

Abstract
Purpose
The risk of severe neutropenia from treatment with irinotecan is related in part to UGT1A1*28, a variant that reduces the elimination of SN-38, the active metabolite of irinotecan. We aimed to identify the maximum-tolerated dose (MTD) and dose-limiting toxicity (DLT) of irinotecan in patients with advanced solid tumors stratified by the *1/*1, *1/*28, and *28/*28 genotypes.
Patients and Methods
Sixty-eight patients received an intravenous flat dose of irinotecan every 3 weeks. Forty-six percent of the patients had the *1/*1 genotype, 41% had the *1/*28 genotype, and 13% had the *28/*28 genotype. The starting dose of irinotecan was 700 mg in patients with the *1/*1 and *1/*28 genotypes and 500 mg in patients with the *28/*28 genotype. Pharmacokinetic evaluation was performed at cycle 1.
Results
In patients with the *1/*1 genotype, the MTD was 850 mg (four DLTs per 16 patients), and 1,000 mg was not tolerated (two DLTs per six patients). In patients with the *1/*28 genotype, the MTD was 700 mg (five DLTs per 22 patients), and 850 mg was not tolerated (four DLTs per six patients). In patients with the *28/*28 genotype, the MTD was 400 mg (one DLT per six patients), and 500 mg was not tolerated (three DLTs per three patients). The DLTs were mainly myelosuppression and diarrhea. Irinotecan clearance followed linear kinetics. At the MTD for each genotype, dosing by genotype resulted in similar SN-38 areas under the curve (AUCs; r2 = 0.0003; P = .97), but the irinotecan AUC was correlated with the actual dose (r2 = 0.39; P < .001). Four of 48 patients with disease known to be responsive to irinotecan achieved partial response.
Conclusion
The UGT1A1*28 genotype can be used to individualize dosing of irinotecan. Additional studies should evaluate the effect of genotype-guided dosing on efficacy in patients receiving irinotecan.
INTRODUCTION
Irinotecan is an active agent in the treatment of several solid tumors. Its pharmacology has been studied for the last 20 years, and the key pathways for irinotecan disposition have been well described. Because irinotecan is a prodrug, its activation to the potent metabolite SN-38 is required for both antitumor activity and the mechanism-related toxicities of the drug, especially neutropenia. The relative importance of intratumoral activation to SN-38, versus hepatic activation, is still unknown. Patients treated with irinotecan have marked variability in toxicity at standard doses.1
UGT1A1*28 is a biomarker of neutropenia that is mentioned in the irinotecan package insert. This germline genetic variant results in reduced expression of UGT1A1, the main metabolizing enzyme that inactivates SN-38 through glucuronidation to SN-38 glucuronide (SN-38G).2 In several studies, the variability in systemic exposure to SN-38 has been associated with the risk of neutropenia.3–6 Thus, the presence of UGT1A1*28 could be an indicator of a patient's risk of myelosuppression.4,7,8 According to the package insert,9 a reduced first dose of irinotecan should be considered in patients homozygous for this allele (*28/*28), but the extent of dose reduction is not indicated. Whether the standard dose of irinotecan is appropriate for patients who do not have the *28/*28 genotype remains to be established.
Despite the known variability in irinotecan disposition, the drug is still dosed according to body surface area (BSA), a strategy without a pharmacologic rationale. Because UGT1A1*28 is associated with the risk of irinotecan-related myelosuppression, a phase I study of single-agent irinotecan in patients with refractory cancer was designed to find the safe doses of irinotecan according to the UGT1A1*28 genotypes of patients. We hypothesized that patients with the *1/*1 genotype would tolerate a higher dose than the standard dose and that patients with the *28/*28 genotype would require dose reduction.
PATIENTS AND METHODS
Patient Eligibility
Patients with histologically confirmed solid tumors or lymphoma refractory to standard therapy were enrolled. Eligibility criteria included UGT1A1 *1/*1, *1/*28, and *28/*28 genotypes (patients carrying the rare UGT1A1*36 and UGT1A1*37 alleles captured by the assay were not eligible); Eastern Cooperative Oncology Group (ECOG) performance status of 0 to 1; measurable or assessable disease; life expectancy greater than 3 months, age 18 years or older; leukocyte count greater than 3,000/μL; absolute neutrophil count (ANC) at least 1,500/μL; platelet count at least 100,000/μL; total bilirubin within normal institutional limits; ALT and AST levels at less than 2.5 times the upper limit of normal; and a serum creatinine level within normal institutional limits or a glomerular filtration rate greater than 50 mL/min/1.73 m2 for patients with creatinine levels above institutional normal as calculated by the modified Modification of Diet in Renal Disease equation recommended by the National Kidney Disease Education Program.
