

Abstract
Objective:
We evaluated the relationship of amyloid, seen on Pittsburgh compound B (PiB)-PET, and metabolism, seen on [18F]-fluorodeoxyglucose (FDG)-PET, in normal subjects to better understand pathogenesis and biomarker selection in presymptomatic subjects.
Methods:
Normal participants (aged 70–95 years; 600 with PiB-PET, FDG-PET, and MRI) were included. We performed a cross-sectional evaluation and subcategorized participants into amyloid-negative (<1.4), high-normal (1.4–1.5), positive (1.5–2.0), and markedly positive (>2.0) PiB standardized uptake value ratio groups representing different levels of amyloid brain load. Associations with metabolism were assessed in each group. Relationships with APOE ε4 carriage were evaluated.
Results:
Hypometabolism in “Alzheimer disease (AD)-signature” regions was strongly associated with PiB load. Hypometabolism was greater with more positive PiB levels. Additional, more-diffuse cortical hypometabolism was also found to be associated with PiB, although less so. No hypermetabolism was seen in any subset. No significant incremental hypometabolism was seen in APOE-positive vs -negative subjects.
Conclusions:
Hypometabolism in PiB-positive, cognitively normal subjects in a population-based cohort occurs in AD-signature cortical regions and to a lesser extent in other cortical regions. It is more pronounced with higher amyloid load and supports a dose-dependent association. The effect of APOE ε4 carriage in this group of subjects does not appear to modify their hypometabolic “AD-like” neurodegeneration. Consideration of hypometabolism associated with amyloid load may aid trials of AD drug therapy.
Hypothetical models of the temporal sequence of brain-imaging biomarker findings that may reflect the Alzheimer disease (AD) pathologic cascade have been proposed by our group and others.1–4 Understanding the relationship of biomarker abnormalities at their earliest stages will help explain the pathologic mechanisms of AD, aid in targeted therapy approaches, and aid in determining the utility of biomarkers in patients with preclinical AD pathology and in patients with early dementia.
Most reports to date have only considered the relationship of amyloid-positive vs -negative normal subjects and hypometabolism. This does little to elucidate the effect of different levels of amyloid on brain metabolism in presymptomatic subjects. To further elucidate the association between amyloid load and metabolism, we cross-sectionally examined a very large group of cognitively normal (CN) elderly subjects.
METHODS
Standard protocol approvals, registrations, and patient consents.
Participants were drawn from the Mayo Clinic Study of Aging as described previously.3 [18F]-fluorodeoxyglucose (FDG)-PET and Pittsburgh compound B (PiB)-PET were performed on the same day. All participants provided written consent with approval of the Mayo Clinic and Olmsted Medical Center Institutional Review Boards. MRI scans were performed a median of 16 days before the PET scans. A total of 606 Mayo Clinic Study of Aging participants considered CN based on a consensus diagnosis after thorough clinical and cognitive assessment by neurologists, geriatricians, neuropsychologists, and study nurses5 completed the above imaging between December 8, 2004 and September 2012, and were available for analysis. Six subjects were excluded because of imaging/APOE-quality issues (3) or brain lesions (3). Our consensus diagnostic process uses quantitative data from a brief mental status examination, 9 neuropsychological tests, and the Clinical Dementia Rating scale, but uses a consensus approach rather than rigid adherence to cut-scores to arrive at a diagnosis of CN.5
PET imaging methods.
MRIs were performed at 3 tesla using an 8-channel phased array coil (GE, Milwaukee, WI). Image acquisition and image analysis are described in detail elsewhere.6 PiB-PET global cortical uptake normalized to cerebellar gray matter (standardized uptake value ratio [SUVR]) was then used to assign subjects to groups of different levels of PiB uptake. FDG-PET cortical-to-pons ratios were calculated for each brain region (SUVR). No partial volume correction was used. Subjects were classified according to PiB SUVR into 4 groups: amyloid-negative (<1.4), high-normal (1.4–1.5), positive (1.5–2.0), and markedly positive (>2.0) (table 1).
