

Abstract
Glycoside hydrolases catalyze the selective hydrolysis of glycosidic bonds in oligosaccharides, polysaccharides, and their conjugates. β-glucosidases occur in all domains of living organisms and constitute a major group among glycoside hydrolases. On the other hand, the benzoxazinoids occur in living systems and act as stable β-glucosides, such as 2-(2,4-dihydroxy-7-methoxy-2H-1,4-benzoxazin-3(4H)-one)-β-D-gluco-pyranose, which hydrolyse to an aglycone DIMBOA. Here, we synthesized the library of novel 1,3-benzoxazine scaffold based aglycones by using 2-aminobenzyl alcohols and aldehydes from one-pot reaction in a chloroacetic acid catalytic system via aerobic oxidative synthesis. Among the synthesized benzoxazines, 4-(7-chloro-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol (compound 7) exhibit significant inhibition towards glucosidase compared to acarbose, with a IC50 value of 11.5 µM. Based upon results generated by in silico target prediction algorithms (Naïve Bayesian classifier), these aglycones potentially target the additional sodium/glucose cotransporter 1 (where a log likelihood score of 2.70 was observed). Furthermore, the in vitro glucosidase activity was correlated with the in silico docking results, with a high docking score for the aglycones towards the substrate binding site of glycosidase. Evidently, the in vitro and in vivo experiments clearly suggest an anti-hyperglycemic effect via glucose uptake inhibition by 4-(7-chloro-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol in the starved rat model. These synthetic aglycones could constitute a novel pharmacological approach for the treatment, or re-enforcement of existing treatments, of type 2 diabetes and associated secondary complications.
Introduction
Diabetes mellitus, particularly its subtype 2 (T2-DM), is considered to be a major and increasing threat to human health and accounts for an estimated more than 300 million cases of diabetes worldwide [1], [2]. Diabetes is associated with a significant number of co morbidities, such as cardiovascular disorders, stroke, diabetic retinopathy and kidney dysfunction, and long-term complications may even lead to limb amputations, among other consequences [2].
In addition to injectable insulin and analogs thereof, four distinct classes of oral hypoglycemic agents are currently used for the treatment of T2-DM. In addition to metformin as an oral drug used for the early control of T2-DM, there are a number of second- and third-line pharmacological agents available, such as sulfonylureas, thiazolidinediones, incretin-based remedies, and α-glucosidase inhibitors. Given the increased perception that handling the early stages of diabetes is of crucial importance, several recent studies and approaches are focusing on agents that can delay or inhibit glucose absorption. Delaying glucose absorption, such as by blocking glycoside hydrolases (particularly α-glucosidases), allows extended time for β-cells to increase insulin secretion, and thereby reduce circulatory glucose levels [3], [4].
α-glucosidases are membrane-bound enzymes that catalyze the selective hydrolysis of glycosidic bonds in oligosaccharides, polysaccharides, and their conjugates to release glucose and the respective monosaccharides. Although they occur throughout living organisms, most of them are located in the brush border of the small intestine to facilitate glucose uptake [4], [5]. Thus, the use of α-glucosidase inhibitors (AGIs) for the treatment of T2-DM delays the release of glucose and halts glucose absorption, thereby lowering the postprandial blood glucose level and improving pre-diabetic conditions. The currently most prominent AGIs are acarbose (Glucobay), a natural compound from an Actinoplanes strain, and the N-hydroxyethyl analogue of 1-deoxynojirimycin, miglitol [4], [6]. However, these compounds have been reported to cause severe gastrointestinal complications.
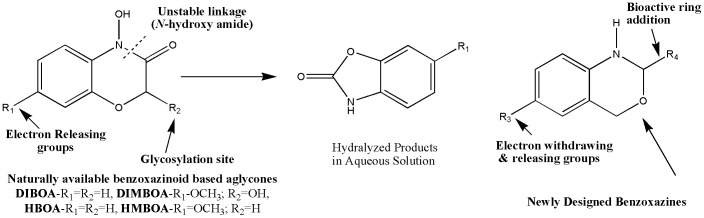
Numerous efforts have hence been made to further develop AGIs in order to improve treatment of pre-diabetic states [7], [8]. Glycosides, the natural substrates for the glycoside hydrolases which are composed of an aglycone and a glycan moiety, have recently attracted particular attention in this field. Stable β-glucosides such as 2-(2,4-dihydroxy-7-methoxy-2H-1,4-benzoxazin-3(4H)-one)-β-D-gluco-pyranoside and desmethoxy derivatives including 6,7-dimethoxy-benzoxazolin-2(3H)-one, 4-hydroxy-2H-1,4-benzoxazin-3(4H)-one, and 4-acetylbenzoxazolin-2(3H)-one, are found in living plants [9]. These glucosides are biologically inactive, but enzymatically converted to active aglycones such as DIMBOA (2,4-dihydroxy-7-methoxy-2H-1,4-benzoxazin-3(4H)-one and the desmethoxy derivative DIBOA, by β-glucosidases [10]. These products have diverse effects, that include anti-auxin, anti-inflammatory, and powerful antibiotic activities. These aglycones are further degraded spontaneously to the corresponding benzoxazolinones, MBOA (6-methoxy-benzoxazolin-2(3H)-one) and the desmethoxy derivative BOA. Degradation is faster if the benzene ring and amide linkage bears electron donating or hydroxyl groups [11], and these chemical variations are what we explored in more detail in the current work. In addition to this, our aim was to remove the relatively labile N-hydroxy amide moiety via replacement by a suitable ring system, masking the glycosylation site of glycoside ( Fig. 1 ). These efforts have the goal of increasing efficacy of the compounds in vivo, in order to lower blood sugar levels by inhibiting glucosidase.
Figure 1. Scheme for the proposed strategy for the synthesis of 1,3-benzoxazine derivatives.
Materials and Methods
Chemicals/ Reagents
Rat intestinal acetone powder was purchased from Sigma Aldrich, St. Louis (USA), Acarbose was purchased from Glucobay Bayer AG (Germany). Porcine pancreatic α-Amylase and Yeast α-Glucosidase were purchased from SRL, Mumbai (India). Glucose oxidase (GOD POD) kit was purchased from Piramal HealthCare Ltd, Mumbai (India). All other chemicals used were of analytical grade and purchased from Sigma Aldrich, St. Louis (USA) and SRL, Mumbai (India). All IR spectra were obtained in KBr disc on a Shimadzu FT-IR 157 Spectrometer. 1H and 13C NMR spectra were recorded on a Bruker WH-200 (400 MZ) spectrometer in CDCl3 or DMSO-d6 as solvent, using TMS as an internal standard and chemical shifts are expressed as ppm. Mass spectra were determined on a Shimadzu LC-MS. The elemental analyses were carried out using an Elemental Vario Cube CHNS rapid Analyzer. The progress of the reaction was monitored by TLC pre-coated silica gel G plates.