Study Objectives
The primary objective was to describe the maximum-tolerated dose (MTD) and dose-limiting toxicity (DLT) of irinotecan in patients with the *1/*1 and *1/*28 genotypes with advanced solid tumors. Secondary objectives included finding the safe dose of irinotecan in patients with the *28/*28 genotype, evaluating the pharmacokinetics of irinotecan and its metabolites, and describing the antitumor response to irinotecan when doses are given according to genotype. The institutional review board of the University of Chicago and NorthShore University Health System approved the study, and each patient signed a written informed consent form before entering the study.
Drug Administration and Dose Escalation
Irinotecan was administered every 3 weeks by intravenous infusion during 90 minutes. In patients with the *1/*1 and *1/*28 genotypes, the starting dose was 700 mg (flat dose equivalent to 390 mg/m2, according to a BSA of 1.8 m2).10 In patients with the *28/*28 genotype, the starting dose was 500 mg (ie, 280 mg/m2). BSA has not been shown to be a predictor of irinotecan pharmacokinetic variability.11 Planned dose escalation in the patients with the *1/*1 and *1/*28 genotypes was by increments of 150 mg.
DLT at cycle 1 was defined as grade 4 neutropenia lasting ≥ 4 days, grade 3 or higher neutropenia on a scheduled treatment day, grade 3 or higher febrile neutropenia, grade 4 anemia or thrombocytopenia, grade 3 or higher diarrhea despite administration of loperamide therapy, grade 3 or higher nonhematologic toxicity, or grade 4 nausea or nausea/vomiting graded by the National Cancer Institute Common Toxicity Criteria, version 3.0.
One treatment cycle was 21 days. Before starting irinotecan, patients were pretreated with ondansetron 16 mg. Diarrhea was treated promptly with loperamide 4 mg at the onset and then with 2 mg every 2 hours, until the patient was diarrhea free for at least 12 hours. Hematopoietic growth factors (eg, granulocyte colony-stimulating factor) and other supportive care measures were administered when deemed clinically appropriate by the treating physician. Atropine 0.25 to 1 mg was administered in case of acute cholinergic syndrome.
The planned dose escalation was to enroll six patients at each dose level and to continue escalating if fewer than two of the six patients experienced DLT. If two or more of the six patients experienced DLT, the dose was declared above the MTD. The next-lower dose level then was expanded to up to 12 patients; if fewer than four of the 12 patients experienced DLT, this dose level was declared the MTD. No intrapatient dose escalation was allowed. In some cohorts, more than 12 patients were enrolled to adequately assess the tolerability of the dose level.
Pharmacokinetics of Irinotecan and UGT1A1*28 Genotyping
We evaluated the pharmacokinetics of irinotecan and metabolites during cycle 1. Serial blood samples were collected into heparinized tubes before drug administration; at 30 minutes; and at 1, 1.5, 1.7, 1.8, 2, 2.25, 2.5, 3, 3.5, 5.5, 6.5, 7.5, 8.5, and 25.5 hours after the start of the irinotecan infusion. The pharmacokinetics of irinotecan and its metabolites SN-38 and SN-38 glucuronide (SN-38G) were determined as previously described12 by using noncompartmental analysis (PK Solutions Software, version 2.0, Summit Research Services, Montrose, CO). Estimated parameters included the area under the concentration-time curve from 0 to infinity (AUC0-∞), clearance (calculated as dose ÷ AUC), and glucuronidation ratio (calculated as AUCSN-38G ÷ AUCSN-38). UGT1A1*28 genotyping from DNA extracted from peripheral blood was conducted as previously reported.13
Efficacy and Toxicity Assessment
Clinical examination and hepatic and renal function tests were performed at baseline and within 48 hours before each irinotecan administration. Computed tomography scans of measurable lesions were assessed at baseline and then repeated every two cycles. Objective tumor response was evaluated every two cycles according to the RECIST criteria,14 and patients who experienced progression before two cycles were not evaluable for tumor response. Blood counts were measured at baseline, weekly during cycle 1, and within 48 hours before each treatment administration in the following cycles. If a patient had grade 4 neutropenia, the blood counts were repeated daily until resolution occurred to determine whether grade 4 neutropenia was a DLT.