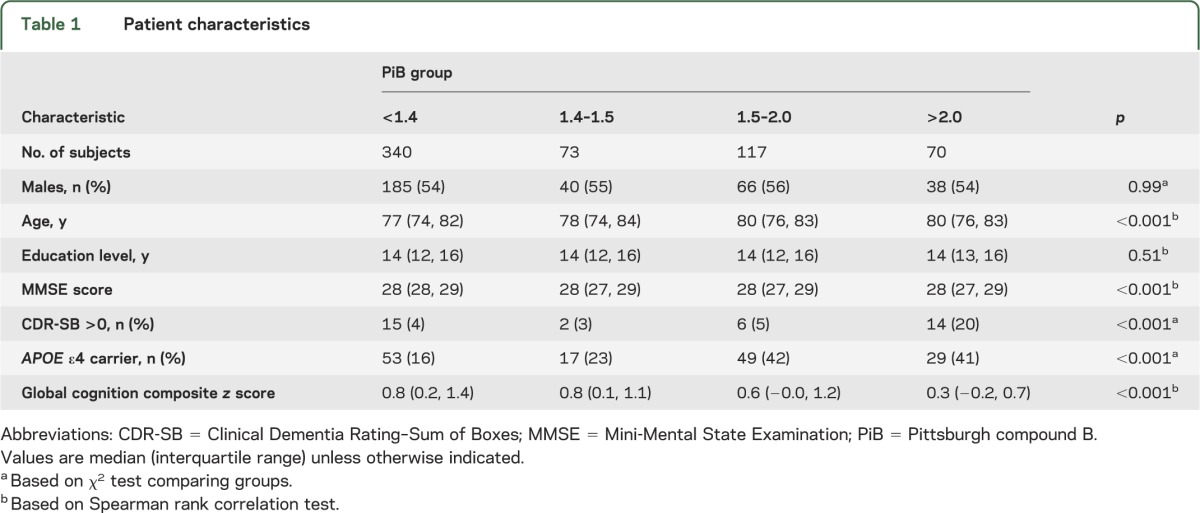
Table 1.
Patient characteristics
Statistical analysis.
In our first analysis, which was performed separately for each of 47 regions of interest (ROIs), we used linear regression to examine the association between regional FDG and global PiB after adjusting for age and sex. We summarize these results by showing the mean FDG within each of the 4 PiB groups. In addition, we report a false discovery rate (FDR)-corrected p value of a “dose-effect” test of the association between regional FDG and the logarithm of PiB after accounting for age and sex. The logarithm transformation was used to reduce skewness and prevent subjects with the highest levels of PiB from having an outsized influence on the model fit.
We followed this analysis with a very similar regression analysis, looking at differences in regional FDG by APOE ε4 carrier status after accounting for age, sex, and PiB. We report 2 sets of uncorrected p values for this analysis. The first set of p values is from a test of APOE ε4 carrier status with age and sex in the model, but not PiB. The second set of p values is from a test of APOE with age, sex, and PiB in the regression model.
In a third analysis, we examined the association between regional FDG and regional PiB after accounting for age and sex. Because the subject groupings based on cortical PiB were not applicable at the regional PiB level, for each ROI we defined a PiB group based on tertiles for that region. Here, we are able to report a dose-effect test of the association between regional FDG and the logarithm of regional PiB after accounting for age and sex.
In a final set of analyses, we used multivariable logistic regression to identify a parsimonious subset of regions that provide independent information for predicting association of elevated PiB, defined here as PiB >1.5. With 47 regions under consideration, we wanted to protect against overfitting, which is dependent on the number of variables considered rather than the number of variables in a final model. We therefore used penalized logistic regression to shrink coefficients toward the null and thus account for possible overfitting.7 The method of penalization we used is named the “elasticnet” and is a combination of a penalty based on the sum of squares of the coefficients (as in ridge regression) and the sum of absolute values of the coefficients (as in the lasso).8 We used version 1.9-1 of the glmnet package in R version 2.15.2 (http://www.-R-project.org) and set the elasticnet mixing parameter to 0.8 so that the lasso penalty was predominant but there remained a degree of ridge regression. In our analyses, age and sex were included in the model but not penalized. This was done by treating age as a continuous variable and indicating in the glmnet software that the age and sex coefficients should be excluded from any penalization. We used leave-one-out cross-validation to select a parsimonious model using the “1-SE rule” in which the final model has cross-validation error within 1 standard error of the optimal model.