Experimental animals
Adult Wistar rats weighing 150–180 g were collected from the University Central Animal Facility and housed under a controlled environment. All animal experiments were approved by the Institutional Animal Ethical Committee (Order No : MGZ/2620/2011-12 Dated 31-01-2012; UOM/IAEC/18/2011), Department of Studies in Zoology, University of Mysore, Mysore and were in accordance with the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA).
Synthesis and characterization of 1,3-benzoxazine derivatives
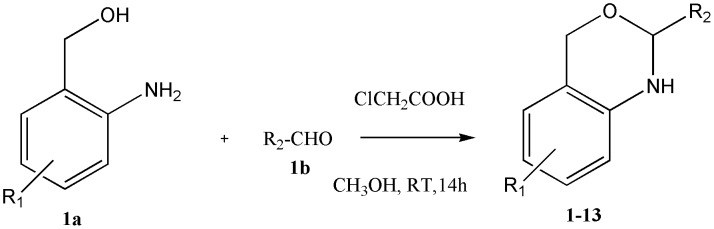
This work is based on the previous synthesis of an oxazine derivative which was able to mimic the pyranoside structure of glycans functionally [12]. In continuation of the glycobiological aspects, the one-pot syntheses of a novel 1,3-benzoxazine scaffold was carried out using 2-aminobenzyl alcohols and different aldehydes in chloroacetic acid via aerobic oxidative synthesis (Fig. 2).
Figure 2. General procedure for the synthesis of novel benzoxazines.
General Procedure for the Synthesis of benzoxazines
To a 50 mL round bottom flask, amino benzyl alcohol (1.0 equiv), aldehyde (1.0 equiv), chloroacetic acid (1.0 equiv) and methanol (10 mL) were added. The reaction mixture was then stirred at room temperature for 16 h. Water was added to the reaction mixture, and extracted with ethyl acetate. The organic layer was dried by using anhydrous sodium sulphate, and then concentrated in vacuum to afford compound benzoxazine as a crystalline solid. The compounds were then purified by column chromatography and characterized via IR, 1H NMR, 13C NMR, mass spectrometry and Elemental Analysis.
Synthesis of 4-(2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol 1
The product 1 was obtained from 2-amino benzyl alcohol (1.23 g,10 mmol), 4-hydroxy benzaldehyde (1.22 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brownish solid in 83% yield. IR νmax (KBR): 3277,2924, 1514, 1220 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) δ 4.13(1H, s, NH) 4.80-4.90 (2H, s, CH2) 5.02 (1H, s, O-H) 5.28 (1H, s,-CH) 6.70-6.72(1H, d, Aromatic -CH) 6.9–7.2 (5H, m, Ar-CH) 8.06 (1H, s, Ar-CH); 13C NMR 70.05, 97.95, 114.64, 120.47, 126.08, 126.13, 129.13, 129.79, 130.22, 138.07, 147.90, 155.51. MASS; m/z found for C14H13NO2 228.2 ([M+1]+).
Synthesis of 2-(1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 2
The product 2 was obtained from 2-amino benzyl alcohol (1.23 g, 10 mmol), indole-3-carbaldehyde (1.45g, 10 mmol) and chloroacetic acid 0.94g (10 mmol). This compound was obtained as black color solid in 82% yield. IR νmax (KBR): 3252, 2898, 1493, 1244 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 4.25 (1H,s,NH), 4.70 (2H,s,- CH2-) 5.12 (1H,s,-CH-), 7.07-7.50(5H,m, Ar-H), 8.01–8.64(4H,m, Ar-H), 11.80 (1H,s,Indole-NH); Anal. Calcd.for C16H14N2O: C, 76.78; H, 5.64; N, 11.19; found C, 76.54; H, 5.51; N, 11.53 %. MASS; m/z found for C16H14ClN2O 251.3 ([M+1]+).
Synthesis of 2-(2-methyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 3
The product 3 was obtained from 2-amino benzyl alcohol (1.23 g, 10 mmol), 2-methyl indole-3-carbaldehyde (1.59 g, 10 mmol) and chloroacetic acid 0.94g (10 mmol). This compound was obtained as black colour solid in 87% yield. IR νmax (KBR): 3212, 2788, 1462, 1238 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 2.11(3H,s, -CH3-), 4.13 (1H,s,-N-H-), 4.83 (2H,s,-CH2-) 5.19 (1H,s-CH-); 7.26(3H,m,-Ar-CH-) 8.06–8.22(5H,m,Ar-CH), 10.12(1H,s,N-H INDOLE) Anal. Calcd.for C17H16N2O; C, 77.25; H, 6.10; N, 10.60; found C, 77.14; H, 6.11; N, 10.43%; MASS; m/z found for C17H16N2O 264.4 ([M+1]+).
Synthesis of 2-(1-(4-(2-cyanophenyl)benzyl-1H-indol-3-yl),2,4-dihydro-1H-benzo(d)(1,3) oxazine 4
The product 4 was obtained from 2-amino benzyl alcohol (1.23 g, 10 mmol), 2-(1-(4-(2-cyanophenyl )1-benzyl-1H-indole-3-carbaldehyde (3.36 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brown colour solid in 86% yield. IR νmax (KBR): 3182, 2918, 1473, 1294 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 4.09 (1H,s, -NH), 4.7 (2H,s,-CH2), 5.4 (3H,s,-CH), 5.4 (3H,s,-CH2) 7.26–8.35 (17H,m,-Ar-CH-) Anal. Calcd.for C30H23N3O; C, 81.61; H, 5.25; N, 9.52; found C, 81.14; H, 5.11; N, 9.43%. MASS; m/z found for C30H23N3O 442.4 ([M+1]+).
Synthesis of 3-(2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)-4H-chromen-4-one 5
The product 5 was obtained from 2-amino benzyl alcohol (1.23 g, 10 mmol), 4-oxo-4H-chromene-3-carbaldehyde (3.21 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brownish solid in 89% yield. IR νmax (KBR): 3302, 2918, 1463, 1215 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 4.11 (1H,s,NH), 4.80 (1H,s,-CH-) 5.32 (2H,s,-CH2-), 7.07–8.32(7H,m, Ar-H) 8.55(H,s, Ar-O-CH); 13C NMR 70.1, 85.5, 110.6, 115.2,116.1, 119.6,122.0,127.6, 127,8, 129.2, 132.6, 133.6, 142.2, 149.1,155.6, 179.2; Anal. Calcd. for C17H13NO3: C, 73.11; H, 4.69; N, 5.02; found C, 73.54; H, 4.51; N, 5.23 %; MASS; m/z found for C17H13NO3 278.3 ([M-1]+).
