

Abstract
In an attempt to dissect the virulence mechanisms of Yersinia ruckeri two adjacent genes, yrpA and yrpB, encoding putative peptidases belonging to the U32 family, were analyzed. Similar genes, with the same genetic organization were identified in genomic analysis of human-pathogenic yersiniae. RT-PCR studies indicated that these genes form an operon in Y. ruckeri. Transcriptional studies using an yrpB::lacZY fusion showed high levels of expression of these genes in the presence of peptone in the culture medium, as well as under oxygen-limited conditions. These two factors had a synergic effect on gene induction when both were present simultaneously during bacterial incubation, which indicates the important role that environmental conditions in the fish gut can play in the regulation of specific genes. LD50 experiments using an yrpA insertional mutant strain demonstrated the participation of this gene in the virulence of Y. ruckeri.
Keywords: Yersinia ruckeri, protease genes, regulation, virulence
Introduction
The gram-negative bacterium Yersinia ruckeri is the etiological agent of enteric redmouth disease, which is globally distributed and mainly affects salmonid fish. This disease causes important economic losses in fish farms. It has been clearly established that the bacterium colonizes the gut,1 but few of the pathogenic mechanisms of Y. ruckeri have been described. The most noteworthy among these involve extracellular factors such as the Yrp1 protease2 and the YhlA hemolysin,3 a high-affinity zinc transporter,4 a response regulator of a two-component system5 and an operon involved in the uptake of cysteine.6
The proteolytic enzymes in bacteria can play a variety of roles: they can provide a source of amino acids by degrading extracellular proteins, intervene in regulation functions and contribute to virulence in pathogenic bacteria. In particular, proteolytic enzymes of Aeromonas salmonicida7 and Listonella anguillarum8 cause alteration in fish tissues and can be considered as virulence factors. However, this is not a general rule, since some proteolytic enzymes of bacteria causing disease in fish, such as the AspA of A. salmonicida9 and Fpp2 of Flavobacterium psychrophilum, are not involved in virulence.10 Therefore, the implication of this type of enzyme in virulence should be assessed in each particular case.
The U32 family of peptidases is a broad family of enzymes with an unknown structure and catalytic mechanism. In bacteria, the prototype enzyme of this family is the PrtC of Porphyromonas gingivalis.11 This is a calcium-dependent metalloprotease, capable of degrading type I collagen but not gelatin or synthetic collagen substrates.12 The U32 peptidases have been described in other pathogenic bacteria such as Proteus mirabilis,13 Helicobacter pylori,14 Salmonella enterica,15 and Aeromonas veronii.16 All of them are putative collagenases and it is generally accepted that they contribute to bacterial infection.11,14
In spite of being present in the three domains of life (MEROPS database) and having a connection with bacterial virulence, the U32 family of peptidases has received relatively little attention. The goal of the present study was to characterize two tandem U32 peptidase genes in Y. ruckeri, not only by analyzing the factors that induce their expression but also by determining their involvement in virulence.
Results
Identification and analysis of YrpA and YrpB encoding genes
The analysis of the sequence adjacent to the 3′ end of the gene iviX (cdsB), previously selected by in vivo expression technology,6,17 allowed the identification of two ORFs, in the same orientation and separated by 12 bp. A putative promoter sequence was identified at the 5′ end of the first ORF and a rho-independent transcription terminator at the 3′ position of the second one (Fig. 1).

Figure 1. Chromosomal arrangement of the region containing genes yrpA and yrpB in Y. ruckeri 150. Arrows indicate the direction of transcription. Putative −10 and −35 promoter and rho-independent terminator sequences are shown. Scp2 and cdsB genes encode for a sterol carrier protein and an l-cysteine desulfidase, respectively. The number of amino acids and the collagenase (COG0826) and U32 peptidase (cIo3113) domains of the products of both genes are shown in the lower part of the figure.
Nucleotide sequence comparison of the two genes by pair-wise BLASTN showed no identity between them. The first ORF consists of 996 bp and encodes a 331-amino-acid protein which exhibits sequence identity with hypothetical proteins of Klebsiella peumoniae (YP_002236432, 88%), Escherichia coli (NP_289734, 88%), and S. enterica (EHC45316, 88%) and with the protein STMproteaseA of P. mirabilis (AAC64577, 85%).13 The second ORF is an 879 bp locus which codes for a 292-amino-acid protein that shares a high sequence identity with hypothetical proteins of Yersinia pseudotuberculosis (YP_001722436, 84%), Yersinia pestis (EIS91064, 84%), Serratia plymuthica (EKF66684, 80%), Dickeya dadantii (YP_003332207, 79%), and Pectobacterium carotovorum (YP_003016184, 79%). Multiple alignments of amino acid sequences of the homologous Y. ruckeri YrpA and YrpB peptidases with human pathogenic yersiniae (Y. enterocolitica, Y. pestis, and Y. pseudotuberculosis) showed a high percentage identity ranging from 92–93% for the first ORF to 84–86% for the second one.
