

Abstract
Connexin 26 (Cx26, GJB2) mutations are the major cause of hereditary deafness and are responsible for >50% of nonsyndromic hearing loss. Mouse models show that Cx26 deficiency can cause congenital deafness with cochlear developmental disorders, hair cell degeneration, and the reduction of endocochlear potential (EP) and active cochlear amplification. However, the underlying deafness mechanism still remains undetermined. Our previous studies revealed that hair cell degeneration is not a primary cause of hearing loss. In this study we investigated the role of EP reduction in Cx26 deficiency-induced deafness. We found that the EP reduction is not associated with congenital deafness in Cx26 knockout (KO) mice. The threshold of auditory brainstem response (ABR) in Cx26 KO mice was even greater than 110 dB SPL, demonstrating complete hearing loss. However, the EP in Cx26 KO mice varied and was not completely abolished. In some cases, the EP could still remain at higher levels (> 70 mV). We further found that the deafness in Cx26 KO mice is associated with cochlear developmental disorders. Deletion of Cx26 in the cochlea before postnatal day 5 (P5) could cause congenital deafness. The cochlea had developmental disorders and the cochlear tunnel was not open. However, no congenital deafness was found when Cx26 was deleted after P5. The cochlea also displayed normal development and the cochlear tunnel was open normally. These data suggest that congenital deafness induced by Cx26 deficiency is not determined by EP reduction and may result from cochlear developmental disorders.
Keywords: Cx26, gap junction, connexin, EP, deafness, inner ear development
Introduction
Approximately 1 child in 1,000 is affected with severe hearing loss in infancy and childhood [1, 2]. Half of these cases are attributed to Connexin 26 (Cx26, GJB2) mutations [3–6]. This is the most common genetic cause for hearing loss and also the highest incidence of genetic diseases caused by single gene mutations in human beings. In the clinic, Cx26 mutations can cause congenital deafness, resulting in a mild-moderate to profound sensorineural hearing loss [3, 6]. However, the mechanism underlying this common nonsyndromic hearing loss is still unclear.
Mouse models show that Cx26 deficiency can cause congenital deafness with cochlear development disorders, hair cell degeneration, endocochlear potential (EP) reduction, and active cochlear mechanics elimination [7–11]. Our previous studies further found that hair cell degeneration is not a primary cause for hearing loss [10]. However, the detailed deafness mechanism still remains undetermined. EP is the driving force for current through the auditory sensory hair cells to produce receptor current and potential [12, 13]. In this study, we investigated the role of EP reduction in Cx26 deficiency-induced hearing loss. We found that the EP reduction is not associated with deafness in Cx26 knockout (KO) mice.
Materials and Methods
Cx26 and Cx30 knockout mice
Cx26 KO mice were generated by crossing Cx26loxP/loxP mice (EM00245, European Mouse Mutant Archive) [7]with Gt(ROSA)26Sortm1(Cre/Esr1)Nat/J (ROAS26Cre/Esr1) mouse strain (stock No. 004847, Jackson Lab, USA), which has a tamoxifen-inducible Cre-mediated recombination system driven by the endogenous mouse Gt(ROSA)26Sor promoter. To activate Cre expression deleting Cx26 expression, 4-hydroxytamoxifen (4-HTMX, H-7904, Sigma-Aldrich) was administrated by intraperitoneal injection with 0.5 mg/10g body weight per day for 3 consecutive days after birth. This procedure was successfully used in the neonatal mouse cochlea with no abnormal cochlear morphology or hearing defects in tamoxifen induced control at various postnatal ages [14]. We also injected the same amount of 4-HTMX in wild-type (WT) littermates as controls. Cx30 KO mice were also purchased from European Mouse Mutant Archive (EM00323) [15]. All experimental procedures were conducted in accordance with the policies of University of the Kentucky Animal Care & Use Committee.
Auditory brainstem response (ABR) measurement
As described in our previous publications [10, 11], ABR was recorded using a Tucker-Davis ABR workstation with ES-1 high-frequency speaker (Tucker-Davis Tech. Alachua, FL). Mice were anesthetized by intraperitoneal injection with a mixture of ketamine and xylazine (8.5 ml saline+1 ml Ketamine+0.55 ml Xylazine, 0.1 ml/10 g). ABR was evoked by clicks in alternative polarity from 80 to 10 dB SPL in a 5 dB step. The signal was amplified (50,000x), filtered (300–3,000 Hz), and averaged by 500 times. The ABR threshold was determined by the lowest level at which an ABR can be recognized. If mice had severe hearing loss, the intensity of stimulation was extended up to 110 dB SPL.
