceae[45]. Given the bidirectional interaction between the microbiota and intestinal inflammation and increased intestinal physiological inflammation upon exposure to stressor [46], it is often suggested that increases in intestinal inflammation during stressful periods could be responsible for changes in gut microbiota. However, inflammatory cytokine gene expression was not significantly changed by either 2 hrs of the SDR stressor or 2 hrs of the stressor repeated on 6 consecutive days. Thus, the results of this study indicate that it is not likely that stressor-induced colonic inflammation is responsible for altering the abundances of the indigenous colonic mucosa-associated microbiota.
The effects of SDR on male mouse immunological, physiological, and behavioral functioning have been well characterized over the past fifteen years [47-49]. The consistent activation of the HPA axis and the SNS makes SDR an ideal method of inducing an acute stress response in male mice. While it would be desirable to determine the effects on female mice as well, aggression in female mice is too low to induce a physiological stress response in this paradigm. Other stressor paradigms, however, have shown that stressor exposure in a lab setting can also affect the microbiota composition of female mice [50,51]. Thus, it is likely that a social stressor in female mice would also impact the lactobacilli, as well as Porphyromonadaceae and Parabacteroides. Interestingly, studies have shown that male and female microbiota can evoke different levels of host sex hormone release and immunological outputs that can then feedback and alter microbial composition [52]. Future studies comparing the microbiota, mucosal immunity and endocrine responses in both male and female mice are likely to demonstrate additional multidirectional interactions between host physiology and the microbiota. This requires further study.
Conclusions
Gut microbes have important effects on the host especially when they are associated with the mucosa. Previous studies have found that stressor exposure can affect the microbiota, in particular the lactobacilli. However, many of these studies focused on microbes that are shed in the stool during prolonged or chronic stressors [19,23,31,50]. It was not previously clear whether the effects of stress on the microbiota were limited to changes in the number of bacteria shed in the stool, nor whether short lasting acute stressors could also impact the microbiota. The present study demonstrates that exposure to as little as 2 hrs of a social stressor is sufficient to significantly affect some populations of the colonic mucosa-associated microbiota. This builds upon previous work by showing that exposure to the social stressor affects multiple regions (e.g. colon, cecum) and niches (e.g. lumen, mucosa-associated) of the GI tract, as well as two different strains of mice, suggesting that the effect of social stresss exposure upon the microbiota is a universal process, as opposed to either an artifact or isolated finding. The mechanisms by which this occurs are not yet understood, but are not likely to involve stressor-induced inflammation in the colon. Future studies should assess the impact of stressor-associated hormones and gut functioning since others have demonstrated that activation of the stress-associated hormone corticotrophin releasing hormone (CRH) alters gut microbiota, an effect that was associated with alterations in intestinal motility [53]. Because the gut microbiota has been associated with diverse diseases that are known to be exacerbated by stressful situations, such as the inflammatory bowel diseases, irritable bowel syndrome, and even multiple sclerosis, it is becoming increasingly important to understand the impact that stress can have on the microbiota and whether stressor-induced alterations in the microbiota are involved in stressor-induced disease exacerbation [54-57].
Methods
Mice
Male CD-1 mice and male C57BL/6, aged 6–8 weeks, were ordered from Charles River Laboratories (Wilmington, MA; Raleigh, NC). The mice were housed in groups of 2 or 3 mice per cage in an approved vivarium and were allowed to habituate to the vivarium for one week prior to testing. The cages were kept in an approved vivarium with a 12:12 hour light–dark schedule with lights on 0600 to 1800. Food and water was available ad libitum. The Ohio State University’s Animal Care and Use Committee approved all experimental procedures (Protocol 2009A0235-R1). CD-1 mice were used in cytokine and bacterial qPCR experiments. C57BL/6 mice were only used in the 454 pyrosequencing analysis and the cytokine and bacterial qPCR experiments.
Social disruption stressor
The SDR experiments were performed as described previously [58]. Briefly, an aggressive male CD-1 retired breeder, termed the aggressor, was placed into a cage with younger C57BL/6 resident mice at 1700 hours, which represents the beginning of the mouse active cycle. The aggressor repeatedly attacked and defeated the C57BL/6 test mice over the course of the 2 hour SDR cycle. If the aggressor did not begin to attack the test mice within 10 min of placement in the cage, it was removed and another aggressor was put in. Wounding was monitored throughout the SDR cycle. Only slight superficial wounds were allowed. If wounds that penetrated the cutaneous layer developed over the course of SDR, those test mice were removed from the study. While it would be desirable to test the effects of the stressor on both male and female mice, female mice do not fight in this paradigm. Thus, all analyses were limited to male mice.
