

Abstract
X-ray crystallography and solution NMR of detergent-reconstituted OmpA (outer membrane protein A from E. coli) had shown that this protein forms an 8-stranded transmembrane β-barrel, but only limited information was obtained for the extracellular loops. In NMR studies of OmpA in two different detergent micelles, “NMR-invisible” amino acid residues in-between the extracellular loops and the β-barrel prevented complete structural characterization. Here, we show that this NMR-invisible ring around the β-barrel of OmpA is present also in lipid bilayer nanodiscs and in mixed micelles with a third detergent, suggesting that the implicated rate processes have a functional role rather than representing an artifact of the protein reconstitution. In addition to sequence-specific NMR assignments for OmpA in the nanodiscs, the present results are based on a protocol of micro-coil TROSY- and CRINEPT-type NMR diffusion measurements for studying the hydrodynamic properties and the foldedness of [2H,15N]-labeled membrane proteins in nanodiscs, which can be applied under closely similar conditions to those used for NMR structure determinations or crystallization trials.
Keywords: Membrane proteins, structural biology, NMR spectroscopy, membrane protein reconstitution, nanodiscs
Introduction
The outer membrane protein A (OmpA) is an abundant integral membrane protein (IMP) in E. coli. It is a key factor for bacterial survival and virulence,[1,2] and it also plays a crucial role in E. coli infections by mediating cell-adhesion,[3] host invasion,[4] biofilm formation[5] and immune evasion.[2,6] OmpA consists of an N-terminal transmembrane (TM) domain and a C-terminal periplasmic domain. Mutagenesis showed that most of the functions of OmpA are to be assigned to the TM domain,[7] for which structure determinations have been reported by X-ray crystallography[8,9] and solution NMR.[10,11] Both methods determined an 8-stranded TM β-barrel, but the structure of the extracellular portion of OmpA has been evasive. No electron density was seen for most of the extracellular loops in the crystal structures,[8,9] and “NMR-invisible” amino acid residues at the interface of the TM-barrel and the extracellular loops made it impossible to establish links between the loops and the β-strands in structure determinations by NMR.[10,11] Slow conformational dynamics seems to be the reason why part of the NMR signals are broadened beyond detection. The influence on these implicated rate processes by different membrane-mimicking systems for OmpA reconstitution has attracted much interest, in part because MD-simulations suggested different OmpA dynamics in detergent micelles and in lipid bilayers.[12] Here we describe studies of OmpA in lipid nanodiscs and in a novel detergent.
Lipo-protein nanodiscs[13] are discoidal particles of about 10 nm diameter, which consist of a central lipid bilayer held in place by association with a membrane scaffold protein (MSP).[14] Nanodiscs have previously been applied for solution NMR characterization of CD4,[15] VDAC-1[16] and KvAP,[17,18] and smaller nanodiscs with shortened MSP variants were applied for a NMR structure determination of OmpX.[19] Here, we explored effects of the detergent-free environment in 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) nanodiscs on the structure and dynamics of OmpA, using newly recorded corresponding measurements with Fos-10-reconstituted OmpA and literature data on studies with different detergents[8–11] as a reference. To this end, micro-coil NMR diffusion measurements[20–22,28] were used, and enzymatic modification of OmpA in the nanodiscs has been implemented.