Patients were treated at the protocol-specified full dose of irinotecan if the laboratory entry criteria were met before each cycle. Any patient who experienced grade 3 or greater toxicity attributable to therapy had treatment held until toxicity resolved to grade 1 or lower. Patients who experienced DLT were treated at the next-lower dose for their genotype (in cycle 2) as long as there was no evidence of progressive disease. Irinotecan was discontinued because of disease progression, patient refusal, or physician recommendation.
Statistical Methods
The MTD for each genotype was determined as described above. Exploratory analyses were conducted on the secondary objectives of the study, and P < .05 was considered statistically significant. The nonparametric Kruskal-Wallis test and analysis of variance were used for group comparisons; these agreed closely, and only the nonparametric results are reported here. Multiple linear regression models were fit to examine the association between ANC nadir and dose, sex, genotype, and pharmacokinetic variables.
RESULTS
Patients, Genotypes, and Dose Range
Sixty-eight patients were assessable for DLT (Table 1). Most patients were men and white. The prevalent tumor types were GI and lung. The distribution of the UGT1A1 genotypes was 31 *1/*1, 28 *1/*28, and nine *28/*28 (P > .05 for the Hardy Weinberg equilibrium). The genotype frequency followed the expected distribution of UGT1A1*28 in populations of European origin. The dose levels of irinotecan administered to patients ranged from 400 to 1,000 mg (flat dose; Table 2).
Table 1.
Patient Characteristics
| Characteristic | No. | % |
|---|---|---|
| No. of evaluable patients | ||
| Enrolled | 68 | |
| Assessable for toxicity | 68 | |
| Assessable for pharmacokinetics | 67 | |
| Assessable for tumor response† | 48 | |
| Age, years | ||
| Median | 63 | |
| Range | 35-80 | |
| Sex | ||
| Male | 45 | 66 |
| Female | 23 | 34 |
| Ethnicity | ||
| White | 56 | 82.4 |
| Black | 9 | 13.2 |
| Hispanic | 3 | 4.4 |
| Body surface area, m2 | ||
| Median | 1.9 | |
| Range | 1.4-2.4 | |
| Previous chemotherapy | ||
| Yes | 66 | 97 |
| No | 2 | 3 |
| ECOG PS | ||
| 0 | 27 | 39.7 |
| 1 | 40 | 58.8 |
| 2 | 1 | 1.5 |
| Tumor type‡ | ||
| Gastrointestinal | 30 | 44 |
| Lung | 30 | 44 |
| Other | 8 | 12 |
| UGT1A1*28 genotype§ | ||
| 1/1 | 31 | 46 |
| 1/28 | 28 | 41 |
| 28/28 | 9 | 13 |
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Twenty patients were not assessed for response, because they were taken off study during cycle 1. One patient had only three blood draws and, therefore, could not undergo pharmacokinetic evaluation.
Other tumor types included breast, maxillary sinus, endocrine, thymic carcinoid, carcinoid, and Merkel cell.
The UGT1A1*28 genotypes did not deviate from the Hardy-Weinberg equilibrium (P ≥ .05).
Table 2.
Dose Escalations and DLTs by Dose Level and UGT1A1*28 Genotype
| Irinotecan Dose, mg† | Total No. of Patients |
1/1 Genotype |
1/28 Genotype |
28/28 Genotype |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No. of DLTs | Type of DLT | No. of Patients | No. of DLTs | Type of DLT | No. of Patients | No. of DLTs | Type of DLT | No. of Patients | ||
| 400 | 6 | — | — | 1 | G3 neutropenic fever | 6 | ||||
| 500 | 3 | — | — | 3 | G3 infection, G4 neutropenia > 4 days, G3 diarrhea | 3 | ||||
| 700 | 31 | 1 | G4 neutropenia > 4 days and G3 diarrhea | 9 | 5 | G4 neutropenia (n = 2), G3 (n = 2) or G4 (n = 1) febrile neutropenia | 22 | — | ||
| 850 | 22 | 4 | G3 febrile neutropenia (n = 2), G4 febrile neutropenia and G4 diarrhea, G4 neutropenia > 4 days | 16 | 4 | G3 nausea, G3 nausea and vomiting, G5 neutropenia, G4 neutropenia > 4 days and thrombocytopenia | 6 | — | ||
| 1,000‡ | 6 | 2 | G3 diarrhea and febrile neutropenia, G3 diarrhea and G4 neutropenia > 4 days | 6 | — | — | ||||
Abbreviations: DLTs, dose-limiting toxicities; G, grade.