Both genes are present in a similar genetic organization in K. pneumoniae (KPN_03566 and KPN_03567), Enterobacter aerogenes (ST548_p3910 and ST548_p3911), Citrobacter koseri (CKO_04553 and CKO_04554), E. coli (EC042_3447 and EC042_3448), Klebsiella oxytoca (KOX_03610 and KOX_03615), Shigella flexneri (S3416 and S3417), and Shigella dysenteriae (SDY_3337 and SDY_3338). In addition to this, the cluster is also present in different species of the genus Yersinia, including Y. enterocolitica (YE0449 and YE0450), Y. pestis (YPN_0607 and YPN_0608), and Y. pseudotuberculosis (YPTB0494 and YPTB0495).
In silico analysis showed that both proteins include the domains COG0826 and cIo3113, corresponding to collagenases and peptidases type U3218 (Fig. 1). Both domains are present in the STM proteaseA of P. mirabilis13 and the PrtC collagenase of P. gingivalis.11,12 Based on these identities the loci were named yrpA and yrpB (Yersinia ruckeri protease A and B, respectively). Both the YrpA and YrpB proteins lack a signal peptide, according to the SignalP program. Comparison of the primary structure of YrpA and YrpB revealed a low shared protein identity, but indicated a conserved region with maximum identity of 26% from amino acid residues 96 to 181.
The phylogenetic tree reveals a clear relation between all the Yersinia species clustered within the tree for both YrpA and YrpB peptidases (Figs. S1 and S2). These clusters are also, in both cases, close to that constituted by the Serratia species (Figs. S1 and S2). Interestingly, all the Enterobacteria species harboring the genes are pathogens or opportunistic pathogens and all of them are obligate or facultative anaerobes.
RT-PCR analysis
The small size of the intergenic space and the absence of a putative promoter sequence upstream yrpB gene suggested that yrpA and yrpB might be transcribed as a single unit. RT-PCR analysis confirmed the prediction that both genes form an operon. The results obtained with this analysis are shown in Figure 2. A 743 bp fragment corresponding to the overlapping region of yrpA and yrpB genes was amplified when RNA from the parental strain was used, confirming that both genes are co-transcribed (Fig. 2).
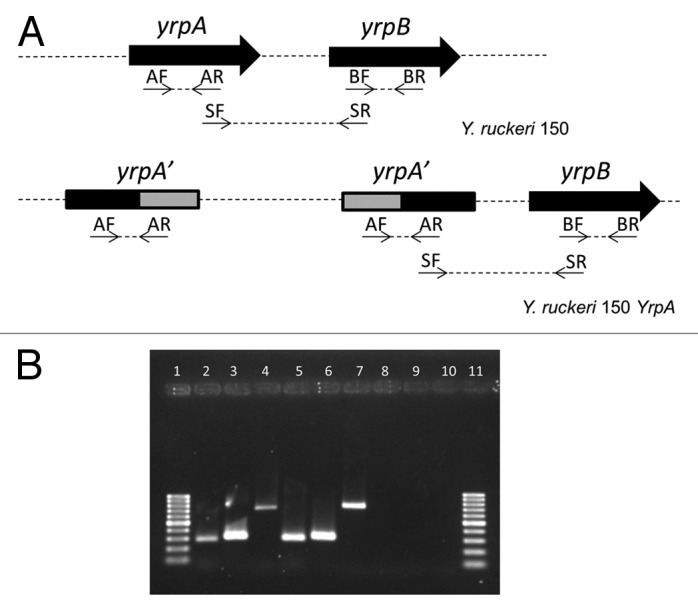
Figure 2. Agarose gel electrophoresis of the RT-PCR amplification products. (A) The positions of the primers used, within the yrpAB operon, are indicated both in the parental (Y. ruckeri 150) and in the yrpA mutant strains (Y. ruckeri 150 YrpA). Gray areas represent the internal fragment of the yrpA used to carry out the insertional mutagenesis of this gene. (B) RT-PCR results. Lanes 1 and 11: molecular weight marker corresponding to sizes ranging from 1000 to 100 bp. Lanes 2–4: PCR products obtained from RT-PCR experiments with Y. ruckeri 150 using primers: AF and AR (lane 2), BF and BR (lane 3), and SF and SR (lane 4). Lanes 5–7: PCR products obtained from RT-PCR experiments with Y. ruckeri yrpA; AF and AR primers (lane 5), BF and BR primers (lane 6), SF and SR (lane 7). Lanes 8 and 9: control reactions to assess DNA contamination in Y. ruckeri 150 RNA (lane 8) and Y. ruckeri yrpA RNA (lane 9). For this purpose SF and SR primers were used and the reverse transcription step was omitted. Lane 10: negative control of RT-PCR using 1 µL of water instead of RNA.