Results
NMR diffusion measurements used to monitor OmpA NMR sample optimization
The quality of NMR data recorded in solutions of reconstituted membrane proteins depends critically on the solution conditions, as illustrated in Figure 1.[20,23] Here, following previous work with membrane proteins in detergent micelles,[20,21] we used a micro-scale NMR protocol based on TROSY and CRINEPT principles[24,25] to study the impact of varying pH, temperature and salt concentration on the foldedness of [2H,15N]-labeled OmpA in nanodiscs and on the hydrodynamic properties of the protein-containing nanodiscs (Table 1). To monitor OmpA reconstitution, we acquired 2D [15N,1H]-CRINEPT-HMQC-[1H]-TROSY[25] and 2D [15N,1H]-TROSY[24] spectra of OmpA/Fos-10 and OmpA/nanodisc samples The close similarity of the spectra in Figure 1A, C shows that both samples contained intact, OmpA-loaded nanodiscs (for all these experiments, the OmpA-loaded nanodiscs were taken from the same homogeneous stock solution (Fig. 1E), and the presence of intact nanodiscs was independently also evidenced by diffusion measurements; see below). The quality of the TROSY-type experiments, which was the technique used for obtaining NMR assignments of OmpA, changed quite dramatically between different solution conditions (Figure 1B, D). The hydrodynamic properties of the OmpA solutions were investigated by measuring rotational and translational diffusion coefficients, Dr and Dt (Table 1). 1H-TRO-STE experiments[26] were used to determine the translational diffusion coefficients based on NMR observation of both the protein and the lipid or detergent signals (Figures S1 and S2). For OmpA/Fos-10 micelles, different diffusion rates were obtained from the protein and detergent signals. This is due to averaging of the detergent signals with those of Fos-10 monomers and empty Fos-10 micelles, which are smaller in size and thus diffuse faster than OmpA-containing Fos-10 micelles,[22] whereas the protein signals report exclusively on the diffusion of OmpA-containing micelles. In contrast, the lipid and protein Dt values of DMPC nanodisc samples are identical within experimental error, both for OmpA-containing and “empty” nanodiscs (Table 1). This observation can be rationalized by current models of nanodisc-reconstituted IMPs, which assume that the solutions contain only very small populations of free lipid.[14] Combining 1D [15N,1H]-TRACT[27] measurements of rotational diffusion coefficients with the translational diffusion data allowed us to determine the hydrodynamic radii, Rh, for all samples which contained 15N-labeled protein (Table 1). For OmpA/Fos-10, the hydrodynamic radius was 3.0 nm, which is in agreement with values for other Fos-10 reconstituted outer membrane proteins of comparable size.[21,28] The OmpA/DMPC nanodisc preparations showed significantly larger hydrodynamic radii of about 4.8 nm, which is in close agreement with the values for MSP1D1(−)-nanodiscs.[14]
Figure 1.

Optimization of the solution conditions for [80%-2H,u-15N]-OmpA in DMPC nanodiscs monitored by 700 MHz 1.7 mm micro-coil NMR. A) and B): Initial solution: 0.3 mM OmpA in DMPC nanodiscs, 50 mM HEPES at pH 7.5, 150 mM NaCl, T = 298 K. A) 2D [15N,1H]-CRINEPT-HMQC-[1H]-TROSY spectrum,[25] 1024 scans, 26 h measurement time. B) 2D [15N,1H]-TROSY spectrum,[24] 256 scans, 16 h measurement time. C) and D): Optimized solution: 0.5 mM OmpA in DMPC nanodiscs, 20 mM sodium phosphate at pH 6.0, 50 mM NaCl, T = 313 K. C) 2D [15N,1H]-CRINEPT-HMQC-[1H]-TROSY spectrum,[25] 512 scans, 13 h measurement time. D) 2D [15N,1H]-TROSY spectrum,[24] 256 scans, 16 h measurement time. Acquisition and processing parameters: A) and C): data size 128 (t1) × 2,048 (t2) complex points; t1max = 22.6 ms; t2max = 86.0 ms. B) and D): 200 (t1) × 2,048 (t2) complex points; t1max = 35.2 ms; t2max = 86.0 ms. Before Fourier transformation the time domain data matrices were multiplied with an exponential window function in the acquisition dimension, and with a 90°-shifted sine bell window [39] in the indirect dimension. E) Size exclusion chromatogram of the stock solution of [80%-2H,u-15N]-His6-TEV-OmpA in DMPC nanodiscs, from which 50 μl aliquots were taken to prepare the samples used for the 1.7 mm micro-coil NMR measurements.
Table 1.
NMR diffusion measurements with [80%-2H,u-15N]-OmpA in DMPC nanodiscs and Fos-10 micelles, T = 313 K.