Synthesis of 2-(2-butyl-4-chloro-1H-imidazol-5-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 6
The product 6 was obtained from 2-amino benzyl alcohol (1.23 g, 10 mmol), 2-butyl-4-chloro-1H-imidazole-5-carbaldehyde (1.86 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as black colour solid in 86% yield. IR νmax (KBR): 3225, 2892, 1433, 1214 cm−1 1H NMR (400 MHz, CDCl3 δ in ppm) 0.90 (3H,t,CH3), 1.30 (2H,m,- CH2-) 1.60 (2H,m,-CH2-), 2.60 (2H,t,CH2), 4.30 (1H,s,- NH-) 4.60 (2H,s,-CH2-), 5.60 (1H,s,- CH-) 6.60–7.60 (4H,m, Ar-H), 8.2 (1H,s, imidazole N-H) Anal. Calcd.for C15H18ClN3O: C, 61.75; H, 6.22; N, 14.40; found C, 61.54; H, 6.51; N, 14.53 %. MASS; m/z found for C15H18ClN3O 292.2 ([M+1]+).
Synthesis of 4-(7-chloro-2,4-dihydro-1H-benzo[d][1,3] oxazin-2-yl)phenol 7
The product 7 was obtained from 2-amino-4-chloro benzyl alcohol (1.57 g, 10 mmol), 4-hydroxy benzaldehyde (1.22 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brownish solid in 87% yield. IR νmax (KBR): 3258, 2897, 1487, 1269 cm−1 1H NMR (400 MHz, CDCl3 δ in ppm) δ 4.13(1H,s,NH) 4.80-4.90 (2H,s,CH2) 5.02 (1H,s,O-H) 5.28 (1H,s,-CH) 6.70-6.72(2H,d, Aromatic -CH) 6.9–7.2 (4H,m,Ar-CH) 8.06 (1H,s,Ar-CH). 13C NMR 68.97, 94.73, 110.80, 115.66, 119.08, 129.97, 130.40, 133.39, 134.24, 139.12, 149.38, 159.12. Anal. Calcd.for C14H12ClNO2; C, 64.25; H, 4.62; N, 5.35; found C, 64.14; H, 4.21; N, 5.53%. MASS; m/z found for C14H12ClNO2 262.2 ([M+1]+).
Synthesis of 7-chloro-2-(1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 8
The product 8 was obtained from 2-amino-5-chloro benzyl alcohol (1.57 g,10 mmol), indole-3-carbaldehyde (1.45 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brownish solid in 85% yield. IR νmax (KBR): 3213, 2978, 1448, 1236 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 4.25 (1H,s,NH), 4.70 (2H,s,- CH2-) 5.12 (1H,s,-CH-), 7.07-7.50(5H,m, Ar-H), 8.01–8.64(3H,m, Ar-H), 11.80 (1H,s,Indole-NH); Anal. Calcd. for C16H13ClN2O: C, 67.49; H, 4.60; N, 9.84; found C, 66.54; H, 4.51; N, 9.53%; MASS; m/z found for C16H13ClN2O 285.8 ([M+1]+).
Synthesis of 6-chloro-2-(2-phenyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 9
The product 9 was obtained from 2-amino-5-chloro benzyl alcohol (1.57 g, 10 mmol), 2-phenyl indole-3-carbaldehyde (2.21 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as colourless solid in 89% yield. IR νmax (KBR): 3232, 2843, 1452, 1243 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 2.31(2H,s, -CH2-), 2.39 (3H,s,CH3), 5.11 (1H,s,-C-H), 4.2 (1H,s,-N-H-) 6.26–8.41 (12H,m,Ar-CH-); 10.00 (1H,s,N-H) Anal. Calcd.for C22H17N2OCl; C, 73.23; H, 4.72; N, 7.76; found C, 73.34; H, 4.31; N, 7.43%; MASS; m/z found for C22H17ClN2O 361.5 ([M+1]+).
Synthesis of 3-(6-chloro-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)-4H-chromen-4-one 10
The product 10 was obtained from 2-amino -5-chloro benzyl alcohol (1.57 g,10 mmol), 4-oxo-4H-chromene-3-carbaldehyde (1.74 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as brownish solid in 83% yield. IR νmax (KBR): 3219, 2797, 1488, 1285 cm−1 1H NMR (400 MHz, CDCl3 δ in ppm) 4.11 (1H,s,NH), 4.80 (1H,s,-CH-) 5.32 (2H,s,-CH2-), 7.77–8.55(7H,m, Ar-H); Anal. Calcd.for C17H12ClNO3: C, 65.08; H, 3.86; N, 4.46; found C, 66.54; H, 3.51; N, 4.23%; MASS; m/z found for C17H12ClNO3 314.0 ([M+1]+).
Synthesis of 6-methyl-2-(2-methyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 11
The product 11 was obtained from 2-amino-5-methyl benzyl alcohol (1.37 g,10 mmol), 2-methyl indole-3-carbaldehyde (1.59 g, 10 mmol) and chloroacetic acid 0.94 g (10 mmol). This compound was obtained as light yellow colour solid in 80% yield. IR νmax (KBR): 3219, 2818, 1513, 1284 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 2.40(3H,s, -CH3-), 2.5 (3H,s,-CH3), 4.6 (2H,s,-CH2), 5.25 (1H,s,-CH) 6.67–7.32 (7H,m,-Ar-CH-) 10.15(1H,s,N-H), Anal. Calcd.for C18H18N2O; C, 77.67; H, 6.52; N, 10.06; O, 5.75; found C, 77.14; H, 6.11; N, 10.43%. MASS; m/z found for C18H18N2O 278.2, 280.2 ([M+1]+).
Synthesis of 6-methyl-2-(2-phenyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1,3]oxazine 12
The product 12 was obtained from 2-amino-5-methyl benzyl alcohol (1.37 g, 10 mmol), 2-phenyl indole-3-carbaldehyde (2.21 g, 10 mmol) and chlor acetic acid 0.94 g (10 mmol). This compound was obtained as colourless solid in 86% yield. IR νmax (KBR): 3210, 2817, 1433, 1224 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 2.31(2H, s, -CH2-), 2.39 (3H, s, CH3), 5.11 (1H, s, -C-H), 4.2 (1H, s, -N-H-) 6.26–8.41 (12H, m, Ar-CH-); 10.00 (1H, s, N-H) Anal. Calcd. for C23H20N2O; C, 81.15; H, 5.92; N, 8.23; found C, 81.34; H, 5.71; N, 8.43%; MASS; m/z found for C23H20N2O 341.2 ( [M+1]+).