Flat dose.
All patients who received 1,000 mg were men.
Assessment of MTD and DLT by Genotype
In patients with the *28/*28 genotype treated at the starting dose of 500 mg, three DLTs were observed in three patients, and no additional patients were enrolled at this dose level. The protocol subsequently was amended to treat the patients with the *28/*28 genotype at 400 mg, and one DLT was observed in six patients. The MTD in patients with the *28/*28 genotype, therefore, was determined to be 400 mg.
In patients with the *1/*28 genotype, two DLTs were observed in six patients treated at 850 mg (in addition to two grade 3 nausea and/or vomiting subsequently amended not to be considered as DLTs). Because five DLTs were observed in 22 patients treated at 700 mg, the MTD in patients with the *1/*28 genotype was determined to be 700 mg.
In patients with the *1/*1 genotype, one DLT was observed in nine patients treated at 700 mg. Two DLTs were observed in six men treated at 1,000 mg. At 850 mg, only two of 11 men experienced DLT, whereas two of five women experienced DLT. The protocol, therefore, was amended to escalate the irinotecan dose to 1,000 mg only in men; two of these six patients experienced DLT. Because, four DLTs were observed overall in 16 patients treated at 850 mg, the MTD in patients with the *1/*1 genotype was determined to be 850 mg.
As summarized in Table 2, the predominant DLT was myelosuppression, with neutropenic DLTs accounting for 75% of DLTs (16 of 20) and severe diarrhea accounting for 25% of DLTs (five of 20). One patient (a man with lung cancer, *1/*28 genotype, and treatment at 850 mg) died as a result of complications of pneumonia (regarded as probably related to treatment) in the presence of grade 4 neutropenia.
Pharmacokinetics of Irinotecan and Antitumor Activity
Sixty-seven patients were assessable for pharmacokinetics. In the 400 to 1,000 mg dose range, irinotecan clearance followed linear kinetics (Table 3). SN-38 AUC displayed the same linear behavior (results not shown). As expected, dose-adjusted irinotecan AUC was independent of the UGT1A1*28 genotype (P = .62), whereas dose-adjusted SN-38 AUC increased if patients had the *1/*28 or *28/*28 genotype relative to those with the *1/*1 genotype (P = .01; Fig 1).
Table 3.
Pharmacokinetic Parameters of Irinotecan, SN-38, and SN-38G
| Dose, mg | No. of Patients | Irinotecan AUC0-∞ (h · mg/L) |
Irinotecan CL (L/h) |
SN-38 AUC0-∞ (h · mg/L) |
SN-38G AUC0-∞ (h · mg/L) |
GR |
|||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | Mean | SD | Mean | SD | ||
| 400 | 6 | 17.8 | 4.7 | 24.2 | 8.4 | 0.867 | 0.708 | 1.37 | 0.59 | 2.63 | 1.95 |
| 500 | 3 | 35.0 | 17.3 | 18.4 | 12.7 | 1.196 | 0.662 | 3.41 | 2.19 | 2.65 | 0.61 |
| 700 | 30 | 33.8 | 11.8 | 23.3 | 8.5 | 0.808 | 0.655 | 2.76 | 1.96 | 5.17 | 5.06 |
| 850 | 22 | 42.8 | 12.3 | 21.5 | 6.0 | 0.868 | 0.761 | 3.63 | 2.15 | 5.43 | 3.24 |
| 1,000 | 6 | 44.5 | 12.1 | 24.0 | 7.2 | 0.665 | 0.290 | 2.87 | 0.81 | 4.81 | 1.63 |
NOTE. One patient treated at 850 mg had almost identical SN-38G concentrations at 8.5 and 25 hours, resulting in an SN-38G AUC0-∞ of 196.56 h · mg/L and a GR of 178.69, which have been removed from the calculations of the means and SDs in the 850-mg–dose group.