To compare the relative strength of associations between “AD-signature” regions and “non–AD-signature” regions and amyloid load, we averaged the FDG uptake in the angular gyrus and posterior cingulum regions and evaluated whether FDG hypometabolism in this composite AD-signature region was more strongly associated with global PiB than the FDG hypometabolism in several non–AD-signature regions (8 frontal regions; superior, middle, superior orbital, middle orbital, inferior orbital, medial superior, and supplementary motor). To do this, we performed a formal test of dependent overlapping correlations.9 We next averaged the regional PiB levels in this composite AD-signature region and compared the strength of associations between the composite FDG hypometabolism and composite PiB uptake vs regional FDG and regional PiB in the non-AD regions using a test of dependent nonoverlapping correlations.10 To account for age and sex, we regressed these factors out of the PiB measures before performing our correlation analyses.
RESULTS
Table 1 summarizes the clinical characteristics of a sample of 600 CN subjects and indicates that higher PiB was associated with increasing age, worse cognitive performance, and APOE ε4 carrier state.
Hypometabolism was associated with global PiB on ROI analysis (figure 1). The strongest associations were seen in regions that are typical AD-signature regions (angular gyrus, posterior cingulate, temporal and parietal regions). Associations were seen in many other regions with decreasing significance.
Figure 1. Global cortical PiB vs regional FDG.

Age- and sex-adjusted mean FDG within each PiB group for the 47 regions of interest. The regions are sorted by level of significance. The estimates assume an 80-year-old subject and represent the average FDG for both men and women. The right side of the plot indicates the FDR-corrected p value for a “dose-effect” test of whether the log of PiB is associated with regional FDG after accounting for age and sex. FDG = [18F]-fluorodeoxyglucose; FDR = false discovery rate; PiB = Pittsburgh compound B.
Regarding APOE, FDG was generally not lower among APOE ε4 carriers compared with noncarriers after accounting for PiB level. A hypothesis-generating minor exception was the posterior cingulum, in which carriers tended to average lower FDG by approximately 0.03 SUVR units. After adjusting for PiB, they had a p = 0.04 without FDR correction. With FDR correction, that and all other p values are not significant (figure 2). This suggests that most of the APOE-related differences in hypometabolism are mediated by PiB.
Figure 2. ROI analysis of hypometabolism stratified by APOE status.

Age- and sex-adjusted mean FDG for APOE ε4 carriers and noncarriers by region. The regions are sorted by level of significance based on unadjusted p values. The estimates assume an 80-year-old subject with PiB of 1.4 and represent the average FDG for both men and women. The first column of p values on the right side of the plot is FDR-corrected p values from a test of APOE after adjusting for age, sex, and PiB. The second column shows unadjusted p values. An asterisk is used to indicate regions in which APOE was significant at the p < 0.10 level after adjusting for age and sex but not PiB. FDG = [18F]-fluorodeoxyglucose; PiB = Pittsburgh compound B; ROI = region of interest.
Regional hypometabolism and regional PiB (figure 3) show strong associations in typical AD-signature regions (angular gyrus, posterior cingulate, precuneus, temporal and parietal regions). The effect is relatively widespread, but non–AD-signature regions such as frontal regions show much less association of hypometabolism with PiB.
Figure 3. Regional hypometabolism associated with regional PiB.

Age- and sex-adjusted mean FDG within each regional PiB tertile. The regions are sorted by level of significance. The estimates assume an 80-year-old subject and represent the average FDG for both men and women. The right side of the plot indicates the FDR-corrected p value for a “dose-effect” test of whether the log of regional PiB is associated with regional FDG after accounting for age and sex. FDG = [18F]-fluorodeoxyglucose; FDR = false discovery rate; PiB = Pittsburgh compound B.
Penalized logistic regression identified a parsimonious subset of regions that provide independent information for predicting PiB >1.5 while at the same time protecting against overfitting due to considering a total of 47 regions. We found the key hypometabolic regions to be the angular gyrus and posterior cingulum, with each contributing independent predictive information.
The correlation between AD-signature regions and global PiB was −0.22, significantly higher than the correlation between non–AD-signature regions of FDG and global PiB (−0.06 to −0.14; p < 0.001 for all regions). The correlation between AD-signature regions and regional PiB was −0.23, also significantly higher than the correlation between non–AD-signature regions of FDG and regional PiB (−0.02 to −0.12; p < 0.001 for all regions) (table e-1 on the Neurology® Web site at Neurology.org).