EP recording
Mice were anaesthetized as described above. Body temperature was maintained at 37–38 °C. EP was measured by a lateral-wall access as we previously reported [11]. The cochlea was exposed by a ventral approach and the bone over the spiral ligament was gently picked to form a small hole. A glass pipette filled with a K+-based intracellular solution was inserted into the hole. The DC potential was continually recorded as the electrode pipette penetrated through the lateral wall by MultiClamp 700A amplifier (Molecular Devices, CA) under the current-clamp configuration and digitized utilizing a Digidata 1322A (Molecular Devices, CA) [11].
Cochlear tissue preparation and immunofluorescent staining
The cochlear tissue preparation and immunofluorescent staining were performed as previously reported [16, 17]. Briefly, the cochlear cryostat cross-sections were fixed with 4% paraformaldehyde in 0.1 M PBS (pH 7.4) for 30 min. After being incubated in a blocking solution (10% goat serum and 1% BSA in the PBS) with 0.1% Triton X-100 for 30 min at room temperature, the tissue was incubated with monoclonal mouse anti-Cx26 and polyclonal rabbit anti-Cx30 antibodies (1: 400, Catalog No. 33-5800 and 71-2200, respectively, Invitrogen, CA) in the blocking solution at 4°C overnight. After washing out with PBS, the tissues were incubated in a 1:600 dilution of secondary Alexa Fluor® 488 and 568 conjugated antibodies (Invitrogen, CA) in the blocking solution at room temperature for 1 hr. The section was then washed out, mounted with a fluorescence mounting medium (H-1000, Vector Lab, CA), and observed under a fluorescence microscope (Nickon T2000) or a confocal microscope (Leaca TCS SP2).
Results
Cx26 KO mice show congenital deafness (Fig. 1). During postnatal development, ABR is recordable at postnatal day 14 (P14) in WT mice. The hearing function was matured around P20 and the ABR threshold decreased to ~30 dB SPL (Fig. 1B). However, no ABR was evoked in Cx26 KO mice and the ABR thresholds were greater than 110 dB SPL throughout postnatal development (Fig. 1B), demonstrating complete hearing loss.
Fig. 1.

Complete and congenital deafness in Cx26 KO mice as measured by ABR recording. A: ABR waveforms evoked by click stimulations in different postnatal developmental ages in WT and Cx26 KO mice. B: ABR thresholds in the mouse postnatal development. The ABR thresholds in Cx26 KO mice are consistently greater than 110 dB SPL, demonstrating complete hearing loss.
EP in Cx26 KO mice was decreased but not absent. Fig. 2 shows that the EPs in WT and Cx26 KO mice were 99.6±7.41 mV (mean±SD, n=15) and 57.7±19.4 mV (mean±SD, n=10), respectively. However, the EP reduction in Cx26 KO mice is irrelative to hearing loss and shows wide variation (Fig. 2A). Unlike complete hearing loss, EP in Cx26 KO mice was not completely abolished and could still remain at higher levels (> 70 mV), even though the ABR thresholds were even greater than 110 dB SPL (Fig. 2B). On the other hand, EP reduction is considered as a major cause for Cx30 deficiency induced deafness [15]. Different from Cx26 KO mice, EP in Cx30 KO mice was almost completely abolished and absent (3.58±4.11 mV, mean±SD, n=5). Consistently, the ABR thresholds were even greater than 110 dB SPL (Fig. 2).
Fig. 2.

EP reduction is disproportional to complete deafness in Cx26 KO mice. A: EPs in Cx26 KO, Cx30 KO, and WT mice. Red lines represent the average levels. EP in Cx26 KO mice is reduced but not absent. B: EP reduction in Cx26 KO mice is not proportional to hearing loss. ABR thresholds are even greater than 110 dB SPL, whereas EPs have a large variation and can be greater than 70 mV. Inset: The relationship between EPs and ABR thresholds in WT and Cx30 KO mice. Cx30 KO mice have complete deafness with complete absence of EPs (<10 mV).