RT-PCR analysis achieved with total RNA from yrpA mutant indicated that the insertion of pJP5603 in this gen resulted in a non-polar mutation allowing expression of yrpB in this strain. Thus, when RNA of yrpA mutant was used as template to test if this mutation exerted a polar effect upon yrpB, an internal fragment of 306 bp from yrpB and the 743 pb fragment corresponding to the yrpA-yrpB overlapping region were amplified (Fig. 2). These results indicated that transcription was not interrupted by insertion of the pJP5603 plasmid. However, this mutagenesis approach leads to non-functional proteins because of the insertion of a plasmid in the middle of the coding sequence.
Regulation of the yrpAB operon
A transcriptional fusion between the yrpAB promoter and the lacZY genes from the pIVET8 plasmid was generated (Fig. 3). This construction was used to study how the expression of this operon is regulated in response to different environmental signals. The results obtained by β-galactosidase activity determination in cultures grown under different conditions showed that both peptone and microaerobic conditions (5–7% O2) exert an important influence on the transcription levels of yrpAB. Thus, the level of β-galactosidase activity was 666 ± 102 Miller units (MU) and 973 ± 150 MU when the cells were incubated in M9CG medium and M9CG supplemented with 5% (w/v) peptone, respectively. When casein was used, a similar result was obtained with values of 680 ± 107 MU (M9CG) and 1041 ± 130 MU (M9CG+5% w/v, casein). In the same way, when microaerobic conditions in M9CG medium were used during bacterial growth, a total of 1100 ± 132 MU of enzymatic activity were obtained in comparison to the 622 ± 107 MU found in the same medium but in aerobic conditions. Interestingly, when both conditions, the presence of 5% peptone and microareobiosis, existed simultaneously in the culture, a synergistic effect occurred, with values of 3168 ± 135 MU vs. 670 ± 108 MU. The presence in the M9CG medium of amino acids such as leucine, glutamine, and proline at 30 and 100 mM, and also that of NH4Cl at 0.22 M and glucose (0.5% w/v), did not cause any effect on the promoter activity. In the same way, the effect of environmental factors such as temperature (18 °C and 28 °C), osmotic stress and different pH (from 5 to 8), did not change the promoter expression.
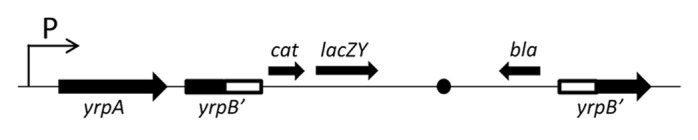
Figure 3. Chromosomal organization of the transcriptional fusion between the yrpAB operon and the lacZY genes in the ∆yrpB strain. Arrows indicate the direction of the transcription and white areas represent the internal fragment of the yrpB used to carry out the insertional mutagenesis of this gene. yrpB′, truncated yrpB gene; cat, chloramphenicol acetyltransferase gene (promoterless); lacZY, genes for lactose fermentation (promoterless); bla, ampicillin resistance gene; P, putative promoter. The black dot represents the origin of replication of the pJP5603 plasmid.
Phenotypic characterization of ΔyrpA and ΔyrpB strains
Two isogenic mutants in the yrpA and yrpB genes were obtained by insertional mutagenesis in order to determine the role of these genes in the bacterial physiology and virulence. Both mutants showed a growth curve similar to that of the parental strain in M9CG, M9CG containing 5% of peptone and CN media (data not shown).
When LD50 experiments were performed in order to define the involvement of this operon in virulence it was found that whereas the parental strain had a LD50 value of 1.73 × 102 colony forming units (CFU), the ∆yrpA strain LD50 was 7.1 × 104 CFU (Table 1).