Tissue removal
After exposure to the SDR stressor (a single 2 hr cycle for pyrosequencing, a single 2 hr cycle or 2 hr cycles repeated on 6 consecutive days for qPCR), or at the equivalent time in non-stressed HCC mice, mice were euthanized using CO2 asphyxiation. Colons were removed aseptically. Luminal contents were removed by cutting into the tissue where liquid and/or solid contents rested, and gently extracting with forceps. This represented the non-mucosa associated populations. Scraping/tissue squeezing was not performed so as not to disturb the mucosa-associated populations, which are adhered to an easily disturbed mucous layer. The colonic tissue and remaining adherent microbiota populations were placed into a microcentrifuge tube and snap frozen in liquid nitrogen. All samples were stored at -80°C until processing. Studies involving 454 pyrotag sequencing utilized the C57BL/6 colonic tissue that was collected after a single 2 hr cycle of SDR, with n = 5 per group (HCC vs. SDR). Animals were kept 3 and 2 mice/cage to minimize cage effects. Studies involving bacterial quantification PCR utilized colonic tissue collected at 0 days of SDR (n = 16 C57BL6, n = 21 CD-1), following a single 2 hr cycle of SDR (n = 7 C57BL/6, n = 15 CD-1) as well as samples collected after SDR repeated on 6 consecutive days (n = 8 C57BL/6, n = 6 CD-1). All experiments were n = 3 or 4 per group. The single 2 hour cycle of SDR in C57BL/6 was replicated once, and six repeated exposures of SDR on C57BL/6 was replicated twice. The single 2 hour cycle of SDR in CD1 mice was replicated four times, and six repeated exposures of SDR on CD1 mice was replicated once. Successive replicates were performed to reduce high variability between samples in bacterial qPCR. No-SDR-exposure groups were sacrificed alongside both single 2 hr cycle of SDR and 6 repeated cycles of SDR groups and combined during statistical analyses, resulting in an increased number of replications over test groups.
bTEFAP pyrosequencing
Amplicon pyrosequencing (bTEFAP®) was originally described by Dowd et al. on C57BL/6 mice colonic samples and has been utilized in describing a wide range of environmental and health related microbiomes including the intestinal populations of a variety of sample types and environments [59-61]. In this protocol, a 1-mm segment was used from the center of the entire colonic length. In a modified version of this process, 16S universal Eubacterial primers 530 F 5’GTGCCAGCMGCNGCGG and 1100R 5’GGGTTNCGNTCGTTR were used in a single-step 30 cycle PCR using HotStarTaq Plus Master Mix Kit (Qiagen, Valencia, CA). The following conditions were used: 94°C for 3 minutes, followed by 28 cycles of 94°C for 30 seconds; 53°C for 40 seconds and 72°C for 1 minute; after which a final elongation step at 72°C for 5 minutes was performed. Following PCR, all amplicon products from different samples were mixed in equal concentrations and purified using Agencourt Ampure beads (Agencourt Bioscience Corporation, MA, USA). Samples were sequenced utilizing Roche 454 FLX titanium instruments and reagents and following manufacturer’s guidelines.
Microbial community analysis
The returned sequences were processed and analyzed using Quantitative Insights Into Microbial Ecology (QIIME) (version 1.4.0) [62] according to standard protocols. Filtering passed sequences based on: length between 200 and 1000 bp, zero ambiguous bases or primer mismatches, and no homopolymer runs greater than 6. 97.9% of the sequences passed quality filtering for further analysis. After filtering, there was an average of 10496 sequences per sample. Sequences with 97% similarity were grouped into OTU using UCLUST [63]. Representative sequences from each OTU were aligned against the Greengenes core reference alignment using PyNAST [64,65]. The RDP Classifier was used to assign taxonomy to each representative sequence against the RDP database using standard options [66]. The minimum confidence threshold for taxonomic assignment was 0.80. OTUs were considered unclassified if there was not a strong match within this confidence interval for the representative sequence within the RDP database. A phylogenetic tree was built using FastTree from the aligned OTU representative sequences for determining UniFrac distances between samples [67].
Quantification of bacterial groups using quantitative polymerase chain reaction (qPCR)
DNA extraction
50 mg of tissue, taken from the ascending colon portion of the colonic tract, were homogenized in liquid nitrogen in a mortar and pestle and transferred to a microcentrifuge tube. The samples were incubated for 75 min in lysozyme buffer (20 mg/mL lysozyme, 20 mM Tris–HCl, 2 mM EDTA, 1.2% Triton) at 37°C to weaken the cell wall of Gram-positive bacteria. Following this, DNA was isolated using a modified version of the Qiagen DNA Mini Isolation Kit protocol (Qiagen, Germany). In short, 100 μL of Buffer ATL and 20 μL of proteinase K were added to the microcentrifuge tube and incubated for 3.5 hr at 56°C, with manual vortexing at 30 minute intervals for the duration of the incubation. The samples were heated at 85°C for 10 min to inactivate proteinase K and then transferred into a new tube containing 20 μL of proteinase K and 200 μL of Buffer AL. The samples were incubated at 56°C for 30 min followed by 95°C for 10 min. 200 μL of absolute ethanol was then added to the sample and the sample was added to a QIAamp spin column. After sample washing, the DNA from each sample was recovered into 100 μL of Buffer AE using a QIAamp spin column per manufacturer’s protocols. The DNA extracts were stored at -80°C until analysis was performed. Genomic DNA from pure bacterial cultures was isolated essentially as described above, but using a Qiagen protocol for the isolation of genomic DNA from Gram-positive bacteria and shorter (45 minutes instead of 75 minutes) incubation in the lysozyme buffer.