Deletion of Cx26 also causes cochlear developmental disorders [7, 9–10]. We further tested whether cochlear developmental disorders are associated with congenital deafness induced by Cx26 deficiency. We injected tamoxifen into tamoxifen-inducible Cx26-Esr1/Cre mice to delete Cx26 at different postnatal ages. Fig. 3 shows that injection of tamoxifen before P10 could almost completely delete Cx26 expression in the cochlea. However, the expression of co-expressed Cx30 appeared normal. No apparent difference in Cx30 expression between WT and Cx26 conditional KO (cKO) mice is visible.
Fig. 3.
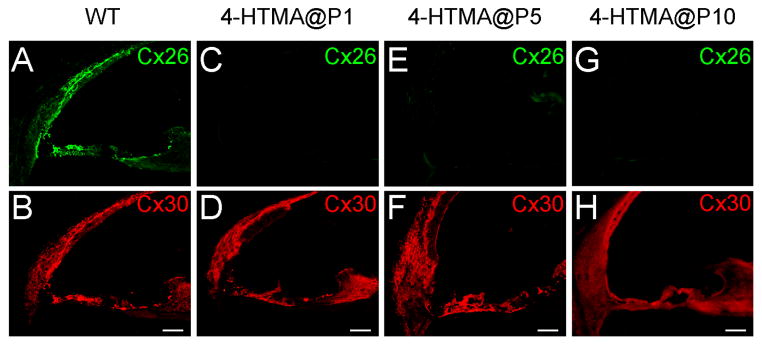
Immunofluorescent staining for Cx26 (green) and Cx30 (red) in the cochlea for injection of 4-HTMX at different postnatal ages to delete Cx26. No intense Cx26 labeling in the cochlea is visible in Cx26 conditional KO mice. Mice were P30–45 old. Scale bar = 50 μm.
However, these Cx26 cKO mice demonstrated different phenotypes for deletion of Cx26 at different postnatal developmental stages (Fig. 4). The ABR recording at P30 shows that the mice had severe hearing loss after injection of tamoxifen to delete Cx26 before P4; the ABR threshold was greater than 100 dB SPL (Fig. 4D). However, injection of tamoxifen to delete Cx26 after P6 could not result in congenital deafness. The mice had normal hearing at P30. The ABR thresholds were ~30 dB SPL at the same level as that in WT mice (Fig. 4D). The middle point for deletion of Cx26 to generate congenital deafness was at P4.2 (Fig. 4D). Morphological examination shows that deletion of Cx26 in the cochlea before P4 could result in cochlear developmental disorders (Fig. 4A). The cochlear tunnel was not developed and the under-tectorial-membrane space was absent. However, the cochlea dispayed normal development for deletion of Cx26 after P5. The cochlear tunnel was open and not collapsed (Fig. 4B-C).
Fig. 4.

Developmental changes in the cochlea for deletion of Cx26 at different postnatal days and associated hearing loss. A–C: The effect on cochlear development for injection of 4-HTMX to delete Cx26 in the cochlea at different postnatal developmental stages. White arrows indicate the cochlear tunnel, which is open following injection of 4-HTMX to delete Cx26 in the cochlea after P5. Empty triangles in panel A indicate the collapse of the under-tectorial-membrane space. Mice were P45–80 old. D: ABR thresholds measured at P30 for injection of 4-HTMX to delete Cx26 at different postnatal developmental ages. Mice show congenital deafness for deletion of Cx26 in the cochlea before P3 but retain normal hearing for deletion of Cx26 after P5. A red line represents data fitting to the sigmoid function. The middle point is at P4.2.
Discussion
In this experiment, we found that hearing loss in Cx26 KO mice is not associated with EP reduction (Fig. 2), indicating that EP reduction does not play a primary role in Cx26 deficiency induced hearing loss. EP is a driving force for hair cell transduction process [12, 13]. EP reduction can induce hearing loss, which is considered as a major cause of deafness in Cx30 KO mice [15]. Cx30 KO mice had complete deafness and the ABR threshold was greater than 110 dB SPL (Fig. 2B). Consistently, the EP in Cx30 KO mice was completely absent (Fig. 2). However, the EP in Cx26 KO mice was 57.7±19.4 mV and not completely abolished (Fig. 2). In some cases, the EP could still remain at higher level (> 70 mV), even as Cx26 KO mice showed complete deafness and the ABR threshold was even greater than 110 dB SPL as that in Cx30 KO mice (Fig. 1, also see ref. 10). These data indicate that the EP reduction in Cx26 KO mice is unlikely to be a determined cause of deafness.