| Sample | Dt (10−11 m2 s−2)[a] | Dr (106 s−1)[b] | Rh (nm)[c] |
|---|---|---|---|
| 0.5 mM OmpA/nanodiscs before TEV-cleavage | 6.0 ± 0.2 (proteins)[d] 6.3 ± 0.2 (lipid) |
2.0 ± 0.2 | 4.8 ± 0.1 |
| 0.3 mM OmpA/nanodiscs after TEV-cleavage | 6.9 ± 0.2 (proteins)[d] 7.1 ± 0.2 (lipid) |
2.4 ± 0.2 | 4.7 ± 0.1 |
| 0.4 mM ‘empty’ nanodiscs | 6.6 ± 0.2 (scaffold protein) 6.3 ± 0.2 (lipid) |
- | - |
| 1.0 mM OmpA/Fos-10 before TEV-cleavage | 8.6 ± 0.2 (protein) 15.1 ± 0.2 (detergent) [e] |
7.3 ± 0.2 | 3.0 ± 0.1 |
Determined using 1H-TRO-STE NMR experiments.[26] Self-diffusion constants were obtained independently from measurements of the integral over the protein signals between 7.7 and 9.0 ppm (Figure S1), and of the integrals of three lipid or detergent resonances, respectively, between 0.0 and 2.0 ppm (Figure S2).
Determined with the 1D TRACT NMR experiment.[27]
Rh = (3Dt/4Dr)1/2
In the OmpA nanodiscs the contributions from the unlabeled scaffold protein and the 2H,15N-labeled OmpA are superimposed, but could be separated with the use of a 15N-filter. Within the experimental accuracy, observation of the individual protein components and their sum yielded identical results.
OmpA solutions for structural studies by NMR
Solutions of [80%-2H,u-13C,u-15N]-OmpA reconstituted in Fos-10 detergent micelles (OmpA/Fos-10) were prepared for the NMR experiments. The OmpA/Fos-10 NMR sample used for obtaining NMR assignments contained 1 mM OmpA, 150 mM Fos-10 in 5 mM sodium phosphate at pH 6.8, 10 mM NaCl and 10% D2O.
The preparation of an OmpA/nanodisc sample for NMR assignment with TROSY-type triple resonance experiments was based on the results of optimizing the solution conditions, as described above and in the Experimental Section (Figure 1, Table S1). The OmpA/nanodisc sample used for the structural studies contained 0.7 mM OmpA, 1.4 mM scaffold protein (MSP1D1(−)), 120 mM DMPC in 20 mM sodium phosphate at pH 6.0, 50 mM NaCl and 10% D2O.
Polypeptide backbone NMR assignment of OmpA in DMPC-nanodiscs and in Fos-10 micelles
The 2D [15N,1H]-TROSY correlation maps showed well-dispersed sets of resonances for both, the OmpA/Fos-10 and OmpA/nanodisc samples (Figure 2). Comparison with earlier studies of OmpA reconstituted in different detergents[10,11] indicated that the spectra in the new environments manifest a β-barrel fold. Similar to the previous studies, [10,11] only about 130 of the 167 expected backbone amide resonances were observed in the new preparations.
Figure 2.

2D [15N,1H]-TROSY correlation NMR spectra of [80%-2H,u-13C,u-15N]-OmpA at T = 313 K recorded in standard 5 mm sample tubes at 800 MHz. A) 1.0 mM OmpA in Fos-10 micelles. B) 0.7 mM OmpA in DMPC nanodiscs. Acquisition and processing parameters: data size 256 (t1) × 2,048 (t2) complex points; t1max = 45.1 ms; t2max = 71.0 ms; 16 (A) and 64 (B) scans per t1-increment; overall measurement times 1 h (A) and 5 h (B). Identical data handling as in Figure 1. Apparent differences, specifically for the Trp indole resonances at 1H chemical shifts between 10 and 11 ppm, are due to a combination of two factors. Firstly, the signal intensities in panel B are lower than the corresponding lines in panel A. Secondly, differences are to be attributed to the different environments of OmpA in the Fos-10 micelles and the DMPC nanodiscs
For a detailed comparison of OmpA in the different environments, we obtained sequence-specific resonance assignments in Fos-10 micelles and in DMPC nanodiscs. TROSY-type HNCA, HN(CO)CA, ct-HNCA, HNCACB, HNCO and HN(CA)CO experiments[29,30] were recorded with the OmpA/Fos-10 sample (Figure 3A), which yielded assignments for 60% of the polypeptide backbone (Figure 4). These assignments were transferred to the OmpA/nanodisc sample by comparison of 2D [15N,1H]-TROSY and 3D [15N,1H]-TROSY-HNCA data sets (Figures 2 and 3). These tentative assignments were validated with the use of a 3D [1H,1H]-NOESY-[15N,1H]-TROSY spectrum recorded for the OmpA/nanodisc sample. Following this strategy, we were able to obtain polypeptide backbone assignments to nearly the same extent in nanodiscs as in Fos-10 micelles (Fig. 4). In addition to enabling these polypeptide backbone resonance assignments, the NMR data provided evidence that OmpA in Fos-10 micelles as well as in DMPC nanodiscs has the same β-barrel structure as was previously observed in crystals[9] and in solution after reconstitution with different detergents.