Table 1. Median lethal doses of parental and ∆yrpA strains of Y. ruckeri.
| LD50 (CFU) 95% confidence limit | |||
|---|---|---|---|
| Bacterial strain | LD50 (CFU)a | Lower | Upper |
| Parental strain 150 | 1.73 × 102 | 0.18 × 102 | 6.78 × 102 |
| ∆yrpA | 7.1 × 104 | 5.7 × 103 | 3.9 × 106 |
a LD50 values were determined from data involving groups of 10 fish for each dilution containing between 10 and 107 CFU.
Discussion
The yrpA and yrpB genes encode two proteins whose domains and homologies indicate that they must be included in the U32 peptidase family of proteases, a group of enzymes whose catalytic center is still unknown. The prototype of these enzymes is the PrtC collagenase of P. gingivalis.11 However, YrpA shows only a 27% identity to PrtC, whereas it shares 85% identity with a protease of P. mirabilis belonging to the U32 family.13 These differences in identity indicate heterogeneity among the members of this family of proteases which are also present in gram-positive bacteria such as Streptococcus mutants19 and Streptococcus agalactiae.20 However, these two enzymes, YrpA and YrpB, exhibit a high degree of identity with other bacterial proteases from the enterobacteria group. For both proteases, the highest identity is shown with the homologous proteins of other Yersinia species, making up a cluster in each case, indicating they are conserved in the genus.
Nucleotide sequence comparison between yrpA and yrpB genes showed no homology, showing that the presence of these genes in the bacteria is not a duplication phenomenon.
In silico analysis postulated the absence in both YrpA and YrpB of signal peptides and suggested localization in the bacterial cytoplasm. This result is in agreement with what was found when the proteases of P. gingivalis11,12 and P. mirabilis13 were examined. However, the deduced cytoplasmic localization of all these enzymes does not match the results found by Kato et al.11 and Zhao et al.13 These authors found that the proteases were secreted into the milieu. This was also the case of a collagenase of Helicobacter pylori belonging to the U32 family.14 This paradox implies the absence of agreement between the in silico analysis and the results found in experimentation. This could be due to the presence of a specific and particular secretion system for this family of proteases, as was suggested by Kavermann et al.14 for the H. pylori collagenase.
The presence of casein and casein peptone in the culture media was able to induce the yrpAB operon but casamino acids did not have the same effect. According to Keil-Dlouha et al.,21 this induction could be dependent on the presence of the peptide bond in the substrate, together with the tertiary structure of the fragments.
More interesting was operon induction under microaerobic conditions. All bacteria possessing an operon homologous to the yrpAB were anaerobic or facultatively anaerobic. Interestingly, this operon is situated next to the cdsAB operon in the genome of Y. ruckeri, and this is also induced under oxygen limitation.6 This low oxygen environment is a constant in the gut of many animals22 and seems to be a trigger for the expression of some virulence genes in pathogenic bacteria colonizing the gut such as V. cholerae23 and Shigella,24 although the mechanisms involved in this regulation have not been defined yet. Taking into account that the gut of the rainbow trout is the main organ affected during the Y. ruckeri infection,1 together with the presence of a microaerobic environment in this organ, it is more than likely that the peptidases YrpA and YrpB are induced specifically in this tissue. The fact that a synergic effect on operon induction was found when both environmental conditions, the presence in the culture media of peptone and microaerobic atmosphere were simultaneously established, indicate a complex regulation mechanism with different elements involved in its expression.
Generally, most bacterial proteases are negatively regulated by glucose and ammonium,25,26 and as one would expect, the Yrp1 protease of Y. ruckeri was also repressed by these compounds.27 The fact that they had no effect on the yrpAB operon activity indicates that it is not under a regulation system similar to that of other bacterial proteases and suggests that it could play an additional function apart from a nutritional one, or even have a specific role during the infection process. This is also supported by the absence of differences in growth between the parental and the mutant strains in M9CG media containing peptone. A differential rate of growth would be expected if the yrpAB operon were involved in peptone degradation.
The LD50 experiments confirmed that at least one of these peptidases is involved in the infection process, since the ∆yrpA strain was attenuated in its virulence. This characteristic is also shared by the U32 peptidases of P. mirabilis13 and H. pylori,14 which are considered essential virulence factors for gut and gastric colonization, respectively. The presence of these genes in the genome of Yersinia species that are important from a public health point of view, such as Y. pestis, Y. pesudotuberculosis, and Y. enterocolitica, makes their study especially interesting.