We found that deletion of Cx26 before P4 could result in congenital deafness with cochlear developmental disorders (Figs. 3–4), whereas deletion of Cx26 in the cochlea after P5 had little influence on the cochlear development with normal hearing (Fig. 4), demonstrating that cochlear developmental disorders are associated with congenital deafness. Cx26 deficiency could arrest the cochlear tunnel development; the tunnel is not open [9, 10]. The cochlear tunnel plays a critical role in the cochlear function. It has been found that outer hair cells (OHCs) in Cx26 deficient mice still retain electromotility [10, 18]. However, active cochlear mechanics as measured by distortion product otoacoustic emission (DPOAE) was eliminated [10, 19]. Recently, we found that Cx26 expression in the cochlear supporting cells plays a critical role in active cochlear amplification [11]. Targeted-deletion of Cx26 in the OHC supporting cells can eliminate active cochlear amplification in vivo. Active cochlear amplification is required for normal hearing. Elimination of active cochlear amplification can induce hearing loss [20, 21].
We found that deletion of Cx26 before P5 can induce congenital hearing loss with developmental disorders (Figs. 3–4). This is consistent with previous findings that deletion of Cx26 at the embryonic period or P0-1 can arrest the postnatal development of the organ of Corti [9–10, 22]. This also further suggests that Cx26 expression in the cochlea at the early postnatal development stage (<P5) is critical for cochlear postnatal development and maturation.
We previously reported that cell degeneration is not a primary cause for Cx26 deficiency induced congenital deafness [10] and that Cx26 deficiency can reduce active cochlear amplification [11]. In this study, we also found that congenital deafness in Cx26 deficient mice is not determined by EP reduction (Figs. 1–2) and is associated with cochlear developmental disorders (Figs. 3–4). These new findings provide important information not only for understanding the mechanism underlying Cx26 deficiency associated deafness but also for developing new therapeutic strategies and approaches for protection and treatment of this common hereditary hearing loss.
Highlights.
The deafness mechanism of Cx26 deficiency induced hearing loss was investigated
Deafness in Cx26 KO mice is associated with cochlear developmental disorders
Cx26 KO before P5 can induce congenital deafness and cochlear developmental disorders
Endocochlear potential reduction in Cx26 KO mice is irrelative to deafness
Acknowledgments
This work was supported by NIH R01 DC 05989.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mason JA, Herrmann KR. Universal infant hearing screening by automated auditory brainstem response measurement. Pediatrics. 1998;101:221–228. doi: 10.1542/peds.101.2.221. [DOI] [PubMed] [Google Scholar]
- 2.Petersen MB, Willems PJ. Non-syndromic, autosomal-recessive deafness. Clin Genet. 2006;69:371–392. doi: 10.1111/j.1399-0004.2006.00613.x. [DOI] [PubMed] [Google Scholar]
- 3.Zhao HB, Kikuchi T, Ngezahayo A, White TW. Gap junctions and cochlear homeostasis. J Memb Biol. 2006;209:177–186. doi: 10.1007/s00232-005-0832-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dobrowolski R, Willecke K. Connexin-caused genetic diseases and corresponding mouse models. Antioxid Redox Signal. 2009;11:283–295. doi: 10.1089/ars.2008.2128. [DOI] [PubMed] [Google Scholar]
- 5.Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 6.Castillo FJ, Castillo I. The DFNB1 subtype of autosomal recessive non-syndromic hearing impairment. Front Biosci. 2011;17:3252–3274. doi: 10.2741/3910. [DOI] [PubMed] [Google Scholar]