[10,11] Thus, the assigned residues have similar chemical shifts in the nanodiscs and in Fos-10 micelles, with only 5 residues having values of Δδave = [(Δδ1H)2 + (Δδ15N/5)2 + (Δδ13C/3)2]1/2 slightly larger than 0.3 ppm (Figure S3). Furthermore, the 3D [1H,1H]-NOESY-[15N,1H]-TROSY spectra (Figure S4) show long-range NOEs between neighboring β-strands which are compatible with short distances between the corresponding hydrogen atoms in the previously reported structures.[9–11] It is then interesting to notice that in both the nanodiscs and the Fos-10 micelles, the signal intensities decreased toward the edges of the OmpA β-barrel, and residues with NMR-unobservable 15N–1H moieties formed a ring around the β-barrel on the extracellular side, i.e., at the interface of the β-barrel and the extracellular loops. This again coincides with what was previously seen for OmpA reconstituted in the detergents DPC[10] and DHPC.[11]Overall, the structure-related data obtained here with OmpA in DMPC nanodiscs match very closely the findings for OmpA reconstituted in three different detergent micelles, i.e., Fos-10, DPC[10] and DHPC.[11] Previously, similar chemical shifts have been reported for OmpX in lipid nanodiscs and in detergent micelles,[19] so that the studies with both OmpA and OmpX provide evidence for near-identical polypeptide backbone conformations of these β-barrel membrane proteins in detergent micelles and lipid nanodiscs.
Figure 3.

Polypeptide backbone assignments of [80%-2H,u-13C,u-15N]-OmpA using the 3D [15N,1H]-TROSY-HNCA NMR experiment. A) 1.0 mM OmpA in Fos-10 micelles. B) 0.7 mM OmpA in DMPC nanodiscs. [ω2(13C), ω3(1H)]-strips at the indicated 15N frequencies are shown. Sequence-specific resonance assignments are indicated at the top of each strip, and red lines indicate sequential connectivities. 70(t1) × 60(t2) × 2048(t3) complex points were accumulated, with t1max(15N) = 11.9, t2max(13C) = 4.7, t3max(1H) = 71.3 ms. 64 scans per increment were acquired, resulting in a total measuring time of 69 h per experiment.
Figure 4.

Crystal structure of OmpA (PDB-ID:1QJP) and survey of the polypeptide backbone NMR assignments obtained for OmpA in Fos-10 micelles and in DMPC nanodiscs. The ribbon representation shows the crystal structure to the extent that it was reported in the final refinement.[9] The dotted lines indicate the lengths of the extracellular loop segments which were not determined in the crystal structure. The color-code indicates the extent of the NMR assignments. Green: assignments obtained in both Fos-10 micelles and DMPC nanodiscs (68 residues); red: additional assignments obtained only in Fos-10 micelles (26 residues); blue: unassigned residues (73 residues). There remained 27 unassigned peaks in the 2D [15N,1H]-TROSY spectrum recorded with OmpA in Fos-10 micelles, and 41 unassigned peaks in the 2D [15N,1H]-TROSY spectrum recorded with OmpA in DMPC nanodiscs.
Discussion
This paper extends the structural characterization of the integral membrane protein OmpA from E. coli with studies using detergent-free lipid-bilayer nanodiscs as well as a new detergent. Sequence-specific NMR assignments for OmpA in DMPC nanodiscs and in Fos-10 micelles showed that OmpA adopts similar folds in both environments. This in turn enabled validation of earlier results obtained with OmpA reconstituted in the detergents DPC[10] and DHPC,[11] which also contained NMR-unobservable residues at the interface between the β-barrel and the extracellular loops. The coincidence of the data in three different detergents and the detergent-free nanodisc lipid-bilayer environment supports that the observed NMR line broadening at the interface between the β-barrel and the extracellular loops is due to function-related conformational dynamics of OmpA rather than to reconstitution artifacts.