DISCUSSION
These data demonstrate regional hypometabolism on FDG-PET is associated with PiB retention in normal subjects. Notably, this hypometabolism becomes evident in low PiB SUVR ranges widely used as a cutoff for distinguishing normal and abnormal PiB (approximately 1.5).6,11 In the groups with greatest PiB accumulation, greater hypometabolism occurs. This implies that these 2 pathologic biomarker events are linked closely in a dose-related manner. The present observations are consistent with prior reports but also demonstrate hypometabolism associated with minimal amyloid burden as well as an incremental dose relationship of the biomarkers in a population-based study that is generalizable to the population at large. In addition, we have shown that the association of hypometabolism and amyloid accumulation also occurs in a regional, individual ROI basis and is more significant in AD-signature regions.
Prior data published on hypometabolism in CN subjects have been inconsistent. For example, hypometabolism has been shown to occur in “at-risk” normal subjects through the spectrum of disease to subjects with AD in AD signature patterns (posterior cingulate, precuneus, parietal and temporal regions).12–17 Some reports have demonstrated that hypometabolism may be more closely associated with APOE status than with amyloid deposition.18 Others have described increased metabolism associated with increased amyloid accumulation in AD vulnerable regions (e.g., posterior cingulum and precuneus) in normal subjects and in subjects with mild cognitive impairment. They have suggested that hypermetabolism may be the initial metabolic perturbation on the AD neurodegenerative cascade.19,20 The finding of increased metabolism is difficult to reconcile given other data showing that typical AD patterns of hypometabolism occur in high-risk normal subjects even at young ages and that hypometabolism in normal subjects has been shown to be a predictor of progression to AD in pathologically verified AD cases.21–23 We were unable to identify any consistent hypermetabolic associations in our population on our ROI analysis. Some of the discrepant findings between these prior studies showing hypermetabolic associations and the present work could possibly be attributable to increased noise introduced by a limited number of subjects. None of the previous work had more than 52 normal subjects included.
This work is limited by the fact that the eventual disease outcome of the normal subjects is unknown. Amyloid-positive normal subjects may die without any dementia or they may develop different neurodegenerative diseases.24,25 Another weakness of this work is that by defining our “negative” PiB population as those with an SUVR of 1.4, we are limited in our ability to assess hypometabolism that might occur already in this group. In addition, this work is not intended to define or validate a PiB cutoff. Unraveling these issues will require longitudinal study to provide disease outcome and autopsy correlation.
Prior investigations have demonstrated subtle but detectable hypometabolism in AD-signature regions in CN very young subjects (20–39 years) who are APOE ε4 carriers when they may be unlikely to have amyloid accumulation, although amyloid status was not documented in this group.23 In small samples, others have not been able to identify amyloid-positive scans in similar young subjects in their 30s, supporting the idea that amyloid accumulation may be a later event.26 In addition, in APOE ε4 carriers, amyloid accumulation appears to be related to gene dose with homozygotes demonstrating greater brain amyloid, suggesting that the ε4 allele may be driving amyloid accumulation27 and may be associated with hypometabolism. Our data cannot confirm the APOE ε4 gene dose effect because we had only 10 APOE homozygotes in our sample, but increasing frequency of APOE ε4 carrier status does occur with increasing PiB dose in our population (table 1). We likewise cannot confirm amyloid PET findings in the young. Nevertheless, these prior observations suggest that hypometabolism occurs in association with APOE ε4 carriage, possibly without, or before, amyloid accumulation. Our data show that the association of hypometabolism and brain amyloidosis is not contingent on APOE ε4 status in normal subjects. In fact, in the current study, the subjects with APOE ε4 do not have significantly more hypometabolism than those without, suggesting that in this elderly cohort, after accounting for amyloid load, APOE ε4 does not significantly worsen hypometabolism in the AD-signature brain regions in this population.