Synthesis of 3-(6-methyl-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)-4H-chromen-4-one 13
The product 13 was obtained from 2-amino-5-methyl benzyl alcohol (1.37 g, 10 mmol), 4-oxo-4H-chromene-3-carbaldehyde (1.74 g, 10 mmol) and chloro acetic acid 0.94 g (10 mmol). This compound was obtained as yellow color solid in 88% yield. IR νmax (KBR): 3241, 2828, 1413, 1224 cm−1, 1H NMR (400 MHz, CDCl3 δ in ppm) 2.20(3H,s, -CH3-), 4.11-4.13 (1H,s,-N-H), 4.11-4.13 (1H, s, -C-H), 4.79 (1H, s, -CH2-) 5.26 (1H, s, -CH2-); 6.6–8.3 (7H, m, -Ar-CH-) 8.5(1H, s, O-CH), Anal. Calcd. for C18H15NO3; C, 73.71; H, 5.15; N, 4.78; found C, 73.14; H, 5.11; N, 4.43%; Mass; m/z found for C18H15NO3 294.1 ([M+1]+).
Inhibitory activity against α-Amylase
The α-amylase inhibition assay was performed according to a previous report [13]. Porcine pancreatic α-amylase (3 units/mL) was dissolved in 0.1 M phosphate buffered saline, pH 6.9. The various concentrations of 1,3-benzoxazine derivatives (0–100 µM) were pre-incubated with enzyme independently for 10 min at 37°C. The reaction was initiated by adding substrate solution (0.1% starch) to the incubation medium. After 10 min incubation, the reaction was stopped by adding 250 µL dinitrosalicylic (DNS) reagent (1% 3, 5-dinitrosalicylic acid, 0.2% phenol, 0.05% Na2SO3 and 1% NaOH in aqueous solution) to the reaction mixture. The reaction was terminated by keeping the reaction mixture in boiling water bath for 10 min. Thereafter, 250 µL of 40% potassium sodium tartarate solution was added to the mixtures to stabilize the colour. After cooling to room temperature in a cold water bath, the absorbance was recorded at 540 nm using a Varioskan multimode plate reader (Thermo Scientifics, USA). Acrabose was used as positive control. The percentage of inhibition was calculated using Abs Contol – Abs Sample ×100/Abs Control. Where Abs Control was the absorbance without sample, Abs sample was the absorbance of enzyme with compound. Concentration-response assays were used to determine the potency (IC50) of 1,3-benzoxazine derivatives based on the logistic analysis of the concentration-response curve using Microsoft Excel.
Inhibitory activity against α-Glucosidase
The α-glucosidase inhibitory activities of synthesized compounds were evaluated using the method developed by Tsujii et al. [14]. Briefly, α-glucosidase was dissolved in phosphate buffer (50 mM, pH 6.9) and pre-treated with various concentrations of 1,3-benzoxazine derivatives (0–100 µM) independently for 10 min at 37°C. The reaction was initiated by the addition of 50 µL of 5 mM p-nitrophenyl- α -D-glucopyranoside solution in phosphate buffer (50 mM, pH 6.9). The enzyme reaction was carried out at 37°C for 30 min. The reaction was terminated by the addition of Na2CO3 (1 M). The enzymatic activity of α-glucosidase was quantified by measuring the absorption at 405 nm using a Varioskan multimode plate reader (Thermo Scientifics, USA). The inhibitory effect of compounds was defined as inhibitory activity (%) = (Abs Control – Abs Compound treated)/Abs Control ×100. Concentration-response assays were used to determine the potency (IC50) of 1,3-benzoxazine derivatives based on the logistic analysis of the concentration-response curve using Microsoft Excel.
Intestinal α-glucosidase inhibitory assay
Intestinal α-glucosidase activity was determined by measuring the amount of glucose hydrolyzed from maltose or sucrose [15]. Briefly, rat intestinal acetone powder was homogenized in 0.9% saline and the suspension was centrifuged at 10,000 g for 30 min at 4°C and the supernatant obtained was used as enzyme source. The enzyme solution was pre-incubated with various concentrations of compound 7 (5–25 µM) or acarbose (5 µM) or combined [5 µM acarbose with increasing concentrations of compound 7 (1–10 µM)] in 100 mM phosphate buffer pH 6.9 at 37°C for 10 min and the reaction was started by adding maltose (37 mM) or sucrose (56 mM) and incubated at 37°C for 30 min (for maltase) and 60 min (for sucrase). After the respective incubation period, reaction was terminated by keeping the samples in boiling water bath for 10 min. The concentration of glucose released from the reaction mixtures was determined by using Glucose oxidase (GOD POD) kit according to the manufacturer’s protocol. Results were expressed as percentage inhibition of intestinal maltase/sucrase activity.
Oral maltose and sucrose tolerance test
The experimental animals were randomly divided into 11 groups each consisting of 5 rats. Following overnight fasting, animals were assigned to the following groups and treated with the respective compounds through oral gavage: Group - I Saline control (0.9% saline); Group - II Maltose control (3 g/kg); Group - III Sucrose control (3 g/kg); Group -IV Acarbose (3 mg/kg) + Maltose; Group - V Acarbose (3 mg/kg) + Sucrose; Group - VI Compound 7 (50 mg/kg) + Maltose; Group - VII Compound 7 (100 mg/kg) + Maltose; Group - VIII Compound 7 (50 mg/kg) + Sucrose; Group - IX Compound 7 (100 mg/kg) + Sucrose; Group - X Acarbose (3 mg/kg) + Compound 7 (50 mg/kg) + Maltose; Group –XI Acarbose (3 mg/kg) + Compound 7 (50 mg/kg) + Sucrose. 5 minutes following compound 7 or Acarbose administration either maltose (3 g/kg) or sucrose (3 g/kg) solution were administered to the respective groups. Blood was collected from the tail vein to the tubes containing anticoagulant (2.5% trisodium citrate and 1.37% citric acid in the ratio 1∶5; anti-coagulant: blood) at time point 0 (just before sucrose/maltose administration), and subsequently at 30, 60, 90, 120 and 180 min after substrate (sucrose/maltose) administration. Plasma was separated by centrifuging the samples at 2000 rpm for 10 min and stored at −20°C until analysis. Plasma glucose concentration was determined by using the Glucose oxidase (GOD POD) kit according to the manufacturer’s protocol.