A micro-coil NMR protocol for efficient characterization and optimization of IMP/nanodisc preparations under NMR solution conditions and with minimal sample volumes (≤50 μl)[21,22,28] was instrumental in obtaining ‘structure-grade’ samples of OmpA (Figure 1, Tables 1, S1). This approach should readily be applicable to other IMPs reconstituted in nanodiscs, which opens new avenues for studies of IMP-structures by solution NMR. The finding that OmpA/nanodiscs and “empty” nanodiscs exhibited nearly identical diffusion rates (Table 1) indicates that the overall size and the hydrodynamic properties of DMPC/nanodiscs are only slightly affected by the embedded IMP. Since the quality of solution NMR spectra is directly linked to the diffusion rates, there is an indication that high-quality spectra in nanodiscs (Figures 2 and 3) might be obtained also for larger IMPs than OmpA. Overall, the present paper shows that similar results are obtained in nanodiscs and in detergent micelles and thus supports that detergent micelles are a good environment for structural studies of β-barrel integral membrane proteins, and it also indicates applications of nanodisc reconstitution for this class of proteins in situations where conventional detergent-based systems fail to provide a suitable environment.
Advantages of nanodisc reconstitution for functional studies of IMPs have been impressively documented.[31–36] The presently used enzymatic cleavage of the His6-TEV-OmpA fusion construct in nanodiscs (Figure 5) now indicates a potential advantage of nanodisc reconstitution also for sample preparation in structural biology. While certain detergents, in particular Fos-10, are known to rapidly inactivate TEV protease,[37] TEV protease-processing of OmpA/nanodiscs was successful because the nanodiscs did not interfere with the functional protein–protein interaction.
Figure 5.
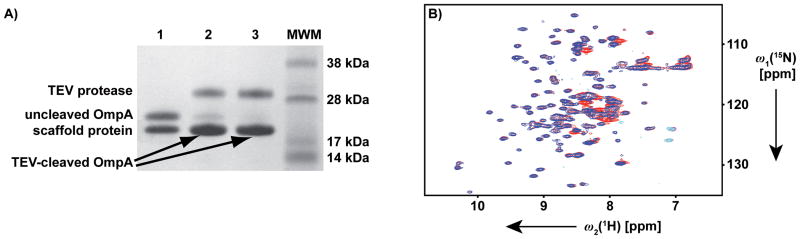
TEV protease cleavage of the N-terminal expression tag MGSSHHHHHHSSGENLYFQ in [80%-2H,u-15N]-His6-TEV-OmpA reconstituted in DMPC nanodiscs. A) SDS PAGE. 1: Reaction mixture before addition of the TEV protease; 2 and 3: Reaction mixture after incubation at 4°C for 24 h and 48 h, respectively, following the addition of the TEV protease; MWM: Molecular weight markers. Assignments of the protein bands are indicated on the left. Arrows highlight the position of cleaved OmpA, which is partially overlapped with the scaffold protein band. B) Overlay of the 2D [15N,1H]-TROSY correlation NMR spectra of OmpA in DMPC nanodiscs purified before (red) and after (blue) TEV protease cleavage for 48 h at 4 °C.
Experimental Section
Sample preparation
For reconstitution into Fos-10 detergent micelles, OmpA (residues 1–176) was expressed, purified, and refolded according to established protocols.[10,11,21,28] Purified OmpA/Fos-10 was exchanged into NMR buffer (5 mM sodium phosphate at pH 6.8, 10 mM NaCl) by repeated dilution/concentration cycles, using Amicon Ultra concentrators (Millipore) with a 10 kDa molecular weight cutoff. The final OmpA/Fos10 concentration was 1.0 mM.
A search for NMR-favorable conditions was started with a solution of OmpA/nanodiscs in 50 mM HEPES at pH 7.5 with 150 mM NaCl, which is a typical solvent for nanodisc preparations of membrane proteins.[36] NMR data thus obtained showed the formation of intact nanodiscs containing folded OmpA (Figure 1A), but the spectral quality of [15N,1H]-TROSY-correlation experiments (Figure 1C) was insufficient for structural studies. Based on experience with other β-barrel proteins[21,22,28] we therefore changed the solution conditions to improve the NMR properties of OmpA in nanodiscs. Specifically, we decreased the pH-value to 6.0, lowered the NaCl concentration to 50 mM, changed the buffering agent from HEPES to 20 mM sodium phosphate, and increased the temperature from 298K to 313K. This combination of changes of the sample conditions resulted in marked improvements of the NMR spectra (Figure 1B, D).