The previous findings of hypometabolism from young APOE ε4 carrier populations and our aging population are not discrepant but complementary and thought-provoking in our view. We have demonstrated that APOE ε4 carriers demonstrate hypometabolism closely linked with amyloid accumulation and that hypometabolism is associated with PiB positivity in APOE ε4 noncarriers that is similar to that seen in carriers. Much of the association we see between FDG and APOE ε4 is attributable to the relationship between PiB and APOE ε4. We interpret these data to likely mean that APOE ε4 status alone can induce hypometabolism, and that amyloid deposition alone can induce similar hypometabolism, especially in AD-signature cortical regions. Any potentiation of prior APOE ε4–related, nonamyloid-induced hypometabolism with an entirely different mechanism could be possible but we do not have data to make that distinction. We see that the hypometabolic effect of amyloid clearly occurs independently of APOE ε4 positivity in APOE ε4–negative normal elderly subjects and speculate whether distinct mechanisms may be at work to induce the hypometabolism seen in young amyloid-negative, APOE ε4–positive vs elderly amyloid-positive, APOE ε4–positive subjects.
Previous basic science work on the mechanisms of hypometabolism and work in APOE ε4 interaction has shown that both amyloid and APOE ε4 can independently affect metabolism. In neuronal cell lines, amyloid accumulation alone can cause mitochondrial dysfunction with impaired energy metabolism.28 In addition, human amylin, amyloid protein deposited in the pancreas in patients with diabetes, also perturbs mitochondrial function.29 Likewise, APOE ε4 fragments alone have been associated with reduced expression of genes encoding for cytochrome oxidase leading to mitochondrial dysfunction with impaired oxidative metabolism as shown histochemically in the posterior cingulate.30,31 Direct effects of APOE on the endoplasmic reticulum could also lead to decreased glucose uptake in cells.32
Additional ideas relative to hypometabolism mechanisms associated with amyloid, which could be especially pertinent to our aging subjects, may be that chronic diseases seen frequently in aging and associated with AD can lead to mitochondrial dysfunction through amyloid deposition in target tissues. Examples of this include diabetes29,33 and hypertension.34 This could implicate an intermediary step whereby chronic disease–linked amyloid deposition induces metabolic abnormalities. This concept could add merit to a multifactorial cascade argument for AD in which any one or a combination of these diseases could be causative. Evaluations of each of these individual disease contributions to, and relationships with, early hypometabolism and amyloid deposition are ongoing avenues of investigation in our population.
In recent hypothetical models of AD biomarker pathology that we and others initially proposed describing the temporal evolution of AD biomarkers, the alterations in brain metabolism that occur on the AD cascade had been postulated to occur years after amyloid accumulation had reached a “pathologic” PiB (SUVR of 1.5) threshold.35,36 The present data support a closer association of hypometabolism and amyloid deposition at lower levels and help to validate our more recent hypothetical model.2 Our present data suggest that amyloid accumulation and hypometabolism are more likely to occur simultaneously throughout the spectrum of brain amyloid levels. Figure 4 illustrates this relationship by using the PiB threshold of 1.5 as the “positive” level, with coexisting hypometabolism shown as predicted by the data in the present work, and projects the change in PiB and FDG measurements over time based on our prior work determining the rates of change of PiB at 0.07 SUVR per year37 and FDG-PET (in PiB-positive subjects) at 0.017 SUVR per year38 in serially studied subjects in our population. We also incorporated the idea that hypometabolism continues to worsen throughout life in patients with AD (even while amyloid levels appear to plateau), as supported by prior work.39 Other authors have estimated that it takes 9 years for amyloid accumulation to progress from the normal SUVR of 1.2 (the average low PiB-PET value in their normal population) to an SUVR of 1.5 that is considered pathologic.4 In addition, we must consider that low amyloid (below 1.5) or very low hypometabolism may not be detectable with today's technology. Also, we have no data determining which occurs first at these lowest levels.
Figure 4. Theoretical relationship of brain amyloid load and hypometabolism in AD.