Abbreviations: AUC0-∞, area under the concentration-time curve from zero to infinity; CL, clearance rate; GR, glucuronidation ratio; SD, standard deviation.
Fig 1.
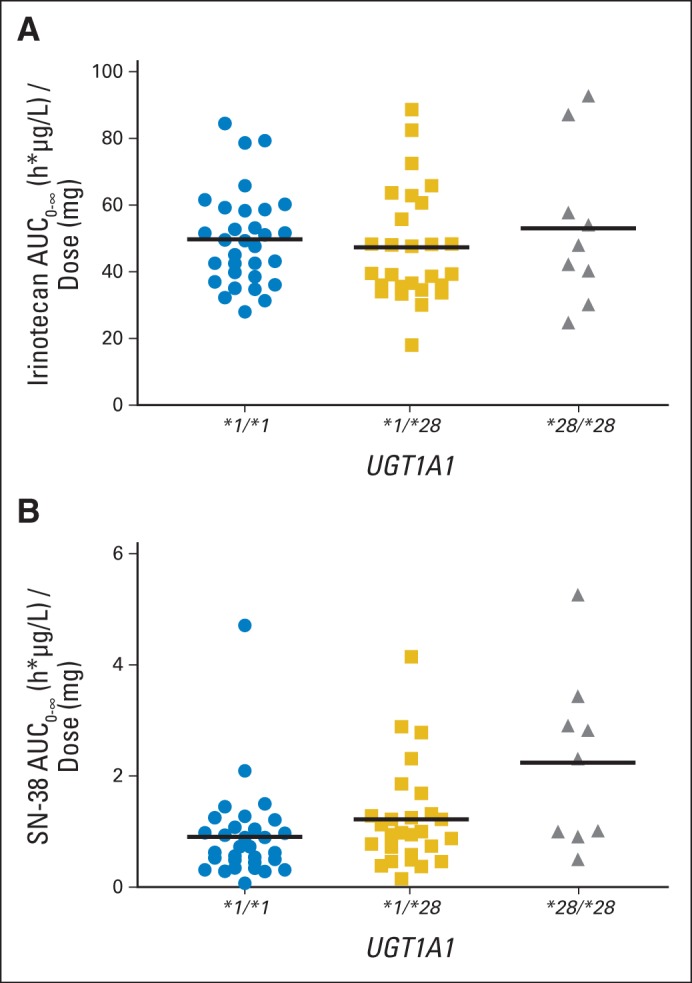
Relationship between the UGT1A1*28 genotype and dose-adjusted areas under the curve (AUCs) of irinotecan (A; P = .62) and SN-38 (B; P = .01). Horizontal bars represent the means.
The MTD was 850 mg in patients with the *1/*1 genotype, 700 mg in those with the *1/*28 genotype, and 400 mg in those with the *28/*28 genotype. Individualizing dose by genotype resulted in similar mean SN-38 AUCs across all MTDs (r2 = 0.0003; P = .97; Fig 2). Conversely, at the MTD for each genotype, irinotecan AUC increased significantly with dose (r2 = 0.39; P < .001; Fig 2).
Fig 2.