Glucose uptake study
Porcine diaphragm was purchased from a slaughter house, and cleaned using ice cold 0.9% saline several times to remove blood stains. This diaphragm was used for glucose uptake and inhibition by compound 7. Diaphragm (100 mg) was suspended in a 24 well culture plate containing 500 µL saline. In order to initiate the reaction, 5 mM glucose was added to each well. To enhance the glucose uptake by the diaphragm, 1 unit of insulin was used in each well, and the volume was made up to 1 ml with saline. For inhibition studies, compound 7 at a concentration of 50 & 100 µM was used. From each well 100 µL of the assay mixture was aspirated at different time intervals (0, 5, 10, 20, 30 and 60 min). From this, glucose concentration was measured using Glucose oxidase (GOD POD) kit according to the manufacturer’s protocol.
Molecular docking studies
The software Insight II/Discovery Studio 2.5 from Accelrys was used for docking and visualization of the results as described earlier [16]. The crystal structure of amylase was retrieved (PDBID: 3TOP). Before performing the Ligand fit protocol of Discovery Studio, the protein was cleaned, and the size and spatial orientation of the active site was identified. All energy calculations were performed using the CHARMM force field. Each energy-minimized final docking position of the ligands was evaluated using the interaction score function in the Ligand Fit module of Discovery Studio as reported previously [17].
Chem-informatics analysis
Utilizing the available amount of bioactivity data, we also rationalized the modes-of-action for the experimentally tested benzoxazines using in silico target prediction approaches. To this end, the Parzen-Rosenblatt Window classifier was employed with the smoothing parameter set to 0.9, using approximately 190,000 bioactive compounds covering 477 human protein targets as the training dataset. For details on the method, dataset and validation see reference 15.
Statistical analysis
Results are expressed as mean values ± SEM of three independent experiments. Data were compared by analysis of variance (ANOVA) followed by the Tukey “honestly significantly different” (HSD) post hoc analysis. Significance was accepted at p<0.05 (*), p<0.01 (**) and p<0.001 (***).
Results and Discussion
This work is based upon the previous synthesis of an oxazine derivative which was able to mimic the pyranoside structure of glycans functionally [12]. In continuation of the glycobiological aspects, the one-pot syntheses of novel 1,3-benzoxazine scaffold was carried out using 2-aminobenzyl alcohols and different aldehydes in chloroacetic acid via aerobic oxidative synthesis ( Fig. 2 ). The above design principles led to the synthesis of 13 compounds ( Table 1 ) whose α-glucosidase inhibitory activity was validated both in vivo as well as in vitro, and supported by computational approaches as described below. The synthesized aglycones inhibited both α-glucosidase and α-amylase activity, with overall relatively similar IC50 values between 11 µM and 60 µM ( Table 1 ). Among the tested derivatives, compound 7{(4-(7-chloro-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol} exhibiting strong inhibition of both glucosidases, with an IC50 values of 11 µM and 11.5 µM for α-amylase and α-glucosidase respectively. The addition of phenolic and halogen substituents to the 1,3-benzoxazine ring was found to increase the inhibitory potency (compound 7), whilst the incorporation of a flavone moiety decreases the inhibitory potency (compounds 5, 10, and 13). 1,3-benzoxazines bearing an electron withdrawing chlorine substituent were found to be more potent against α-glucosidase (compound 7), whereas the electron donating methyl group was not particularly favoured (compound 11). Introduction of an imidazole ring (compound 6), to give 1,3-benzoxazine, resulted in an enhanced inhibition (IC50 = 16 µM), while a chromene moiety decreased the activity (compound 13).
Table 1. Physical characteristics and inhibitory activities (α-glucosidase and α-amylase) of novel 1,3-benzoxazines.
| Sl No. | Benzoxazines | Yield | Melting Point | α-Glucosidase IC50 (µM) | α-Amylase IC50 (µM) |
| 1 | 4-(2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol | 83% | 160-162°C | 17.1±0.1 | 20.4±0.2 |
| 2 | 2-(1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 82% | 65-67°C | 32.0±0.2 | 59.2±1.0 |
| 3 | 2-(2-methyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 87% | 70-72°C | 21.6±0.1 | 10.6±0.3 |
| 4 | 2-(1-(4-(2-cyanophenyl)benzyl-1H-indol-3-yl),2,4-dihydro-1H-benzo(d)(1,3) oxazine | 86% | 122-124°C | NS | NS |
| 5 | 3-(2,4-dihydro-1H-benzo[d][1], [3]oxazin-2-yl)-4H-chromen-4-one | 89% | 142-144°C | NS | 23.3±0.5 |
| 6 | 2-(2-butyl-4-chloro-1H-imidazol-5-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 86% | 80-82°C | 16.7±1.0 | 18.5±0.1 |
| 7 | 4-(7-chloro-2,4-dihydro-1H-benzo[d][1], [3]oxazin-2-yl)phenol | 87% | 139-141°C | 11.5±0.1 | 11.0±0.3 |
| 8 | 7-chloro-2-(1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 85% | 125-127°C | 27.7±1.0 | 26.4±0.5 |
| 9 | 6-chloro-2-(2-phenyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 89% | 220-222°C | 27.8±0.2 | 22.2±0.1 |
| 10 | 3-(6-chloro-2,4-dihydro-1H-benzo[d][1], [3]oxazin-2-yl)-4H-chromen-4-one | 83% | 141-143°C | 31.9±0.8 | NS |
| 11 | 6-methyl-2-(2-methyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 80 % | 102-104°C | 23.8±0.1 | NS |
| 12 | 6-methyl-2-(2-phenyl-1H-indol-3-yl)-2,4-dihydro-1H-benzo[d][1], [3]oxazine | 86% | 220-222°C | 20.5±0.5 | 17.6±0.3 |
| 13 | 3-(6-methyl-2,4-dihydro-1H-benzo[d][1], [3]oxazin-2-yl)-4H-chromen-4-one | 88% | 202-204°C | NS | 51.0±0.3 |
| Acarbose | 4.3±0.04 | 4.4±0.02 | |||
NS-not significant.