Based on the result of Figure 1D, we selected the following procedure to prepare the nanodisc sample for the structural studies. OmpA was subcloned into a modified pET28b vector (Novagen) that contained an N-terminal tobacco etch virus (TEV) protease-cleavable His6-purifaction tag (His6-TEV: MGSSHHHHHHSSGENLYFQG). Introducing an affinity tag was essential to enable efficient enrichment of OmpA-containing nanodiscs over ‘empty’ nanodiscs. Expression, purification and Fos-10 refolding of His6-TEV-OmpA was carried out as for untagged OmpA, to yield His6-TEV-OmpA/Fos-10. Subsequently, His6-TEV-OmpA/Fos-10 was mixed with membrane scaffold protein (MSP1D1(−)) and DMPC (Avanti Polar Lipids) in a molar ratio of 1:10:800, and incubated at room temperature for 1 h with gentle agitation. To initiate nanodisc assembly, one equivalent (w/v) of Amberlite XAD-2 resin (Sigma-Aldrich) was added, and the resulting suspension was shaken at room temperature for 15 h. After removal of the resin by filtration, His6-TEV-OmpA/nanodiscs were separated from ‘empty’ nanodiscs by immobilized metal affinity chromatography (IMAC) on an ÅKTA FPLC system (GE Healthcare), making use of the His6-purification tag on OmpA. In the thus enriched solution of OmpA-loaded nanodiscs, the purification tags were removed by addition of TEV protease, and the cleavage reaction was monitored by SDS PAGE and NMR spectroscopy (Figure 5). Complete cleavage was achieved within 48 hours at 4 °C. The reaction mixture was then subjected to reverse IMAC to further purity the “tag-free” OmpA/nanodiscs. Sample purity and monodispersity was confirmed by SDS-PAGE and analytical size exclusion chromatography (aSEC, Figure 1E). Prior to NMR measurements, the nanodisc preparations were exchanged into NMR buffer (20 mM sodium phosphate at pH 6.0, 50 mM NaCl) and concentrated using Amicon Ultra concentrators (Millipore) with a 10 kDa molecular weight cutoff. The final concentration of OmpA/nanodiscs was 0.7 mM.
NMR spectroscopy
NMR diffusion experiments were recorded on a Bruker DRX-700 spectrometer equipped with a 1.7 mm TXI microprobe (Bruker). Dr of the OmpA preparations were measured using the TRACT experiment.[27] Relaxation-weighted one-dimensional spectra were recorded in a two-dimensional manner, using a variable relaxation delay ranging from 1 to 32 ms. 15N-relaxation rates, Rα and Rβ, were obtained by fitting the integrals over the spectral region from 9.5 to 8.7 ppm (Figure S1) to a single-parameter exponential decay. Dt was obtained using transverse relaxation-optimized stimulated echo (1H-TRO-STE) spectroscopy.[26] Diffusion-weighed one-dimensional spectra were acquired in a two-dimensional fashion, using a pair of gradient pulses of 4.5 ms duration separated by a delay of 50 ms, with gradient strengths ranging from 1 to 50 G cm−1. The TRACT and 1H-TRO-STE data were analyzed with in-house TopSpin (Bruker) macros in combination with the program XMGRACE (http://plasma-gate.weizmann.ac.il).
3D TROSY-type HNCA, ct-HNCA and HNCACB experiments[29,30] were acquired on a Bruker Avance800 spectrometer equipped with a 5 mm TXI probe, and 3D TROSY-type HN(CO)CA, HNCO and HN(CA)CO experiments[29,30] were measured on a Bruker Avance 600 spectrometer with a 5 mm TCI cryoprobe. Details of the experimental setups are given in the figure captions. Spectra were processed using the software TopSpin 1.3, and analyzed with CARA[38] (http://cara.nmr.ch) to interactively obtain sequence-specific resonance assignments.