Sample derived standards
The method of Chen et al. [68] was adapted for the generation of sample-derived qPCR standards [68]. Briefly, equivalent mass amounts of colonic DNA from each sample were pooled as the template for PCR to obtain even bacterial representation in order to generate standards for bacterial copy number quantification. DNA was quantified using the Quant-it PicoGreen kit (Life Technologies, Grand Island, New York). Amplification PCR targeting specific genera or species-level 16S rRNA was performed to obtain standard amplicons based upon the representative populations within each sample. PCR was achieved using an ABIPrism 7000 thermocycler. The primer sets are shown in Table 5. The standard amplification PCR thermoprofile for the genus Lactobacillus consisted of initial denaturation at 94°C for 5 min, followed by 35 cycles of 94°C for 30 sec, 58°C for 30 sec, and 72°C for 40 sec, and final extension at 72°C for 10 min. For L. reuteri PCR, the thermoprofile used was the same as those for the genus-specific except for a higher primer annealing temperature (60°C instead of 58°C). The Bacteroides-Prevotella-Porphyromonas standard amplification thermoprofile was 1 cycle at 95°C for 5 min, followed by 35 cycles of 95°C for 20 s, 68°C for 30 s, and 72° for 55 s, with 1 final annealing cycle of 72°C for 5 min. The Parabacteroides distasonis standard amplification thermoprofile was 1 cycle at 94°C for 5 min, followed by 30 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 40 s, with 1 final annealing cycle at 72°C for 8 min. After the amplification PCR, the PCR product was purified using the Qiagen PCR Purification Kit and quantified by Quant-it PicoGreen kit to determine copy number of the community standards.
Table 5.
PCR primers and probes
|
Lactobacillus genus |
Forward |
AGCAGTAGGGAATCTTCCA |
| Reverse |
CACCGCTACACATGGAG |
|
|
Lactobacillus reuteri |
Forward |
CAGACAATCTTTGATTGTTTAG |
| Reverse |
GCTTGTTGGTTTGGGCTCTTC |
|
|
Parabacteroides distasonis |
Forward |
TGCCTATCAGAGGGGGATAAC |
| Reverse |
GCAAATATTCCCATGCGGGAT |
|
|
Porphyromonas-Bacteroides-Prevotella |
Forward |
GGTGTCGGCTTAAGTGCCAT |
| Reverse |
CGGA(C/T)GTAAGGGCCGTGC |
|
| TNF-α |
Forward |
CTGTCTACTGAACTTCGGGGTGAT |
| Reverse |
GCTCTGGGCCATAGAACTGATG |
|
| Probe |
ATGAGAAGTTCCCAAATGGCCTCCCTC |
|
| IL-1β |
Forward |
GGCCTCAAAGGAAAGAATCTATACC |
| Reverse |
GTATTGCTTGGGATCCACACTCT |
|
| Probe |
ATGAAAGACGGCACACCCACCCTG |
|
| iNOS |
Forward |
CAGCTGGGCTGTACAAACCTT |
| Reverse |
TGAATGTGATGTTTGCTTCGG |
|
| Probe | CGGGCAGCCTGTGAGACCTTTGA |
A standard curve for absolute quantification was created from the previously amplified standards to encompass 101-108 copies per reaction. The population of total lactobacilli and L. reuteri was separately quantified using respective specific primers and SYBR Green against these dilution series as described previously [69]. qPCR thermoprofiles for Lactobacillus and L. reuteri were 1 cycle at 94°C for 5 min, followed by 40 cycles of 94°C for 30 s, specific annealing temperature for 30 s, and 72°C for 40 s. Annealing temperature for Lactobacillus was 58°C and L. reuteri was 60°C. qPCR thermoprofile for P. distasonis was 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 10 min, then 40 cycles at 95°C for 15 s and 60°C for 60 s. qPCR thermoprofile for Bacteroides-Prevotella-Porphyromonas was 1 cycle at 95°C for 5 min, followed by 40 cycles at 95°C for 15 s, 68°C for 20 s, and 72°C for 30 s. Abundance of bacterial groups (copies of 16 s rRNA gene per gram/sample) was computed based on the copies of qPCR reaction and the number of reactions that could be performed with the DNA derived from 1 g of each tissue sample. The detection limit were as follows: total lactobacilli and Bacteroides-Prevotella-Porphyromonas was ~4.0 log copies/gram of wet tissue (log10), L. reuteri was ~4.5 log copies/gram of wet tissue (log10), P. distasonis was ~5.0 log copies/gram of wet tissue. Primer sequences have been previously published and are listed in Table 5[70-74].