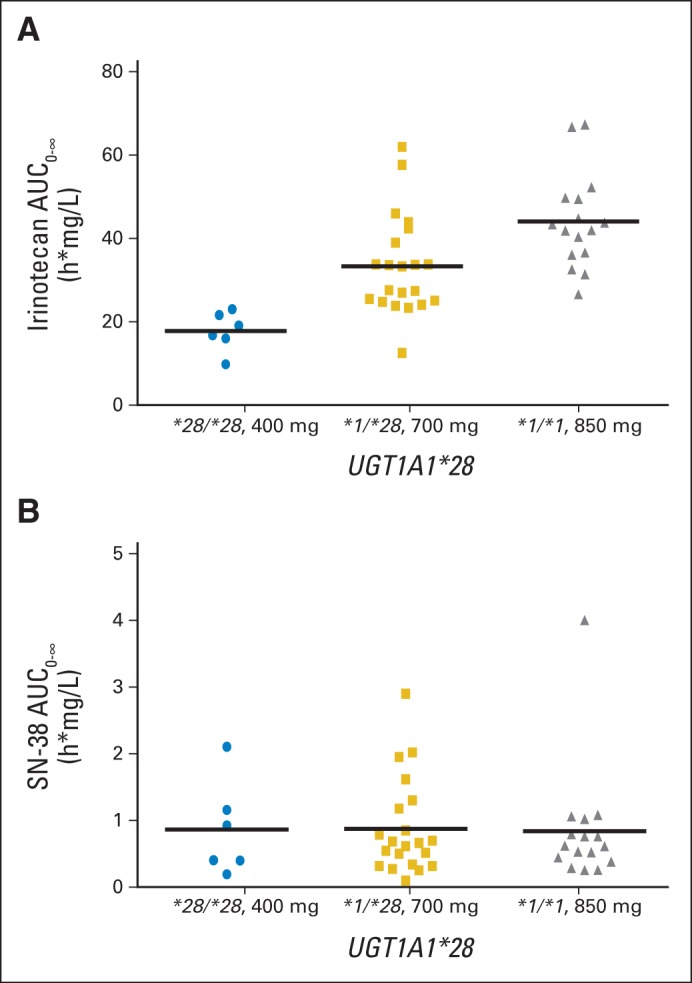
Area under the curve (AUC) of irinotecan (A; r2 = 0.39; P < .001) and SN-38 (B; r2 = 0.0003; P = .97) at the maximum-tolerated dose (MTD) for each genotype. Horizontal bars represent the means.
After 28 patients had been enrolled, an interim analysis was performed, and a multivariable model for ANC nadir that included dose, genotype, sex, BSA, and SN-38 AUC was developed. Both SN-38 AUC and sex were associated with ANC nadir (P < .01). By using the complete data from the study, this model (r2 = 0.24) also detected a significant effect of SN-38 AUC (r2 = 0.14; P = .001) but only a trend for sex (r2 = 0.01; P = .098), which suggests lower nadirs for women compared with men; dose and genotype were not statistically significant.
Forty-eight patients were assessable for antitumor response. Four partial responses were observed in patients with non–small-cell lung cancer (700 mg and *1/*28 genotype), gastric cancer (850 mg and *1/*1 genotype), small bowel cancer (850 mg and *1/*28 genotype), and small-cell lung cancer (850 mg and *1/*1 genotype).
DISCUSSION
The current landscape of early drug development is changing, and selecting patients by predictive biomarkers is becoming a common practice to reduce the size of trials and achieve end points faster and at a lower cost. Dose-finding studies should be guided by genetic markers of safety, because they can effectively optimize and tailor dosing. To our knowledge, this is the first single-agent, phase I study that has used germline genetics to individualize dosing of a cancer therapy. This study represents a model for performing genotype-directed safety studies with genetic information. These results remind us about the importance of the germline constitutive genome as the basis for dosing and the risk of adverse reactions, because there are clearly subgroups of patients that tolerate different doses of drug therapies.15 If these results are taken in the context of the various cancer genome sequencing efforts,16–18 it is likely that somatic genetic markers will direct treatment selection and that germline genetic also might contribute to determination of resistance and safe administration doses, in a truly individualized approach.
For irinotecan, we demonstrated that three distinct groups of patients can be identified according to the common variant UGT1A1*28 and that the tolerable doses of irinotecan range from 400 to 850 mg. This is a difference of more than two-fold; the ability of the genotype to predict dosing of a cytotoxic agent is remarkable. We also demonstrate that the toxicity profile of individualized irinotecan dosing does not change. Neutropenia is the dominant DLT, and diarrhea contributes to a lesser extent. This is expected, as these adverse effects are likely pharmacodynamic consequences of higher exposure to SN-38, which is in part determined by the UGT1A1*28 variant.3–6 Our results conclusively identify its role as a major determinant of safe dosing of irinotecan.
The patients with the homozygous *28/*28 genotype are at the highest risk of toxicity, particularly at the higher doses of irinotecan8 used in the schedule of administration once every 3 weeks. Pre-emptive genotyping of UGT1A1*28 is not commonly performed to identify these patients, because most guidelines (including the package insert) do not clearly indicate the extent to which the dose should be reduced.2 In the patients with the *28/*28 genotype in this trial, a 20% dose reduction to 500 mg (the equivalent of 278 mg/m2) is not safe: All three patients treated at this dose level experienced a DLT. Instead, a 40% dose reduction to 400 mg (220 mg/m2) seemed to be tolerated (only one grade 3 neutropenic fever among six treated patients; Table 2). In the FOLFIRI regimen (irinotecan with infusional fluorouracil and leucovorin), a 30% dose reduction appears safe for patients with the *28/*28 genotype.19 Our study, for the first time to our knowledge, provides information about the safe doses of irinotecan in the every-3-week regimen in the patients with the highest risk of severe toxicity.