3D [1H,1H]-NOESY-[15N,1H]-TROSY datasets were recorded on a Bruker Avance800 spectrometer, using a mixing time of 100 ms and a sample temperature of 313 K. 300(t1) × 80(t2) × 2048(t3) complex points were accumulated, with t1max (1H) = 10.4, t2max (15N) = 14.1, t3max(1H) = 71.3 ms. 16 scans per increment were acquired, resulting in a total measuring time of 128 h.
Supplementary Material
Acknowledgments
This work was supported by the NIH Roadmap initiative grant P50 GM073197 for technology development and by the Skaggs Institute for Chemical Biology at The Scripps Research Institute. K.W. is the Cecil H. and Ida M. Green Professor of Structural Biology at The Scripps Research Institute. L.S. is supported by a Boehringer Ingelheim Fonds PhD Fellowship, and by the German Academic Exchange Service (DAAD).
References
- 1.Smith SG, Mahon V, Lambert MA, Fagan RP. FEMS Microbiol Lett. 2007;273:1–11. doi: 10.1111/j.1574-6968.2007.00778.x. [DOI] [PubMed] [Google Scholar]
- 2.Weiser JN, Gotschlich EC. Infect Immun. 1991;59:2252–2258. doi: 10.1128/iai.59.7.2252-2258.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Torres AG, Kaper JB. Infect Immun. 2003;71:4985–4995. doi: 10.1128/IAI.71.9.4985-4995.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prasadarao NV, Wass CA, Weiser JN, Stins MF, Huang SH, Kim KS. Infect Immun. 1996;64:146–153. doi: 10.1128/iai.64.1.146-153.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orme R, Douglas CW, Rimmer S, Webb M. Proteomics. 2006;6:4269–4277. doi: 10.1002/pmic.200600193. [DOI] [PubMed] [Google Scholar]
- 6.Prasadarao NV, Blom AM, Villoutreix BO, Linsangan LC. J Immunol. 2002;169:6352–6360. doi: 10.4049/jimmunol.169.11.6352. [DOI] [PubMed] [Google Scholar]
- 7.Bremer E, Cole ST, Hindennach I, Henning U, Beck E, Kurz C, Schaller H. Eur J Biochem. 1982;122:223–231. doi: 10.1111/j.1432-1033.1982.tb05870.x. [DOI] [PubMed] [Google Scholar]
- 8.Pautsch A, Schulz GE. Nat Struct Biol. 1998;5:1013–1017. doi: 10.1038/2983. [DOI] [PubMed] [Google Scholar]
- 9.Pautsch A, Schulz GE. J Mol Biol. 2000;298:273–282. doi: 10.1006/jmbi.2000.3671. [DOI] [PubMed] [Google Scholar]
- 10.Arora A, Abildgaard F, Bushweller JH, Tamm LK. Nat Struct Biol. 2001;8:334–338. doi: 10.1038/86214. [DOI] [PubMed] [Google Scholar]
- 11.Fernandez C, Hilty C, Bonjour S, Adeishvili K, Pervushin K, Wüthrich K. FEBS Lett. 2001;504:173–178. doi: 10.1016/s0014-5793(01)02742-9. [DOI] [PubMed] [Google Scholar]
- 12.Bond PJ, Sansom MS. J Mol Biol. 2003;329:1035–1053. doi: 10.1016/s0022-2836(03)00408-x. [DOI] [PubMed] [Google Scholar]
- 13.Bayburt TH, Sligar SG. FEBS Lett. 2010;584:1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Denisov IG, Grinkova YV, Lazarides AA, Sligar SG. J Am Chem Soc. 2004;126:3477–3487. doi: 10.1021/ja0393574. [DOI] [PubMed] [Google Scholar]
- 15.Gluck JM, Wittlich M, Feuerstein S, Hoffmann S, Willbold D, Koenig BW. J Am Chem Soc. 2009;131:12060–12061. doi: 10.1021/ja904897p. [DOI] [PubMed] [Google Scholar]
- 16.Raschle T, Hiller S, Yu TY, Rice AJ, Walz T, Wagner G. J Am Chem Soc. 2009;131:17777–17779. doi: 10.1021/ja907918r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shenkarev ZO, Paramonov AS, Lyukmanova EN, Shingarova LN, Yakimov SA, Dubinnyi MA, Chupin VV, Kirpichnikov MP, Blommers MJ, Arseniev AS. J Am Chem Soc. 2010;132:5630–5637. doi: 10.1021/ja909752r. [DOI] [PubMed] [Google Scholar]