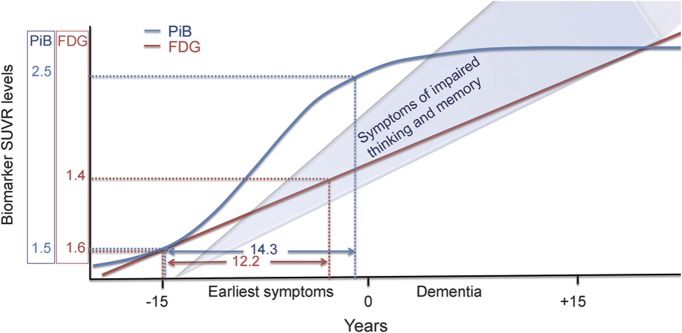
Theoretical dose relationship of brain amyloid load (blue SUVR) and hypometabolism (red SUVR) predicted in normal subjects from these data. The biomarker relationships are estimated for relative to future disease status categorizations as “early symptoms” and “dementia” on the x-axis timeline. The SUVR projections are based on the initial SUVR relationships starting at a PiB SUVR of 1.5 and thereafter projected based on previously determined yearly change rates for PiB and FDG SUVR values in this population. AD = Alzheimer disease; FDG = [18F]-fluorodeoxyglucose; PiB = Pittsburgh compound B; SUVR = standardized uptake value ratio.
This discussion is made more interesting by recent data demonstrating that some level of amyloid plaque burden likely precedes the development of toxic Aβ oligomers that lead to neurotoxicity. A recent report claimed that once a small but critical concentration of amyloid fibrils has accumulated, toxic oligomeric species are predominantly formed from monomeric peptide molecules through a fibril-catalyzed secondary nucleation reaction, rather than through a classic mechanism of homogeneous primary nucleation.40 This is in contrast to the theory that Aβ oligomers form first and then form fibrils. We can hypothesize from these data and our PET imaging data that a pathologic threshold of amyloid fibril accumulation, sufficient to catalyze formation of toxic oligomeric Aβ, is occurring at a PiB SUVR of at least 1.5. Left open to question is whether minimal levels of amyloid lower than 1.5 exert any neurotoxic effect seen as hypometabolism or otherwise, and requires more longitudinal and serial individual data to resolve.
Intervention trials in the normal elderly may benefit from using these data to target therapy to those with amyloid and hypometabolism and in that way have the greatest potential for treating a “functionalized” AD cascade. The corollary to our observations in the aging population and treatment trials is that treatment of AD may need to be individualized in patients depending on the primary cause of neurodegeneration. Diabetes or hypertension may also need to be managed aggressively and monitored in intervention trials. Consideration of these possibilities may enhance the chances of success of future clinical trials.
Supplementary Material
GLOSSARY
- AD
Alzheimer disease
- CN
cognitively normal
- FDG
[18F]-fluorodeoxyglucose
- FDR
false discovery rate
- PiB
Pittsburgh compound B
- ROI
region of interest
- SUVR
standardized uptake value ratio
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Lowe designed the study, took part in data collection, performed and supervised all analyses, generated the first and final drafts, and takes overall responsibility for the data and the manuscript. Mr. Weigand performed data analyses and critically reviewed the manuscript. Mr. Senjem and Dr. Vemuri performed analyses of imaging data and critically reviewed the manuscript. Mr. Jordan acquired image data, performed analyses, and critically reviewed the manuscript. Dr. Kantarci, Dr. Boeve, Dr. Jack, Dr. Knopman, and Dr. Petersen took part in data collection and critically reviewed the manuscript.
STUDY FUNDING
Supported by NIH grants P50 AG16574, U01 AG06786, R01 AG11378, and RO1 AG041851, the Elsie and Marvin Dekelboum Family Foundation, and the Robert H. and Clarice Smith and Abigail Van Buren Alzheimer's Disease Research Program of the Mayo Foundation.