Quantification of colonic cytokine and inflammatory mediator mRNA using quantitative RT-PCR
Total RNA was isolated from whole colonic tissue samples (~120 mg) using TRI-zol (Invitrogen, Carlsbad, California) according to manufacturer’s instructions. The extracted RNA was quantified via spectrophotometry, and converted to cDNA using the Reverse Transcription System (Promega, Madison, WI). qPCR was then completed using a master mix containing 2× Universal TaqMan master mix (Life Technologies, Grand Island, New York), 0.9 μM (each) forward and reverse primers (see Table 5), and 0.250 μg sample cDNA. 18S was used as the housekeeping gene. The PCR was performed using a Prism 7000 sequence detection system with the following thermoprofile: 2 min at 50°C, 10 min at 95°C, and then 40 amplification cycles of 15 s at 95°C and 1 min at 60°C. The relative amount of mRNA was determined using the comparative cycle threshold (C T ) method as previously described [75]. Non-stressed HCC control samples were used as baseline controls and were set at a value of 1. All other samples are based on a fold change from these control samples. Primer sequences have been previously published and are listed in Table 5[76]. All groups were n = 3 – 5, using a single experiment without replicates.
Statistical analyses
Alpha diversity measurements, including Shannon diversity index, equitability (evenness) and Chao (richness) were computed using QIIME on the groups (HCC control vs. SDR Stressor) [77]. Non-parametric T-tests with 999 Monte Carlo permutations were used on these alpha diversity measurements to test for significance at a sampling depth of 4805 sequences per sample.
Taxonomic abundances attained from QIIME were compared between the two groups of mice using non-parametric Mann–Whitney U Tests. PCoA of unweighted UniFrac distance matrices was used to determine clustering between the two groups (HCC control vs. SDR Stressor) [78]. Analysis of Similarity (ANOSIM), a beta-diversity statistic that is available through the vegan package of R and accessible with QIIME, was used to calculate statistical significance between the distance matrices of groups at 999 permutations [79,80]. All of these analyses were performed in QIIME.
Differences in bacterial abundances were determined using non-parametric Kruskal Wallis tests with the cycle of SDR (i.e., 0, 1, or 6 cycles) as the single factor. Mann–Whitney U tests were performed a priori. Changes in cytokine gene expression were determined using a two-factor ANOVA with group (HCC control vs. SDR Stressor) and Day (1 Day vs. 6 Days) as the two independent factors. Protected means comparisons were used as post-hoc tests. In all cases, α was set at 0.05, while tendency was declared at 0.05 < p < 0.10. These tests were performed using SPSS v.21 (IBM, Chicago, IL).
Availability of supporting data
The sequences supporting the results of this article are available in the NCBI Sequence Read Archive under the study accession number SRP035598 (http://www.ncbi.nlm.nih.gov/sra/?term=SRP035598).
Abbreviations
GI: Gastrointestinal; SDR: Social disruption; qPCR: Quantitative real-time polymerase chain reaction; HPA: Hypothalamic-pituitary-adrenal axis; OTU: Operational taxonomic unit; UPGMA: Unweighted pair-group method with arithmetic mean; ANOSIM: Analysis of similarity; HCC: Home cage control; IL-1β: Interleukin 1-beta; TNF-α: Tumor necrosis factor-alpha; iNOS: Inducible nitric oxide synthase; DSS: Dextran sodium sulfate; CRH: Corticotrophin-releasing hormone; bTEFAP: Bacterial tag-encoded FLX amplicon pyrosequencing; QIIME: Quantitative Insights Into Microbial Ecology; PCoA: Principle coordinates analysis.
Competing interests
The authors declare they have no competing interests.
Authors’ contributions
JDG performed all mouse experiments, analyzed 454 sequencing data, and drafted the manuscript. MCN assisted on the 454 sequencing data. ZY assisted with the qPCR method and PCR data analysis. SED performed 454 pyrosequencing. JW assisted with the qPCR method and data analysis. PSK assisted the qPCR method and PCR data analysis. ML designed the mouse experiments. MTB designed the mouse experiments, analyzed 454 sequencing data, and drafted the manuscript. All others edited and approved the final manuscript.
Contributor Information
Jeffrey D Galley, Email: galley.2@osu.edu.
Michael C Nelson, Email: mcnelson.phd@gmail.com.