To study the efficacy of the potent α-glucosidase inhibitor, compound 7 was tested for in vivo maltose and sucrose tolerance test on overnight fasted experimental rats by taking acarbose as positive control as well as glucose uptake by porcine diaphragm by using insulin as enhancer. It can be seen that acarbose (3mg/kg) significantly reduced the plasma glucose concentration at 30, 60, 90 min time intervals in starved rats treated with maltose as substrate compared to maltose control ( Fig. 3A ). In sucrose fed rats differences are less pronounced and were only significant at 60 and 90 min time points ( Fig. 3B ). At a concentration of 50 mg/kg body weight, compound 7 inhibited glucose uptake in rats fed with maltose, which was similar to acarbose treatment at 30 and 60 min. However, compound 7 significantly reduced plasma glucose concentration at the 90 min time point compared to acarbose, indicating a different Pharmacokinetic/Pharmacodynamic (PK/PD) profile of compound 7 on between these substrates. Furthermore, compound 7 significantly reduced the plasma glucose concentration throughout all time points (0–180 min) compared to the acarbose treated group when sucrose was used as a substrate ( Fig. 3B ). At a concentration of 100 mg/kg bodyweight, compound 7 was significantly more effective than the acarbose standard at all time points. In order to establish possible synergistic effects between compound 7 and acarbose, plasma glucose levels were measured in starved rats fed individually with maltose and sucrose at different time points up to 180 minutes. In both cases, synergistic activity of compound 7 (50 mg/kg bodyweight) and acarbose (3 mg/kg bodyweight) prevented substrate utilization, and the plasma glucose concentration remained unchanged when compared to the saline treated group. The inhibitory efficacy of compound 7 on rat intestinal glucosidases (maltase and sucrase) was also evaluated. The compound showed both maltase and sucrase inhibitory activities in a dose-dependent manner ( Fig. 4A and 4B ). Acarbose inhibited both the intestinal glucosidases activity with an IC50 value of 3.5 µM (maltase) and 4 µM (sucrase), while compound 7 was found to be two-fold selective for maltase (IC50 = 10 µM) over sucrase (IC50 = 20 µM). Furthermore, acarbose and compound 7 synergistically inhibited intestinal maltase more efficiently compared to intestinal sucrase ( Fig. 4C and 4D ).
Figure 3. In vivo effect of compound 7 {4-(7-chloro-2,4-dihydro-1H-benzo[d][1,3]oxazin-2-yl)phenol} and acarbose on plasma glucose concentration by oral (A) maltose and (B) sucrose tolerance test.
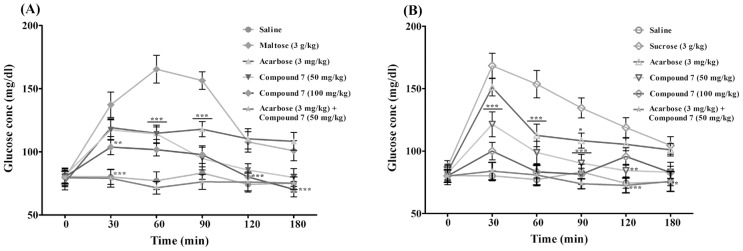
Values are presented as mean ± SEM (n = 5). *p<0.05, **p<0.01, ***p<0.001 significant compared to respective maltose/sucrose alone treated groups.
Figure 4. In vitro effect of compound 7 and its synergistic effect with acarbose on rat intestinal maltase and sucrase activities.

Percentage inhibition of compound 7 on intestinal (A) maltase and (B) sucrose. Percentage inhibition of intestinal maltase (C) and sucrase (D) induced by the compound 7 in presence of acarbose. Percentage inhibition is presented as mean ± SEM of three independent experiments. *p<0.05, **p<0.01, ***p<0.001 significant compared to acarbose.
Further, porcine diaphragm was used in order to understand the effect of compound 7 on insulin mediated glucose uptake via GLUT4. The glucose transporter isoforms GLUT4 and GLUT1 are highly expressed in muscle cells, with GLUT4 being more abundant in an intracellular compartment from which it is quickly translocated to the plasma membrane as a response to insulin challenge. Both insulin-dependent and non-insulin-dependent diabetes were shown to reduce glucose utilization in muscle either due to a defective expression or dysregulation in GLUT4 translocation [3], [18]. In the present study, glucose uptake was found to be normal in the control group treated with no insulin. However as expected, significant increased glucose uptake was observed in diaphragm treated with insulin (1 U) compared to control. We found that compound 7 at a dose of 50 and 100 µM significantly blocked the glucose uptake both in control and insulin treated diaphragm in a dose-dependent manner ( Fig. 5 ).
Figure 5. Effect of compound 7 on glucose transport across porcine diaphragm in the presence and absence of insulin.

Values are presented as presented as mean ± SEM of three independent experiments. ***p<0.001 significant compared to insulin alone treated diapharagm.
In addition to in vivo and in vitro experimental validation of the intended target of α-glucosidase shown above, in silico target prediction [19], [20] was performed with the full set of 13 synthesized compounds, in order to obtain a more comprehensive impression of the bioactivity profile of the synthesized 1,3-benzoxazine derivatives. It was found that only compound 7, the most active in the series, is predicted to target the sodium/glucose co transporters 1 and 2, which may contribute to the in vivo efficacy of this compound. However, no experimental validation of this additional target has been performed.
In order to hypothesize a binding mode, molecular docking has been performed between compound 7 and maltase-glucoamylase ( Fig. 6 ).The crystal structure of MGAM-C (Human maltase-glucoamylase C terminal domain) in complex with its inhibitor Acarbose (PDB ID: 3TOP) was used as a model to determine the molecular interaction between enzyme and the synthesized 1,3-benzoxazine derivatives [21]. In MGAM, both the N- and C-terminal domains (MGAM-N and MGAM-C) carry out the same catalytic function with different substrate specificities. The MGAM-C hydrolyzes linear α-1,4-linked oligosaccharide substrates and plays a pivotal role in the production of glucose in the human lumen and considered as an efficient drug target for T2-DM. Since, there is no information regarding the co-crystal structure of murine glucosidase and acarbose, we have used the co-crystal structure of MGAM-C and acarbose for docking studies.
Figure 6. Interaction map of MGAM-C domain co-crystallized with acarbose.

The labelled key amino acids are represented as a stick model with the carbon atom as green, and other atoms in their parent colours. The binding of acarbose, whose carbon atom is coloured in pink and other atoms with their parent colour. The hydrogen bonding is represented as dark dotted line.