- 18.Shenkarev ZO, Lyukmanova EN, Paramonov AS, Shingarova LN, Chupin VV, Kirpichnikov MP, Blommers MJ, Arseniev AS. J Am Chem Soc. 2010;132:5628–5629. doi: 10.1021/ja9097498. [DOI] [PubMed] [Google Scholar]
- 19.Hagn F, Etzkorn M, Raschle T, Wagner G. J Am Chem Soc. 2013;135:1919–1925. doi: 10.1021/ja310901f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, Horst R, Geralt M, Ma X, Hong WX, Finn MG, Stevens RC, Wüthrich K. J Am Chem Soc. 2008;130:7357–7363. doi: 10.1021/ja077863d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stanczak P, Horst R, Serrano P, Wüthrich K. J Am Chem Soc. 2009;131:18450–18456. doi: 10.1021/ja907842u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horst R, Stanczak P, Serrano P, Wüthrich K. J Phys Chem B. 2012;116:6775–6780. doi: 10.1021/jp212401w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horst R, Stanczak P, Stevens RC, Wüthrich K. Angew Chem Int Ed. 2013;52:331–335. doi: 10.1002/anie.201205474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pervushin K, Riek R, Wider G, Wüthrich K. Proc Natl Acad Sci U S A. 1997;94:12366–12371. doi: 10.1073/pnas.94.23.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Riek R, Wider G, Pervushin K, Wüthrich K. Proc Natl Acad Sci U S A. 1999;96:4918–4923. doi: 10.1073/pnas.96.9.4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horst R, Horwich AL, Wüthrich K. J Am Chem Soc. 2011;133:16354–16357. doi: 10.1021/ja206531c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee D, Hilty C, Wider G, Wüthrich K. J Magn Reson. 2006;178:72–76. doi: 10.1016/j.jmr.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 28.Stanczak P, Zhang Q, Horst R, Serrano P, Wüthrich K. J Biomol NMR. 2012;54:129–133. doi: 10.1007/s10858-012-9658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salzmann M, Pervushin K, Wider G, Senn H, Wüthrich K. Proc Natl Acad Sci U S A. 1998;95:13585–13590. doi: 10.1073/pnas.95.23.13585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Salzmann M, Wider G, Pervushin K, Senn H, Wüthrich K. J Am Chem Soc. 1999;121:844–848. [Google Scholar]
- 31.Boldog T, Grimme S, Li M, Sligar SG, Hazelbauer GL. Proc Natl Acad Sci U S A. 2006;103:11509–11514. doi: 10.1073/pnas.0604988103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alami M, Dalal K, Lelj-Garolla B, Sligar SG, Duong F. EMBO J. 2007;26:1995–2004. doi: 10.1038/sj.emboj.7601661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Whorton MR, Jastrzebska B, Park PS, Fotiadis D, Engel A, Palczewski K, Sunahara RK. J Biol Chem. 2008;283:4387–4394. doi: 10.1074/jbc.M703346200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mi LZ, Grey MJ, Nishida N, Walz T, Lu C, Springer TA. Biochemistry. 2008;47:10314–10323. doi: 10.1021/bi801006s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsukamoto H, Szundi I, Lewis JW, Farrens DL, Kliger DS. Biochemistry. 2011;50:5086–5091. doi: 10.1021/bi200391a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayburt TH, Vishnivetskiy SA, McLean MA, Morizumi T, Huang CC, Tesmer JJ, Ernst OP, Sligar SG, Gurevich VV. J Biol Chem. 2011;286:1420–1428. doi: 10.1074/jbc.M110.151043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lundback AK, van den Berg S, Hebert H, Berglund H, Eshaghi S. Anal Biochem. 2008;382:69–71. doi: 10.1016/j.ab.2008.07.018. [DOI] [PubMed] [Google Scholar]
- 38.Keller R. The Computer Aided Resonance Assignment Tutorial. Cantina Verlag; 2004. [Google Scholar]
- 39.Demarco A, Wüthrich K. J Magn Reson. 1976;24:201–204. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.