Zhongtang Yu, Email: yu.226@osu.edu.
Scot E Dowd, Email: sdowd@mrdnalab.com.
Jens Walter, Email: jwalter1@ualberta.ca.
Purnima S Kumar, Email: kumar.83@osu.edu.
Mark Lyte, Email: mark.lyte@ttuhsc.edu.
Michael T Bailey, Email: Michael.bailey@osumc.edu.
Acknowledgements
These studies were generously supported by NIH grant RO1AT006552-01A1 and NIH/NIDCR grant T32 DE014320.
References
- Turnbaugh PJ, Ley RE, Hamady M, Fraser-Liggett CM, Knight R, Gordon JI. The human microbiome project. Nature. 2007;449(7164):804–810. doi: 10.1038/nature06244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone CA, Stombaugh J, Gonzalez A, Ackermann G, Wendel D, Vazquez-Baeza Y, Jansson JK, Gordon JI, Knight R. Meta-analyses of studies of the human microbiota. Genome Res. 2013;23(10):1704–1714. doi: 10.1101/gr.151803.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lunden G, Petersen LC, Ruf W, Backhed F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature. 2012;483(7391):627–631. doi: 10.1038/nature10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host-microbial relationships in the intestine. Science. 2001;291(5505):881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- Atarashi K, Tanoue T, Shima T, Imaoka A, Kuwahara T, Momose Y, Cheng G, Yamasaki S, Saito T, Ohba Y, Taniguchi T, Takeda K, Hori S, Ivanov II, Umesaki Y, Itoh K, Honda K. Induction of colonic regulatory T cells by indigenous Clostridium species. Science. 2011;331(6015):337–341. doi: 10.1126/science.1198469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Aidy S, van Baarlen P, Derrien M, Lindenbergh-Kortleve DJ, Hooiveld G, Levenez F, Dore J, Dekker J, Samsom JN, Nieuwenhuis EE, Kleerebezem M. Temporal and spatial interplay of microbiota and intestinal mucosa drive establishment of immune homeostasis in conventionalized mice. Mucosal Immunol. 2012;5(5):567–579. doi: 10.1038/mi.2012.32. [DOI] [PubMed] [Google Scholar]
- Williams AM, Probert CS, Stepankova R, Tlaskalova-Hogenova H, Phillips A, Bland PW. Effects of microflora on the neonatal development of gut mucosal T cells and myeloid cells in the mouse. Immunology. 2006;119(4):470–478. doi: 10.1111/j.1365-2567.2006.02458.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, Schreiber S, Rosenstiel P. Nod2 is essential for temporal development of intestinal microbial communities. Gut. 2011;60(10):1354–1362. doi: 10.1136/gut.2010.216259. [DOI] [PubMed] [Google Scholar]
- Couturier-Maillard A, Secher T, Rehman A, Normand S, De Arcangelis A, Haesler R, Huot L, Grandjean T, Bressenot A, Delanoye-Crespin A, Gaillot O, Schreiber S, Lemoine Y, Ryffel B, Hot D, Nùñez G, Chen G, Rosenstiel P, Chamaillard M. NOD2-mediated dysbiosis predisposes mice to transmissible colitis and colorectal cancer. J Clin Invest. 2013;123(2):700–711. doi: 10.1172/JCI62236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Gordon JI. Honor thy symbionts. Proc Natl Acad Sci U S A. 2003;100(18):10452–10459. doi: 10.1073/pnas.1734063100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A. 2005;102(31):11070–11075. doi: 10.1073/pnas.0504978102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5(4):627–638. doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Dowd SE, Galley JD, Hufnagle AR, Allen RG, Lyte M. Exposure to a social stressor alters the structure of the intestinal microbiota: implications for stressor-induced immunomodulation. Brain Behav Immun. 2011;25(3):397–407. doi: 10.1016/j.bbi.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444(7122):1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. Decreased diversity of the fecal Microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197(3):435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- Sekirov I, Tam NM, Jogova M, Robertson ML, Li Y, Lupp C, Finlay BB. Antibiotic-induced perturbations of the intestinal microbiota alter host susceptibility to enteric infection. Infect Immun. 2008;76(10):4726–4736. doi: 10.1128/IAI.00319-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Coe CL. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol. 1999;35(2):146–155. [PubMed] [Google Scholar]
- Mackos AR, Eubank TD, Parry NM, Bailey MT. Probiotic Lactobacillus reuteri attenuates the stressor-enhanced severity of Citrobacter rodentium infection. Infect Immun. 2013;81(9):3253–3263. doi: 10.1128/IAI.00278-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber O, Petersson J, Phillipson M, Perry M, Roos S, Holm L. Lactobacillus reuteri prevents colitis by reducing P-selectin-associated leukocyte- and platelet-endothelial cell interactions. Am J Physiol Gastrointest Liver Physiol. 2009;296(3):G534–G542. doi: 10.1152/ajpgi.90470.2008. [DOI] [PubMed] [Google Scholar]