Patients with the *1/*1 and *1/*28 genotypes represent approximately 90% of the European population.20 In this study, patients with the *1/*28 genotype tolerated 700 mg (390 mg/m2), a dose higher than the standard 350 mg/m2. We also observed that patients with the *1/*1 genotype could tolerate an even higher dose of 850 mg (470 mg/m2); only four DLTs were observed among 16 patients treated at this dose level. An irinotecan dose of 500 mg/m2 has been shown to be tolerable in patients with refractory cancer who have favorable characteristics (eg, good risk, not heavily pretreated),21 but UGT1A1*28 was not used for dose selection in that study. In other UGT1A1*28 genotype–directed studies of FOLFIRI, patients with the *1/*1 genotype tolerated approximately twice the dose recommended by the label,19,22 probably because the therapeutic window is wider in the biweekly regimen of FOLFIRI (standard dose, 180 mg/m2) than in the regimen administered once every 3 weeks (standard dose, 350 mg/m2). However, the patient populations are quite different between our study (heavily pretreated, different tumor types) and the FOLFIRI studies (first-line treatment of metastatic colorectal cancer).19,22 Similar to this study, these studies with FOLFIRI also showed that the patients with the *1/*28 genotype tolerated a lower dose relative to the patients with the *1/*1 genotype, confirming the importance of individualized dosing by genotype. The *1/*28 genotype confers an intermediate-metabolizer phenotype13,23–25 and an intermediate risk of toxicity.4
The pharmacokinetics of irinotecan are informative about the relationship among genotype, dosing, and neutropenia. Although irinotecan pharmacokinetics retains its linearity even at higher doses,26 the UGT1A1*28 status of patients clearly affects the exposure to the active metabolite SN-38, as shown by a linear, direct relationship with the UGT1A1*28 genotypes (Fig 1). Because this relationship already has been demonstrated in many studies,3–6 it is important to show that a strategy of individualizing dosing of irinotecan is able to individualize the exposure to SN-38. As shown in Figure 2, although irinotecan AUC increases according to the different MTDs in each genotype group, the mean SN-38 AUC levels are comparable across the different MTDs in each genotype group. This normalization of SN-38 AUC by genotype-directed dosing may be key to preserving antitumor efficacy, even when patients with the *28/*28 genotype are treated at lower doses. BSA, as expected, is not a determinant of the pharmacokinetic variability, and considerations on dosing by mg/m2 should be made—including a comparison of flat dosing versus BSA dosing at the MTD in additional studies that use genotype-guided dosing of irinotecan.
A single-agent, dose-finding study such as this has pros and cons. Irinotecan is given mostly in combination regimens, and its use as a single agent is limited, although it might have more widespread use as a single agent in countries where expensive biologics are neither reimbursed nor available,27,28 because irinotecan is a generic drug. In Asian countries, UGT1A1*6 should be considered in addition to UGT1A1*28 to inform dosing of irinotecan.6 Single-agent studies have the advantage of being not confounded by overlapping toxicities, and the pharmacokinetic data are not influenced by potential drug-drug interactions. These single-agent studies demonstrate a proof of concept so that they can be followed by additional genotype-directed trials in the combination setting.
These safety studies represent another important step toward precision medicine in oncology. The dose-escalation studies conducted so far,19,22 including ours, clearly show that a significant group of patients treated with irinotecan is probably being under-dosed. However, practice cannot be changed until evidence for improved efficacy of this approach is in fact demonstrated (ie, that increasing dosing by genotype confers higher response and/or longer survival compared with standard dosing). The studies of FOLFIRI in patients treated for first-line metastatic colorectal cancer suggest that higher doses may confer a clinical advantage (measured as time to progression).19,22 These are small studies, and efficacy was a secondary end point. In the highly refractory population of patients with solid tumors included in this trial, four partial responses were observed among 48 evaluable patients with diseases known to respond to irinotecan. Although three of them occurred at a higher dose of 850 mg, it is still premature to conclude that higher doses by genotype are more efficacious. A prospective, phase II study in patients with metastatic colorectal cancer treated with FOLFIRI plus bevacizumab in the first-line setting will evaluate the effect of genotype-directed dosing of irinotecan on survival.