DISCLOSURE
V. Lowe is a consultant for Bayer Schering Pharma and receives research support from GE Healthcare, Siemens Molecular Imaging, AVID Radiopharmaceuticals, the NIH (National Institute on Aging [NIA], National Cancer Institute), the Elsie and Marvin Dekelboum Family Foundation, the MN Partnership for Biotechnology and Medical Genomics, and the Leukemia & Lymphoma Society. S. Weigand, M. Senjem, P. Vemuri, L. Jordan, and K. Kantarci report no disclosures relevant to the manuscript. B. Boeve serves as an investigator for clinical trials sponsored by GE Healthcare; receives royalties from the publication of a book titled Behavioral Neurology of Dementia (Cambridge Medicine, 2009); serves on the scientific advisory board of the Tau Consortium; and receives research support from the NIH and the Mangurian Foundation. C. Jack serves on scientific advisory boards for Siemens; receives research support from the NIH/NIA, and the Alexander Family Alzheimer's Disease Research Professorship of the Mayo Foundation; and holds stock in Johnson & Johnson. D. Knopman served on a data safety monitoring board for Lilly Pharmaceuticals; served as a consultant to TauRx, was an investigator in clinical trials sponsored by Baxter, Elan Pharmaceuticals, and Forest Pharmaceuticals in the past 2 years; and receives research support from the NIH. R. Petersen serves on scientific advisory boards for Pfizer, Inc., Janssen Alzheimer Immunotherapy, Elan Pharmaceuticals, and GE Healthcare; receives royalties from the publication of Mild Cognitive Impairment (Oxford University Press, 2003); and receives research support from the NIH/NIA. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bateman RJ, Xiong C, Benzinger TL, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 2012;367:795–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol 2013;12:207–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jack CR, Jr, Lowe VJ, Weigand SD, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer's disease: implications for sequence of pathological events in Alzheimer's disease. Brain 2009;132:1355–1365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Villemagne VL, Burnham S, Bourgeat P, et al. Amyloid beta deposition, neurodegeneration, and cognitive decline in sporadic Alzheimer's disease: a prospective cohort study. Lancet Neurol 2013;12:357–367 [DOI] [PubMed] [Google Scholar]
- 5.Roberts RO, Geda YE, Knopman DS, et al. The Mayo Clinic Study of Aging: design and sampling, participation, baseline measures and sample characteristics. Neuroepidemiology 2008;30:58–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lowe VJ, Kemp BJ, Jack CR, Jr, et al. Comparison of 18F-FDG and PiB PET in cognitive impairment. J Nucl Med 2009;50:878–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harrell FE. Regression Modeling Strategies: With Applications to Linear Models, Logistic Regression, and Survival Analysis. New York: Springer; 2001 [Google Scholar]
- 8.Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw 2010;33:1–22 [PMC free article] [PubMed] [Google Scholar]
- 9.Meng XL, Rosenthal R, Rubin DB. Comparing correlated correlation coefficients. Psychol Bull 1992;111:172–175 [Google Scholar]
- 10.Raghunathan ET, Rosenthal R, Rubin DB. Comparing correlated but nonoverlapping correlations. Psychol Methods 1996;1:178–183 [Google Scholar]
- 11.Jack CR, Jr, Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain 2008;131:665–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hoffman JM, Guze BH, Baxter LR, Mazziotta JC, Phelps ME. [18F]-Fluorodeoxyglucose (FDG) and positron emission tomography (PET) in aging and dementia: a decade of studies. Eur Neurol 1989;29(suppl 3):16–24 [DOI] [PubMed] [Google Scholar]
- 13.Herholz K. FDG PET and differential diagnosis of dementia. Alzheimer Dis Assoc Disord 1995;9:6–16 [DOI] [PubMed] [Google Scholar]
- 14.Higuchi M, Arai H, Nakagawa T, et al. Regional cerebral glucose utilization is modulated by the dosage of apolipoprotein E type 4 allele and alpha1-antichymotrypsin type A allele in Alzheimer's disease. Neuroreport 1997;8:2639–2643 [DOI] [PubMed] [Google Scholar]
- 15.Frey KA, Minoshima S, Kuhl DE. Neurochemical imaging of Alzheimer's disease and other degenerative dementias. Q J Nucl Med 1998;42:166–178 [PubMed] [Google Scholar]
- 16.Hoffman JM, Welsh-Bohmer KA, Hanson M, et al. FDG PET imaging in patients with pathologically verified dementia. J Nucl Med 2000;41:1920–1928 [PubMed] [Google Scholar]