- Barouei J, Moussavi M, Hodgson DM. Effect of maternal probiotic intervention on HPA axis, immunity and gut microbiota in a rat model of irritable bowel syndrome. PLoS One. 2012;7(10):e46051. doi: 10.1371/journal.pone.0046051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Dowd SE, Parry NM, Galley JD, Schauer DB, Lyte M. Stressor exposure disrupts commensal microbial populations in the intestines and leads to increased colonization by Citrobacter rodentium. Infect Immun. 2010;78(4):1509–1519. doi: 10.1128/IAI.00862-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF. Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun. 2012;26(7):1150–1159. doi: 10.1016/j.bbi.2012.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein CN, Singh S, Graff LA, Walker JR, Miller N, Cheang M. A prospective population-based study of triggers of symptomatic flares in IBD. Am J Gastroenterol. 2010;105(9):1994–2002. doi: 10.1038/ajg.2010.140. [DOI] [PubMed] [Google Scholar]
- Dickhaus B, Mayer EA, Firooz N, Stains J, Conde F, Olivas TI, Fass R, Chang L, Mayer M, Naliboff BD. Irritable bowel syndrome patients show enhanced modulation of visceral perception by auditory stress. Am J Gastroenterol. 2003;98(1):135–143. doi: 10.1111/j.1572-0241.2003.07156.x. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Tack J. Altered intestinal microbiota in irritable bowel syndrome. Neurogastroenterol Motil. 2010;22(5):493–498. doi: 10.1111/j.1365-2982.2010.01482.x. [DOI] [PubMed] [Google Scholar]
- Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90(3):859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134(2):577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- Garrett WS, Lord GM, Punit S, Lugo-Villarino G, Mazmanian SK, Ito S, Glickman JN, Glimcher LH. Communicable ulcerative colitis induced by T-bet deficiency in the innate immune system. Cell. 2007;131(1):33–45. doi: 10.1016/j.cell.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MT, Lubach GR, Coe CL. Prenatal stress alters bacterial colonization of the gut in infant monkeys. J Pediatr Gastroenterol Nutr. 2004;38(4):414–421. doi: 10.1097/00005176-200404000-00009. [DOI] [PubMed] [Google Scholar]
- Carroll IM, Chang YH, Park J, Sartor RB, Ringel Y. Luminal and mucosal-associated intestinal microbiota in patients with diarrhea-predominant irritable bowel syndrome. Gut pathogens. 2010;2(1):19. doi: 10.1186/1757-4749-2-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajaj JS, Hylemon PB, Ridlon JM, Heuman DM, Daita K, White MB, Monteith P, Noble NA, Sikaroodi M, Gillevet PM. Colonic mucosal microbiome differs from stool microbiome in cirrhosis and hepatic encephalopathy and is linked to cognition and inflammation. Am J Physiol Gastrointest Liver Physiol. 2012;303(6):G675–G685. doi: 10.1152/ajpgi.00152.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van den Abbeele P, Van de Wiele T, Verstraete W, Possemiers S. The host selects mucosal and luminal associations of coevolved gut microorganisms: a novel concept. FEMS Microbiol Rev. 2011;35(4):681–704. doi: 10.1111/j.1574-6976.2011.00270.x. [DOI] [PubMed] [Google Scholar]
- Knowles SR, Nelson EA, Palombo EA. Investigating the role of perceived stress on bacterial flora activity and salivary cortisol secretion: a possible mechanism underlying susceptibility to illness. Biol Psychol. 2008;77(2):132–137. doi: 10.1016/j.biopsycho.2007.09.010. [DOI] [PubMed] [Google Scholar]
- Tannock GW, Savage DC. Influences of dietary and environmental stress on microbial populations in the murine gastrointestinal tract. Infect Immun. 1974;9(3):591–598. doi: 10.1128/iai.9.3.591-598.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friswell MK, Gika H, Stratford IJ, Theodoridis G, Telfer B, Wilson ID, McBain AJ. Site and strain-specific variation in gut microbiota profiles and metabolism in experimental mice. PLoS One. 2010;5(1):e8584. doi: 10.1371/journal.pone.0008584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F, Nguyen TL, Brinkman B, Yunta RG, Cauwe B, Vandenabeele P, Liston A, Raes J. Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 2013;14(1):R4. doi: 10.1186/gb-2013-14-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouwknecht JA, Paylor R. Behavioral and physiological mouse assays for anxiety: a survey in nine mouse strains. Behav Brain Res. 2002;136(2):489–501. doi: 10.1016/s0166-4328(02)00200-0. [DOI] [PubMed] [Google Scholar]