- 7.Cohen-Salmon M, Ott T, Michel V, Hardelin JP, Perfettini I, Eybalin M, Wu T, Marcus DC, Wangemann P, Willecke K, Petit C. Targeted ablation of connexin26 in the inner ear epithelial gap junction network causes hearing impairment and cell death. Curr Biol. 2002;12:1106–1111. doi: 10.1016/s0960-9822(02)00904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Y, Tang W, Chang Q, Wang Y, Kong W, Lin X. Connexin30 null and conditional connexin26 null mice display distinct pattern and time course of cellular degeneration in the cochlea. J Comp Neurol. 2009;516:569–579. doi: 10.1002/cne.22117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Y, Chang Q, Tang W, Sun Y, Zhou B, Li H, Lin X. Targeted connexin26 ablation arrests postnatal development of the organ of Corti. Biochem Biophys Res Commun. 2009;385:33–37. doi: 10.1016/j.bbrc.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang C, Zhu Y, Zong L, Lu GJ, Zhao HB. Cell degeneration is not a primary causer for Connexin26 (GJB2) deficiency associated hearing loss. Neurosci Lett. 2012;528:36–41. doi: 10.1016/j.neulet.2012.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Y, Liang C, Chen J, Zong L, Chen GD, Zhao HB. Active cochlear amplification is dependent on supporting cell gap junctions. Nat Commun. 2013;4:1786. doi: 10.1038/ncomms2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wangemann P. Supporting sensory transduction: cochlear fluid homeostasis and the endocochlear potential. J Physiol. 2006;576:11–21. doi: 10.1113/jphysiol.2006.112888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen J, Zhao HB. The role of an inwardly rectifying K+ channel (Kir4.1) in the inner ear and hearing loss. Neuroscience. 2014;265:137–146. doi: 10.1016/j.neuroscience.2014.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chow LM, Tian Y, Weber T, Corbett M, Zuo J, Baker SJ. Inducible Cre recombinase activity in mouse cerebellar granule cell precursors and inner ear hair cells. Dev Dyn. 2006;235:2991–2998. doi: 10.1002/dvdy.20948. [DOI] [PubMed] [Google Scholar]
- 15.Teubner B, Michel V, Pesch J, Lautermann J, Cohen-Salmon M, Sohl G, Jahnke K, Winterhager E, Herberhold C, Hardelin JP, Petit C, Willecke K. Connexin30 (Gjb6)-deficiency causes severe hearing impairment and lack of endocochlear potential. Hum Mol Genet. 2003;12:13–21. doi: 10.1093/hmg/ddg001. [DOI] [PubMed] [Google Scholar]
- 16.Zhao HB, Yu N. Distinct and gradient distributions of connexin26 and connexin30 in the cochlear sensory epithelium of guinea pigs. J Comp Neurol. 2006;499:506–518. doi: 10.1002/cne.21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu YP, Zhao HB. Cellular characterization of Connexin26 and Connnexin30 expression in the cochlear lateral wall. Cell Tissue Res. 2008;333:395–403. doi: 10.1007/s00441-008-0641-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minekawa A, Abe T, Inoshita A, Iizuka T, Kakehata S, Narui Y, Koike T, Kamiya K, Okamura HO, Shinkawa H, Ikeda K. Cochlear outer hair cells in a dominant-negative connexin26 mutant mouse preserve non-linear capacitance in spite of impaired distortion product otoacoustic emission. Neuroscience. 2009;164:1312–1319. doi: 10.1016/j.neuroscience.2009.08.043. [DOI] [PubMed] [Google Scholar]
- 19.Inoshita A, Iizuka T, Okamura HO, Minekawa A, Kojima K, Furukawa M, Kusunoki T, Ikeda K. Postnatal development of the organ of Corti in dominant-negative Gjb2 transgenic mice. Neuroscience. 2008;156:1039–1047. doi: 10.1016/j.neuroscience.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 20.Dallos P. Cochlear amplification, outer hair cells and prestin. Curr Opin Neurobiol. 2008;18:370–376. doi: 10.1016/j.conb.2008.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hudspeth AJ. Making an effort to listen: mechanical amplification in the ear. Neuron. 2008;59:530–545. doi: 10.1016/j.neuron.2008.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S, Sun Y, Lin X, Kong W. Down regulated connexin26 at different postnatal stage displayed different types of cellular degeneration and formation of organ of Corti. Biochem Biophys Res Commun. 2014;445:71–77. doi: 10.1016/j.bbrc.2014.01.154. [DOI] [PubMed] [Google Scholar]