Material and Methods
Bacterial strains, plasmids, and culture conditions
E. coli strains were routinely grown in 2× TY broth and agar, and Y. ruckeri strains in nutrient broth (NB) and on nutrient agar (NA) and M9 medium supplemented with 0.5% glucose (Scharlau Chemie S.A.) and 0.2% Casamino Acids (Becton, Dickinson and Company) (M9CG). Liquid cultures were incubated at 37 °C for E. coli and 18 °C and 28 °C for Y. ruckeri in orbital shakers at 250 rpm. Growth was monitored by determining the OD600. When required, the following compounds were added to the media: 10 µg/mL erythromycin, 50 µg/mL kanamycin, or 100 µg/mL ampicillin, all from Sigma-Aldrich Co. For incubation under microaerobic conditions, 250-mL Erlenmeyer flasks containing 20 mL of medium were inoculated with the bacteria and incubated in a 2.5-L anaerobic jar containing the Anaerocult C system (Merck), used to generate an oxygen depleted (5–7% v/v) and CO2-enriched (8–10% v/v) atmosphere.
Genetic techniques
Routine DNA manipulation was performed by standard procedures. Phage T4 DNA ligase and calf intestinal alkaline phosphatase were purchased from Roche Ltd., restriction enzymes were from Takara Bio Inc. and oligonucleotides were from Sigma-Aldrich Co.
DNA sequencing was performed by the dideoxy chain termination method with the BigDye Terminator version 3.1 (Applied Biosystems) according to the manufacturer’s instructions in an ABI Prism 3130xl DNA sequencer at the University of Oviedo. To obtain the complete sequence of the yrpAB operon adjacent to the cdsB gene,6 genomic DNA from the cdsB mutant was isolated and digested with KpnI. The restriction fragments were re-joined and used to transform cells of E. coli S17–1λpir by electroporation. Transformants were selected on 2 × TY agar medium containing kanamycin. Plasmid DNA was obtained and the DNA fragment situated downstream of the cdsAB operon was sequenced using a primer based on the known sequence of the cdsB gene. Based on the obtained sequence new primers were designed to complete the yrpAB operon sequence. Since the KpnI restriction fragment obtained did not contain the complete sequence of the yrpAB operon the genomic DNA of the cdsB mutant was also digested with SacI. The obtained fragments were re-ligated and transformed as explained above. Sequencing of the DNA inserted into the plasmid DNA from the selected transformants allowed the completion of the yrpAB operon sequence.
Construction of yrpA and yrpB mutants
Internal fragments of 446 and 402 bp of the predicted yrpA and yrpB loci, respectively, were amplified by PCR with the following primers: forward primer yrpA-E (5′-CTGAGAATTC GATGCTCTGA TTCTGGC), with nucleotides 268 to 284 of the yrpA gene in bold type, and reverse primer yrpA-S (5′-TGTAGTCGAC ACCAGATAGC GGCCTTT-3′), with nucleotides 713 to 697 in bold type, to amplify the internal fragment of the yrpA gene; forward primer yrpB-a (5′-AGGAGGATCC GAATTCTTGT TGGAAGC-3′), with nucleotides 265 to 281 in bold type, and reverse primer yrpB-b (5′-ATGCGGATCC GGTCTGAATA CCATTAA-3′), with nucleotides 666 to 650 in bold type, to amplify the internal fragment of yrpB. Primers contained restriction sites for EcoRI (yrpA-E), SalI (yrpA-S), and BamHI (yrpB-a and yrpB-b), in italics, and four additional bases at their 5′ end. The amplified internal fragment from yrpA was digested with EcoRI and SalI and ligated into pJP5603,29 previously digested with the same enzymes. The amplified internal fragment from yrpB was digested with BamHI and ligated into pIVET8,30 previously digested with BglII and dephosphorylated. The ligation mixtures were used to transform electrocompetent cells of E. coli S17–1λpir.28 Selected transformants, containing the plasmid with the insert, were used to conjugate with Y. ruckeri 150 to obtain the Y. ruckeri ∆yrpA (yrpA::pJP5603) and Y. ruckeri ∆yrpB (yrpB::pIVET8) mutants, as previously described.2 The mutations were confirmed by Southern blot analysis after digestion of the parental and mutant strain genomic DNA with EcoRI and BglII (yrpA), and BamHI and PstI (yrpB). The previously amplified internal fragments from yrpA and yrpB were used as probes. Probe labeling, hybridization, and developing were performed with the DIG DNA labeling and detection kit from Roche, following the manufacturer’s instructions.