- Julio-Pieper M, O'Mahony CM, Clarke G, Bravo JA, Dinan TG, Cryan JF. Chronic stress-induced alterations in mouse colonic 5-HT and defecation responses are strain dependent. Stress. 2012;15(2):218–226. doi: 10.3109/10253890.2011.607524. [DOI] [PubMed] [Google Scholar]
- Jang SE, Hyam SR, Han MJ, Kim SY, Lee BG, Kim DH. Lactobacillus brevis G-101 ameliorates colitis in mice by inhibiting NF-kappaB, MAPK and AKT pathways and by polarizing M1 macrophages to M2-like macrophages. J Appl Microbiol. 2013;115(3):888–896. doi: 10.1111/jam.12273. [DOI] [PubMed] [Google Scholar]
- Cuevas M, Flores I, Thompson KJ, Ramos-Ortolaza DL, Torres-Reveron A, Appleyard CB. Stress exacerbates endometriosis manifestations and inflammatory parameters in an animal model. Reprod Sci. 2012;19(8):851–862. doi: 10.1177/1933719112438443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Hill DA, Minkah N, Kirn T, Troy A, Artis D, Bushman F. Community-wide response of the gut microbiota to enteropathogenic Citrobacter rodentium infection revealed by deep sequencing. Infect Immun. 2009;77(10):4668–4678. doi: 10.1128/IAI.00493-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven M, Egan CE, Dowd SE, McDonough SP, Dogan B, Denkers EY, Bowman D, Scherl EJ, Simpson KW. Inflammation drives dysbiosis and bacterial invasion in murine models of ileal Crohn's disease. PLoS One. 2012;7(7):e41594. doi: 10.1371/journal.pone.0041594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupp C, Robertson ML, Wickham ME, Sekirov I, Champion OL, Gaynor EC, Finlay BB. Host-mediated inflammation disrupts the intestinal microbiota and promotes the overgrowth of Enterobacteriaceae. Cell Host Microbe. 2007;2(2):119–129. doi: 10.1016/j.chom.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Collins SM, McHugh K, Jacobson K, Khan I, Riddell R, Murase K, Weingarten HP. Previous inflammation alters the response of the rat colon to stress. Gastroenterology. 1996;111(6):1509–1515. doi: 10.1016/s0016-5085(96)70012-4. [DOI] [PubMed] [Google Scholar]
- Quan N, Avitsur R, Stark JL, He L, Lai W, Dhabhar F, Sheridan JF. Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol. 2003;137(1–2):51–58. doi: 10.1016/s0165-5728(03)00042-0. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R. Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun. 2007;21(4):458–466. doi: 10.1016/j.bbi.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mays JW, Bailey MT, Hunzeker JT, Powell ND, Papenfuss T, Karlsson EA, Padgett DA, Sheridan JF. Influenza virus-specific immunological memory is enhanced by repeated social defeat. J Immunol. 2010;184(4):2014–2025. doi: 10.4049/jimmunol.0900183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangsgaard Bendtsen KM, Krych L, Sorensen DB, Pang W, Nielsen DS, Josefsen K, Hansen LH, Sorensen SJ, Hansen AK. Gut microbiota composition is correlated to grid floor induced stress and behavior in the BALB/c mouse. PLoS One. 2012;7(10):e46231. doi: 10.1371/journal.pone.0046231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Zhang M, Chen CC, Gillilland M Jr, Sun X, El-Zaatari M, Huffnagle GB, Young VB, Zhang J, Hong SC, Chang YM, Gumucio DL, Owyang C, Kao JY. Stress-induced corticotropin-releasing hormone-mediated NLRP6 inflammasome inhibition and transmissible enteritis in mice. Gastroenterology. 2013;144(7):1478–1487. doi: 10.1053/j.gastro.2013.02.038. 1487 e1471-1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markle JG, Frank DN, Mortin-Toth S, Robertson CE, Feazel LM, Rolle-Kampczyk U, von Bergen M, McCoy KD, Macpherson AJ, Danska JS. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science. 2013;339(6123):1084–1088. doi: 10.1126/science.1233521. [DOI] [PubMed] [Google Scholar]
- Park AJ, Collins J, Blennerhassett PA, Ghia JE, Verdu EF, Bercik P, Collins SM. Altered colonic function and microbiota profile in a mouse model of chronic depression. Neurogastroenterol Motil. 2013;25(9):733–e575. doi: 10.1111/nmo.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene BR, Blanchard EB, Wan CK. Long-term monitoring of psychosocial stress and symptomatology in inflammatory bowel disease. Behav Res Ther. 1994;32(2):217–226. doi: 10.1016/0005-7967(94)90114-7. [DOI] [PubMed] [Google Scholar]
- Bennett EJ, Tennant CC, Piesse C, Badcock CA, Kellow JE. Level of chronic life stress predicts clinical outcome in irritable bowel syndrome. Gut. 1998;43(2):256–261. doi: 10.1136/gut.43.2.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, Wekerle H, Krishnamoorthy G. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature. 2011;479(7374):538–541. doi: 10.1038/nature10554. [DOI] [PubMed] [Google Scholar]