- 17.De Leon MJ, Convit A, Wolf OT, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/positron-emission tomography (FDG/PET). Proc Natl Acad Sci USA 2001;98:10966–10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jagust WJ, Landau SM. Apolipoprotein E, not fibrillar beta-amyloid, reduces cerebral glucose metabolism in normal aging. J Neurosci 2012;32:18227–18233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen AD, Price JC, Weissfeld LA, et al. Basal cerebral metabolism may modulate the cognitive effects of Abeta in mild cognitive impairment: an example of brain reserve. J Neurosci 2009;29:14770–14778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oh H, Habeck C, Madison C, Jagust W. Covarying alterations in Abeta deposition, glucose metabolism, and gray matter volume in cognitively normal elderly. Hum Brain Mapp 2014;35:297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mosconi L, Mistur R, Switalski R, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer's disease. Eur J Nucl Med Mol Imaging 2009;36:811–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reiman EM, Caselli RJ, Yun LS, et al. Preclinical evidence of Alzheimer's disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. N Engl J Med 1996;334:752–758 [DOI] [PubMed] [Google Scholar]
- 23.Reiman EM, Chen K, Alexander GE, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer's dementia. Proc Natl Acad Sci USA 2004;101:284–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jack CR, Jr, Knopman DS, Weigand SD, et al. An operational approach to National Institute on Aging–Alzheimer's Association criteria for preclinical Alzheimer disease. Ann Neurol 2012;71:765–775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kantarci K, Lowe VJ, Boeve BF, et al. Multimodality imaging characteristics of dementia with Lewy bodies. Neurobiol Aging 2012;33:2091–2105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rodrigue KM, Kennedy KM, Devous MD, Sr, et al. Beta-amyloid burden in healthy aging: regional distribution and cognitive consequences. Neurology 2012;78:387–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiman EM, Chen K, Liu X, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer's disease. Proc Natl Acad Sci USA 2009;106:6820–6825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Anandatheerthavarada HK, Biswas G, Robin MA, Avadhani NG. Mitochondrial targeting and a novel transmembrane arrest of Alzheimer's amyloid precursor protein impairs mitochondrial function in neuronal cells. J Cell Biol 2003;161:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lim YA, Rhein V, Baysang G, et al. Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 2010;10:1621–1633 [DOI] [PubMed] [Google Scholar]
- 30.Chang S, ran Ma T, Miranda RD, Balestra ME, Mahley RW, Huang Y. Lipid- and receptor-binding regions of apolipoprotein E4 fragments act in concert to cause mitochondrial dysfunction and neurotoxicity. Proc Natl Acad Sci USA 2005;102:18694–18699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Valla J, Yaari R, Wolf AB, et al. Reduced posterior cingulate mitochondrial activity in expired young adult carriers of the APOE epsilon4 allele, the major late-onset Alzheimer's susceptibility gene. J Alzheimers Dis 2010;22:307–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhong N, Ramaswamy G, Weisgraber KH. Apolipoprotein E4 domain interaction induces endoplasmic reticulum stress and impairs astrocyte function. J Biol Chem 2009;284:27273–27280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma T, Du X, Pick JE, Sui G, Brownlee M, Klann E. Glucagon-like peptide-1 cleavage product GLP-1(9-36) amide rescues synaptic plasticity and memory deficits in Alzheimer's disease model mice. J Neurosci 2012;32:13701–13708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langbaum JB, Chen K, Launer LJ, et al. Blood pressure is associated with higher brain amyloid burden and lower glucose metabolism in healthy late middle-age persons. Neurobiol Aging 2012;33:827.e11–827.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jack CR, Jr, Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 2010;9:119–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Forster S, Yousefi BH, Wester HJ, et al. Quantitative longitudinal interrelationships between brain metabolism and amyloid deposition during a 2-year follow-up in patients with early Alzheimer's disease. Eur J Nucl Med Mol Imaging 2012;39:1927–1936 [DOI] [PubMed] [Google Scholar]
- 37.Jack CR, Jr, Wiste HJ, Lesnick TG, et al. Brain beta-amyloid load approaches a plateau. Neurology 2013;80:890–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Knopman DS, Jack CR, Jr, Wiste HJ, et al. Selective worsening of brain injury biomarker abnormalities in cognitively normal elderly persons with β-amyloidosis. JAMA Neurol 2013;70:1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Engler H, Forsberg A, Almkvist O, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer's disease. Brain 2006;129:2856–2866 [DOI] [PubMed] [Google Scholar]
- 40.Cohen SI, Linse S, Luheshi LM, et al. Proliferation of amyloid-beta42 aggregates occurs through a secondary nucleation mechanism. Proc Natl Acad Sci USA 2013;110:9758–9763 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.