RT-PCR
Total RNA was obtained from 1-mL late-exponential-phase cultures of parental strain 150 and mutant 150 yrpA grown in M9GC supplemented with peptone 0.5% (w/v) at 18 °C under microaerobic conditions as it was described above. RNA was isolated by using a High Pure RNA Isolation Kit (Roche) according to the manufacturer’s instructions and it was treated with RNase-free DNase (Ambion) to eliminate traces of DNA. Reverse transcription-PCR (RT-PCR) analyses were performed by using Superscript One-Step with Platinum Taq (Invitrogen Life Technologies). Control PCRs omitting reverse transcription step were performed to determine whether RNA was free of contaminant DNA. The primers used are listed in Table 2.
Table 2. Primers used for RT-PCR.
| Oligonucleotide | Sequence (5′-3′) | Position (nt) | Gene |
|---|---|---|---|
| AF | CCTCTGCAAC TAATGATGAA G | 353–373 | yrpA |
| AR | TCATTCAGAC GAGATTCCAT G | 641–621 | yrpA |
| BF | CTGGCCATGC ACTTAATTGT T | 335–355 | yrpB |
| BR | CTTGCTGATT TTCTTGCGAC A | 640–620 | yrpB |
| SF | CATGGAATCT CGTCTGAATG A | 621–641 | yrpA |
| SR | AACAATTAAG TGCATGGCCA G | 355–335 | yrpB |
In vitro regulation studies
The ∆yrpB strain containing a transcriptional fusion between the yrpAB promoter and the lacZY genes was used to study the regulation of the yrpAB operon. For promoter expression studies, 250-mL flasks containing 20 mL of M9CG medium supplemented with different compounds to be investigated as hypothetical inducers were inoculated with 200 μL of a ∆yrpB overnight culture, followed by incubation in orbital shakers at 250 rpm at 18 °C and 28 °C. When microaerobic conditions were required, the incubation was performed as described above. The influence of the following compounds and conditions on the yrpAB operon expression was assessed: NaCl (0.35 M); pH in the range of 5–6 (50 mM MES) and 7–8 (50 mM HEPES); gelatin (0.5 and 1%, w/v); peptone in the range of 0.5 to 10% (w/v); casamino acids in the range of 0.2 to 5% (w/v); ammonium chloride at 220 mM and different amino acids, such as glutamine, leucine and proline, each of them at 30 mM and 100 mM. For β-galactosidase activity determination, samples of 1 mL from exponential-phase cultures were collected by centrifugation at 13 000 rpm for 10 min and stored at −20 °C. The β-galactosidase activity of the yrpB::lacZY transcriptional fusion was measured in triplicate in three independent experiments by the method of Miller30 using ONPG (o-nitrophenyl-β-d-galactopyranoside) as a substrate. The β-galactosidase activity was expressed in Miller Units = (1000 × OD420) / (t [min] × v [mL] × OD600) where t is the incubation time with the substrate and v the volume of permeabilized cells. After an analysis of variance test, P values of < 0.05 were considered significant.
Animal experiments
Animal experiments were performed in accordance with the European legislation governing animal welfare, and they were authorized and supervised by the Ethics Committee of the University of Oviedo. Rainbow trout (Oncorhynchus mykiss) of about 10–15 g obtained from a commercial fish farm were used in all the experiments. Fish were acclimatized to experimental conditions and randomly selected fish were analyzed to discard the presence of bacteria in spleen, gut and liver. Fish were kept in 60 L tanks at 18 °C ± 1 in dechlorinated water. For LD50 determination, Y. ruckeri parental and yrpA mutant strain cultures were grown to exponential phase, harvested by centrifugation and washed twice with PBS. Cells were re-suspended in PBS and serial dilutions were prepared. Groups of 10 fish were challenged by intraperitoneal injection of 100 µL of dilutions containing 10 to 107 CFU. Mortality was monitored daily over a 7-d period, and LD50 was determined by using IBM SPSS Statistics 20. Two different experiments were performed. Control fishes were injected with an equal volume of PBS.
In silico analysis
Sequences were compared with those in the databases with the BLAST (Basic Local Alignment Search Tool) program. Signal sequence prediction analysis was performed using the SignalP V4.1 server (http://www.cbs.dtu.dk/services/SignalP/). Protein sequences were aligned with MUSCLE (http://www.ebi.ac.uk/Tools/msa/muscle/) using default parameters. Alignment was manually inspected to correct inaccurately situated residues and a maximum likelihood tree was built with MEGA software (http://www.megasoftware.net/).The yrpA and yrpB gene sequences were deposited in GenBank under accession number KF735059.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a grant (AGL2012-35808) from the Spanish Ministerio de Economía y Competitividad (MINECO). R.N., D.P.-P., and D.C. were the recipients of grants from the Spanish Ministerio de Ciencia e Innovación (MICINN).