- Buljevac D, Hop WC, Reedeker W, Janssens AC, van der Meche FG, van Doorn PA, Hintzen RQ. Self reported stressful life events and exacerbations in multiple sclerosis: prospective study. BMJ. 2003;327(7416):646. doi: 10.1136/bmj.327.7416.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RG, Lafuse WP, Powell ND, Webster Marketon JI, Stiner-Jones LM, Sheridan JF, Bailey MT. Stressor-induced increase in microbicidal activity of splenic macrophages is dependent upon peroxynitrite production. Infect Immun. 2012;80(10):3429–3437. doi: 10.1128/IAI.00714-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS. Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP) BMC Microbiol. 2008;8:125. doi: 10.1186/1471-2180-8-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway TR, Dowd SE, Edrington TS, Anderson RC, Krueger N, Bauer N, Kononoff PJ, Nisbet DJ. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci. 2010;88(12):3977–3983. doi: 10.2527/jas.2010-2900. [DOI] [PubMed] [Google Scholar]
- Williams WL, Tedeschi LO, Kononoff PJ, Callaway TR, Dowd SE, Karges K, Gibson ML. Evaluation of in vitro gas production and rumen bacterial populations fermenting corn milling (co)products. J Dairy Sci. 2010;93(10):4735–4743. doi: 10.3168/jds.2009-2920. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol. 2006;72(7):5069–5072. doi: 10.1128/AEM.03006-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26(2):266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Yu Z, Michel FC Jr, Wittum T, Morrison M. Development and application of real-time PCR assays for quantification of erm genes conferring resistance to macrolides-lincosamides-streptogramin B in livestock manure and manure management systems. Appl Environ Microbiol. 2007;73(14):4407–4416. doi: 10.1128/AEM.02799-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson K, Yu Z, Chen J, Jenkins J, Courtney P, Morrison M. Analyses of Bifidobacterium, Lactobacillus, and total bacterial populations in healthy volunteers consuming calcium gluconate by denaturing gradient gel electrophoresis and real-time PCR. Int J Probiotics Prebiotics. 2008;3:31–36. [Google Scholar]
- Walter J, Hertel C, Tannock GW, Lis CM, Munro K, Hammes WP. Detection of Lactobacillus, Pediococcus, Leuconostoc, and Weissella species in human feces by using group-specific PCR primers and denaturing gradient gel electrophoresis. Appl Environ Microbiol. 2001;67(6):2578–2585. doi: 10.1128/AEM.67.6.2578-2585.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig HG, Zoetendal EG, Vaughan EE, Marteau P, Akkermans AD, de Vos WM. Molecular diversity of Lactobacillus spp. and other lactic acid bacteria in the human intestine as determined by specific amplification of 16S ribosomal DNA. Appl Environ Microbiol. 2002;68(1):114–123. doi: 10.1128/AEM.68.1.114-123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97(6):1166–1177. doi: 10.1111/j.1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- Brisbin JT, Gong J, Orouji S, Esufali J, Mallick AI, Parvizi P, Shewen PE, Sharif S. Oral treatment of chickens with lactobacilli influences elicitation of immune responses. Clin Vaccine Immunol. 2011;18(9):1447–1455. doi: 10.1128/CVI.05100-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Liu C, Summanen P, Xu H, Finegold SM. Application of quantitative real-time PCR for rapid identification of Bacteroides fragilis group and related organisms in human wound samples. Anaerobe. 2011;17(2):64–68. doi: 10.1016/j.anaerobe.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Head CC, Farrow MJ, Sheridan JF, Padgett DA. Androstenediol reduces the anti-inflammatory effects of restraint stress during wound healing. Brain Behav Immun. 2006;20(6):590–596. doi: 10.1016/j.bbi.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Allen RG, Lafuse WP, Galley JD, Ali MM, Ahmer BM, Bailey MT. The intestinal microbiota are necessary for stressor-induced enhancement of splenic macrophage microbicidal activity. Brain Behav Immun. 2012;26(3):371–382. doi: 10.1016/j.bbi.2011.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon CE, Weaver W. The mathematical theory of communication. Urbana: University of Illinois Press; 1949. [Google Scholar]
- Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71(12):8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksanen J, Blanchet FG, Kindt R, Legendre P, Minchin PR, O’Hara RB, Simpson GL, Solymos P, Henry M, Stevens HH, Wagner H. Vegan: community ecology package. R package version 2.0-3; 2012. [Google Scholar]
- Development Core Team. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. Coventry, United Kingdom R Foundation for Statistical Computing; 2011. [Google Scholar]