The synthesized 1,3-benzoxazine derivatives have relatively similar IC50 values, Dock Score (column ‘DS’) in particular seems to show higher scores for the more active compounds (7, 1 and 6) as opposed to less active compounds (2, 8, 9 and 10) ( Table 2 ). Structurally, the most active compound 7, binds deep in the MGAM catalytic domain ( Fig. 7 ), in which the chloro- benzoxazine ring stacks into the hydrophobic cluster of Tyr1251, Trp1355, Trp1369, Tyr1427, Phe1559, and Phe1560, along with the phenolic ring stacked to Tyr1251, His1584, Trp1418, and Trp1523. The terminal hydroxyl group of the phenolic component of compound 7 shows hydrogen bonding with Asp1279 and Ile1280, which are also involved in hydrogen bonding with the terminal hydroxyl group of Acarbose in the co-crystal. In addition, the exposed oxygen atom in the benzoxazine ring of compound 7 appears to show ionic interaction with Asp1526 and Arg1510, which is also crucial in the Acarbose-MGAM co-crystal. These results clearly suggest that both acarbose and compound 7 shares similar binding pattern towards MGAM-C.
Table 2. Molecular docking results of MGAM-C with 1,3-benzoxazine derivatives.
| Compounds | Mol Wt | LS1D | LS2D | PLP1 | PLP2 | JAIN | -PMF | -LE | DS |
| 1 | 227.2 | 2.42 | 3.94 | 56.46 | 52.2 | 1 | 166.72 | 2.1 | 56.0 |
| 2 | 250.2 | 3.03 | 4.78 | 58.76 | 59.46 | 2.47 | 181.93 | 3.0 | 50.2 |
| 3 | 264.3 | 1.6 | 4.6 | 70.0 | 66.6 | 2.3 | 176.1 | 2.9 | 51.6 |
| 4 | 441.5 | 2.82 | 5.41 | 79.78 | 76.05 | 2.79 | 162.71 | 6.6 | 64.9 |
| 5 | 279.2 | 2.23 | 4.85 | 71.4 | 66.83 | 1.99 | 187.08 | 3.0 | 50.6 |
| 6 | 291.7 | 3.31 | 5.4 | 74.34 | 73.9 | 2.6 | 156.19 | 3.0 | 56.8 |
| 7 | 261.7 | 2.94 | 4.7 | 64.7 | 59.09 | 1.25 | 168.66 | −2.5 | 58.9 |
| 8 | 284.7 | 1.58 | 4.31 | 64.8 | 63.17 | 1.71 | 168.13 | 3.4 | 52.7 |
| 9 | 360.8 | 1.49 | 4.76 | 73.11 | 71.34 | 2.03 | 188.54 | 6.6 | 58.2 |
| 10 | 313.7 | 3.11 | 5.13 | 75.77 | 72.83 | 2.12 | 191.14 | 2.9 | 52.3 |
| 11 | 278.3 | 1.48 | 4.38 | 63.1 | 62.07 | 2.33 | 164.55 | 3.8 | 51.6 |
| 12 | 340.4 | 1.72 | 4.62 | 68.11 | 69.58 | 3.37 | 181.81 | 6.3 | 57.4 |
| 13 | 293.3 | 2.23 | 4.58 | 72.74 | 68.95 | 1.34 | 198.94 | 1.6 | 51.9 |
| Acarbose | 602.2 | 5.88 | 5.84 | 63.24 | 60.17 | −1.73 | 270.87 | −9.4 | 64.3 |
LS1D and LS2D: LigScore1D and 2D are a fast, simple, scoring function for predicting receptor-ligand binding affinities.
PLP1 and PLP2: Piecewise Linear Potentials 1 and 2 are fast, simple, docking function that has been shown to correlate well with protein-ligand binding affinities.
JAIN: An empirical scoring function (lipophilic interactions, polar attractive interactions, polar repulsive interactions, solvation of the protein and ligand, and an entropy term for the ligand) through an evaluation of the structures and binding affinities of a series of protein-ligand complexes.
PMF: Potential of Mean Force is the scoring function developed based on statistical analysis of the 3D structures of protein-ligand complexes.
LE: Ligand internal Energy, the internal non bonded ligand energy is calculated for each new conformation that is generated.
DS: Dock Score, candidate ligand poses are evaluated and prioritized according to the Dock Score function.
Figure 7. Interaction map of MGAM-C catalytic domain co-crystallized with Compound 7.

The labelled key amino acids are represented as a stick model with carbon atom as green, and other atoms with their parent colour. The binding of compound 7, whose carbon atom is coloured in pink and other atoms with their parent colour. The hydrogen bonding is represented in dark dotted line.
Finally, we applied a metabolite prediction software, namely MetaPrint2D-React [22], to bioactive compound 7 and found the most likely metabolic site to be a glucuronidation site with a (significant) normalised occurrence ratio of less than 0.33 and but greater than 0.15. Hence, this study for the first time demonstrated the design, synthesis, and characterization of novel 1,3-benzoxazine aglycones and their validation in vitro and in vivo. Compound 7 significantly inhibited rat intestinal glucosidases, namely maltase and sucrase, in a dose-dependent fashion and led to decreased blood sugar levels in starved rat model. In addition to this, compound 7 acts synergistically with Acarbose in lowering the blood sugar levels to that of the saline control alone.
Summary
This study demonstrated the novel synthesis of benzoxazine glycones and their effective inhibition towards glucosidases. The newly synthesized 1,3-benzoxazine derivatives showed better IC50 values for both α-glucosidase and α-amylase, ranging from 11–60 µM, and are found to be effective when compared to natural substrate aglycones, such as BOA and derivatives that deteriorate fatter in aqueous solution. The in silico molecular docking studies revealed that benzoxazines bind to the catalytic domain of MGAM-C, correlating with a high DS for the most active compound 7. The docking score of compound 7 and binding poses were found to be similar with the anti-diabetic drug acarbose. Furthermore, studies of in silico target prediction algorithms showed that compound 7 potentially targets the sodium-glucose cotransporter 1. Both in vitro and in vivo experimental results suggested an anti-hyperglycemic effect of compound 7, which significantly inhibits glucose uptake in starved rat model by blocking intestinal maltase and sucrase. Evidently, compound 7 was determined to possess the glucuronidation site, which potentially converts it into a stable glycoside in vivo. The aglycones synthesized in this study could hence constitute a novel pharmacological starting point for the treatment or alleviation of T2-DM and its secondary complications. However, further studies elucidating interaction between compound 7 and specific glucose transporters would be highly exciting.
Acknowledgments
This research was supported by University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, Department of Science and Technology (NO. SR/FT/LS-142/2012) to Basappa. Basappa thanks Karnataka University, INDIA for PAVATE fellowship. KSR thanks to DST and UGC for funding. AB thanks Unilever and SP the Netherlands Organisation for Scientific Research (NWO) and the Prins Bernhard cultuurfonds for funding. MSS thanks UGC-BSR-SAP for the research fellowship.