References
- 1.Méndez J, Guijarro JA. In vivo monitoring of Yersinia ruckeri in fish tissues: progression and virulence gene expression. Environ Microbiol Rep. 2013;5:179–85. doi: 10.1111/1758-2229.12030. [DOI] [PubMed] [Google Scholar]
- 2.Fernández L, Secades P, Lopez JR, Márquez I, Guijarro JA. Isolation and analysis of a protease gene with an ABC transport system in the fish pathogen Yersinia ruckeri: insertional mutagenesis and involvement in virulence. Microbiology. 2002;148:2233–43. doi: 10.1099/00221287-148-7-2233. [DOI] [PubMed] [Google Scholar]
- 3.Fernández L, Prieto M, Guijarro JA. The iron- and temperature-regulated haemolysin YhlA is a virulence factor of Yersinia ruckeri. Microbiology. 2007;153:483–9. doi: 10.1099/mic.0.29284-0. [DOI] [PubMed] [Google Scholar]
- 4.Dahiya I, Stevenson RMW. The ZnuABC operon is important for Yersinia ruckeri infections of rainbow trout, Oncorhynchus mykiss (Walbaum) J Fish Dis. 2010;33:331–40. doi: 10.1111/j.1365-2761.2009.01125.x. [DOI] [PubMed] [Google Scholar]
- 5.Dahiya I, Stevenson RMW. The UvrY response regulator of the BarA-UvrY two-component system contributes to Yersinia ruckeri infection of rainbow trout (Oncorhynchus mykiss) Arch Microbiol. 2010;192:541–7. doi: 10.1007/s00203-010-0582-8. [DOI] [PubMed] [Google Scholar]
- 6.Méndez J, Reimundo P, Pérez-Pascual D, Navais R, Gómez E, Guijarro JA. A novel cdsAB operon is involved in the uptake of L-cysteine and participates in the pathogenesis of Yersinia ruckeri. J Bacteriol. 2011;193:944–51. doi: 10.1128/JB.01058-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gunnlaugsdóttir B, Gudmundsdóttir BK. Pathogenicity of atypical Aeromonas salmonicida in Atlantic salmon compared with protease production. J Appl Microbiol. 1997;83:542–51. doi: 10.1046/j.1365-2672.1997.00247.x. [DOI] [PubMed] [Google Scholar]
- 8.Denkin SM, Nelson DR. Regulation of Vibrio anguillarum empA metalloprotease expression and its role in virulence. Appl Environ Microbiol. 2004;70:4193–204. doi: 10.1128/AEM.70.7.4193-4204.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vipond R, Bricknell IR, Durant E, Bowden TJ, Ellis AE, Smith M, MacIntyre S. Defined deletion mutants demonstrate that the major secreted toxins are not essential for the virulence of Aeromonas salmonicida. Infect Immun. 1998;66:1990–8. doi: 10.1128/iai.66.5.1990-1998.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pérez-Pascual D, Gómez E, Álvarez B, Méndez J, Reimundo P, Navais R, Duchaud E, Guijarro JA. Comparative analysis and mutation effects of fpp2-fpp1 tandem genes encoding proteolytic extracellular enzymes of Flavobacterium psychrophilum. Microbiology. 2011;157:1196–204. doi: 10.1099/mic.0.046938-0. [DOI] [PubMed] [Google Scholar]
- 11.Kato T, Takahashi N, Kuramitsu HK. Sequence analysis and characterization of the Porphyromonas gingivalis prtC gene, which expresses a novel collagenase activity. J Bacteriol. 1992;174:3889–95. doi: 10.1128/jb.174.12.3889-3895.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takahashi N, Kato T, Kuramitsu HK. Isolation and preliminary characterization of the Porphyromonas gingivalis prtC gene expressing collagenase activity. FEMS Microbiol Lett. 1991;68:135–8. doi: 10.1111/j.1574-6968.1991.tb04585.x. [DOI] [PubMed] [Google Scholar]
- 13.Zhao H, Li X, Johnson DE, Mobley HL. Identification of protease and rpoN-associated genes of uropathogenic Proteus mirabilis by negative selection in a mouse model of ascending urinary tract infection. Microbiology. 1999;145:185–95. doi: 10.1099/13500872-145-1-185. [DOI] [PubMed] [Google Scholar]
- 14.Kavermann H, Burns BP, Angermüller K, Odenbreit S, Fischer W, Melchers K, Haas R. Identification and characterization of Helicobacter pylori genes essential for gastric colonization. J Exp Med. 2003;197:813–22. doi: 10.1084/jem.20021531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carlson SA, McCuddin ZP, Wu MT. SlyA regulates the collagenase-mediated cytopathic phenotype in multiresistant Salmonella. Microb Pathog. 2005;38:181–7. doi: 10.1016/j.micpath.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 16.Han H-J, Taki T, Kondo H, Hirono I, Aoki T. Pathogenic potential of a collagenase gene from Aeromonas veronii. Can J Microbiol. 2008;54:1–10. doi: 10.1139/W07-109. [DOI] [PubMed] [Google Scholar]
- 17.Fernández L, Márquez I, Guijarro JA. Identification of specific in vivo-induced (ivi) genes in Yersinia ruckeri and analysis of ruckerbactin, a catecholate siderophore iron acquisition system. Appl Environ Microbiol. 2004;70:5199–207. doi: 10.1128/AEM.70.9.5199-5207.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rawlings ND, Barrett AJ, Bateman A. MEROPS: the peptidase database. Nucleic Acids Res. 2010;38:D227–33. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ioannides M. Detection, cloning, and analysis of a U32 collagenase in Streptococcus mutans GS-5. Ioannides, Marios, Graduate School Theses and Dissertation;http://scholarcommons.usf.edu/etd/1090 Master of Science Thesis. University of South Florida, 2004.
- 20.Carson V. Cloning and analysis of putative collagenases of the U32 family in Streptococcus mutans and Streptococcus agalactiae (group B streptococci). Carson, Valerie, Graduate School Theses and Dissertations; http://scholarcommons.usf.edu/etd/2474 Master of Science Thesis. University of South Florida, 2006.
- 21.Keil-Dlouha V, Misrahi R, Keil B. The induction of collagenase and a neutral proteinase by their high molecular weight substrates in Achromobacter iophagus. J Mol Biol. 1976;107:293–305. doi: 10.1016/S0022-2836(76)80006-X. [DOI] [PubMed] [Google Scholar]
- 22.Dawson AM, Trenchard D, Guz A. Small bowel tonometry: assessment of small gut mucosal oxygen tension in dog and man. Nature. 1965;206:943–4. doi: 10.1038/206943b0. [DOI] [PubMed] [Google Scholar]
- 23.Liu Z, Yang M, Peterfreund GL, Tsou AM, Selamoglu N, Daldal F, Zhong Z, Kan B, Zhu J. Vibrio cholerae anaerobic induction of virulence gene expression is controlled by thiol-based switches of virulence regulator AphB. Proc Natl Acad Sci U S A. 2011;108:810–5. doi: 10.1073/pnas.1014640108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marteyn B, West NP, Browning DF, Cole JA, Shaw JG, Palm F, Mounier J, Prévost MC, Sansonetti P, Tang CM. Modulation of Shigella virulence in response to available oxygen in vivo. Nature. 2010;465:355–8. doi: 10.1038/nature08970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reid GC, Woods DR, Robb FT. Peptone induction and rifampin-insensitive collagenase production by Vibrio alginolyticus. J Bacteriol. 1980;142:447–54. doi: 10.1128/jb.142.2.447-454.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hanlon GW, Hodges NA, Russell AD. The influence of glucose, ammonium and magnesium availability on the production of protease and bacitracin by Bacillus licheniformis. J Gen Microbiol. 1982;128:845–51. doi: 10.1099/00221287-128-4-845. [DOI] [PubMed] [Google Scholar]
- 27.Secades P, Guijarro JA. Purification and characterization of an extracellular protease from the fish pathogen Yersinia ruckeri and effect of culture conditions on production. Appl Environ Microbiol. 1999;65:3969–75. doi: 10.1128/aem.65.9.3969-3975.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Penfold RJ, Pemberton JM. An improved suicide vector for construction of chromosomal insertion mutations in bacteria. Gene. 1992;118:145–6. doi: 10.1016/0378-1119(92)90263-O. [DOI] [PubMed] [Google Scholar]
- 29.Mahan MJ, Tobias JW, Slauch JM, Hanna PC, Collier RJ, Mekalanos JJ. Antibiotic-based selection for bacterial genes that are specifically induced during infection of a host. Proc Natl Acad Sci U S A. 1995;92:669–73. doi: 10.1073/pnas.92.3.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JH. Experiments in molecular genetics. Cold Spring Harbor: Cold Spring Harbor Laboratory; 1972. p. 431-5. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.