Funding Statement
This research was supported by University Grants Commission (41-257-2012-SR), Vision Group Science and Technology, Department of Science and Technology (NO. SR/FT/LS-142/2012) to Basappa. Basappa thanks Karnataka University, INDIA for PAVATE fellowship. KSR thanks DST and UGC for funding. AB thanks Unilever and SP the Netherlands Organisation for Scientific Research (NWO) and the Prins Bernhard cultuurfonds for funding. MSS thanks UGC-BSR-SAP for the research fellowship. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Ceriello A (2005) Postprandial hyperglycemia and diabetes complications: is it time to treat? Diabetes 54: 1–7. [DOI] [PubMed] [Google Scholar]
- 2. Kaul K, Tarr JM, Ahmad SI, Kohner EM, Chibber R (2012) Introduction to diabetes mellitus. Adv Exp Med Biol 771: 1–11. [DOI] [PubMed] [Google Scholar]
- 3. Li JM, Che CT, Lau CB, Leung PS, Cheng CH (2007) Desoxyrhaponticin (3,5-dihydroxy-4'-methoxystilbene 3-O-beta-D-glucoside) inhibits glucose uptake in the intestine and kidney: In vitro and in vivo studies. J Pharmacol Exp Ther 320: 38–46. [DOI] [PubMed] [Google Scholar]
- 4. Israili ZH (2011) Advances in the treatment of type 2 diabetes mellitus. Am J Ther 18: 117–152. [DOI] [PubMed] [Google Scholar]
- 5. Moorthy NS, Ramos MJ, Fernandes PA (2012) Studies on α-glucosidase inhibitors development: magic molecules for the treatment of carbohydrate mediated diseases. Mini Rev Med Chem 12: 713–720. [DOI] [PubMed] [Google Scholar]
- 6. Wardrop DJ, Waidyarachchi SL (2010) Synthesis and biological activity of naturally occurring α-glucosidase inhibitors. Nat Prod Rep 27: 1431–1468. [DOI] [PubMed] [Google Scholar]
- 7. Semighini EP, Resende JA, de Andrade P, Morais PA, Carvalho I, et al. (2011) Using computer-aided drug design and medicinal chemistry strategies in the fight against diabetes. J Biomol Struct Dyn 28: 787–796. [DOI] [PubMed] [Google Scholar]
- 8. Panahi F, Yousefi R, Mehraban MH, Khalafi-Nezhad A (2013) Synthesis of new pyrimidine-fused derivatives as potent and selective antidiabetic α-glucosidase inhibitors. Carbohydr Res 380: 81–91. [DOI] [PubMed] [Google Scholar]
- 9. Macías FA, Marín D, Oliveros-Bastidas A, Molinillo JM (2009) Rediscovering the bioactivity and ecological role of 1,4-benzoxazinones. Nat Prod Rep 26: 478–489. [DOI] [PubMed] [Google Scholar]
- 10. Kato-Noguchi H, Macías FA, Molinillo JM (2010) Structure-activity relationship of benzoxazinones and related compounds with respect to the growth inhibition and alpha-amylase activity in cress seedlings. J Plant Physiol 167: 1221–1225. [DOI] [PubMed] [Google Scholar]
- 11. Kato-Noguchi H (2008) Effects of four benzoxazinoids on gibberellin-induced alpha-amylase activity in barley seeds. J Plant Physiol 165: 1889–1894. [DOI] [PubMed] [Google Scholar]
- 12. Basappa, Murugan S, Kavitha CV, Purushothaman A, Nevin KG, et al. (2010) A small oxazine compound as an anti-tumor agent: a novel pyranoside mimetic that binds to VEGF, HB-EGF, and TNF-α. Cancer Lett 297: 231–243. [DOI] [PubMed] [Google Scholar]
- 13.Adisakwattana S, Ruengsamran T, Kampa P, Sompong W (2012) In vitro inhibitory effects of plant-based foods and their combinations on intestinal α-glucosidase and pancreatic α-amylase. BMC Complement Altern Med Doi: 10.1186/1472-6882-12-110. [DOI] [PMC free article] [PubMed]
- 14. Tsujii E, Muroi M, Shiragami N, Takatsuki A (1996) Nectrisine is a potent inhibitor of alpha-glucosidases, demonstrating activities similarly at enzyme and cellular levels. Biochem Biophys Res Commun 220: 459–466. [DOI] [PubMed] [Google Scholar]
- 15. Adisakwattana S, Chantarasinlapin P, Thammarat H, Yibchok-Anun S (2009) A series of cinnamic acid derivatives and their inhibitory activity on intestinal alpha-glucosidase. J Enzyme Inhib Med Chem 24: 1194–1200. [DOI] [PubMed] [Google Scholar]
- 16.Chandramohanadas R, Basappa, Russell B, Liew K, Yau YH et al.., (2014) Small Molecule Targeting Malaria Merozoite Surface Protein-1 (MSP-1) Prevent Host Invasion of Divergent Plasmodial Species. J Infect Dis. In press. [DOI] [PMC free article] [PubMed]
- 17.Sukhorukov AY, Nirvanappa AC, Swamy J, Ioffe SL, Nanjunda Swamy S, et al.., (2014) Synthesis and characterization of novel 1,2-oxazine-based small molecules that targets acetylcholinesterase. Bioorg Med Chem Lett. In press. [DOI] [PubMed]
- 18. Cazarolli LH, Pereira DF, Kappel VD, Folador P, Figueiredo Mdos S, et al. (2013) Insulin signaling: a potential signaling pathway for the stimulatory effect of kaempferitrin on glucose uptake in skeletal muscle. Eur J Pharmacol 712: 1–7. [DOI] [PubMed] [Google Scholar]
- 19. Koutsoukas A, Simms B, Kirchmair J, Bond PJ, Whitmore AV, et al. (2011) From in silico target prediction to multi-target drug design: current databases, methods and applications. J Proteomics 74: 2554–2574. [DOI] [PubMed] [Google Scholar]
- 20. Koutsoukas A, Lowe R, Kalantarmotamedi Y, Mussa HY, Klaffke W, et al. (2013) In silico target predictions: defining a benchmarking data set and comparison of performance of the multiclass Naïve Bayes and Parzen-Rosenblatt window. J Chem Inf Model 53: 1957–1966. [DOI] [PubMed] [Google Scholar]
- 21. Ren L, Qin X, Cao X, Wang L, Bai F, et al. (2011) Structural insight into substrate specificity of human intestinal maltase-glucoamylase. Protein Cell 2: 827–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Boyer S, Arnby CH, Carlsson L, Smith J, Stein V, et al. (2007) Reaction site mapping of xenobiotic biotransformations. J Chem Inf Model 47: 583–590. [DOI] [PubMed] [Google Scholar]