Glossary Terms
- UGT1A1 (UDP-glucuronosyltransferase 1A1):
an isoform of uridine-diphosphoglucuronate glucuronosyltransferases. UGT1A1 is responsible for the glucuronidation of bilirubin, xenobiotic compounds, and endogenous steroids. Variants of UGT1A1 are known to affect the glucuronidation of SN-38, the active metabolite of irinotecan.
- area under the curve (AUC):
a measure of the amount of drug in the blood over a set period of time (eg, 24 hours) that can be used to determine drug exposure.
- genotype:
the specific genetic makeup of a given individual. Although genotypes give rise to the phenotype of an individual, genotypes and phenotypes are not always correlative. For example, some genotypes are expressed only under specific environmental conditions.
- irinotecan:
a plant alkaloid. A prodrug is converted to a biologically active metabolite 7-ethyl-10-hydroxy-camptothecin (SN-38) by a carboxylesterase-converting enzyme. SN-38 inhibits topoisomerase I activity.
- neutropenic fever:
an oral temperature of at least 100.4°F for at least 1 hour when the absolute neutrophil count is < 0.5 × 109/L.
- pharmacokinetics:
a branch of pharmacology that studies the relationship between drug exposure level, time course of exposure, and the overall response of an organism. Although pharmacokinetics is largely applied to drugs, it is also applicable to other compounds such as nutrients, toxins, hormones, etc. Pharmacokinetics is subdivided into absorption and disposition (distribution, metabolism, and excretion) and is generally referred to as ADME (absorption, distribution, metabolism, excretion). With respect to drugs administered, all processes occur in tandem once a drug dose is administered. In clinical trials, phase I studies will typically study pharmacokinetics and safety of the drug.
- phenotype:
the overall appearance of an organism, or the observable expression of a specific trait, determined by its genotype and environmental factors.
- prodrug:
a drug that is given in an inactive form and is bioactivated to a pharmacologic drug by one or more metabolic processes.
Footnotes
See accompanying editorial on page 2287
Supported by PAAR-Pharmacogenomics of Anticancer Agents Research Group Award No. U01 GM061393 from the National Institute of General Medical Sciences (M.J.R.), The University of Chicago Comprehensive Cancer Center grant No. P30 CA14599 (M.J.R.), and National Cancer Institute grant No. K07CA140390-01 (F.I.).
Terms in blue are defined in the glossary, found at the end of this article and online at www.jco.org.
Authors' disclosures of potential conflicts of interest and author contributions are found at the end of this article.
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Although all authors completed the disclosure declaration, the following author(s) and/or an author's immediate family member(s) indicated a financial or other interest that is relevant to the subject matter under consideration in this article. Certain relationships marked with a “U” are those for which no compensation was received; those relationships marked with a “C” were compensated. For a detailed description of the disclosure categories, or for more information about ASCO's conflict of interest policy, please refer to the Author Disclosure Declaration and the Disclosures of Potential Conflicts of Interest section in Information for Contributors.
Employment or Leadership Position: None Consultant or Advisory Role: None Stock Ownership: None Honoraria: None Research Funding: None Expert Testimony: None Patents, Royalties, and Licenses: Mark J. Ratain, Camptothecin drug combinations and methods with reduced side effects, patent No. 5,786,344; Methods for detection of promoter polymorphism in a UGT gene promoter, patent No. 6,395,481; Methods for detection of promoter polymorphism in a UGT gene promoter, patent No. 6,472,157; Camptothecin drug combination and methods with reduced side effects, patent No. 0768895; Methods and compositions for predicting irinotecan toxicity, patent No. 1629111; Methods for predicting irinotecan toxicity, patent No. 7,807,350 Other Remuneration: None
AUTHOR CONTRIBUTIONS
Conception and design: Federico Innocenti, Richard L. Schilsky, Theodore Karrison
Provision of study materials or patients: Richard L. Schilsky, Robert Marsh, Michael L. Maitland, Ravi Salgia
Collection and assembly of data: Federico Innocenti, Jacqueline Ramírez, Linda Janisch, Larry K. House, Soma Das, Michelle Turcich, Michael L. Maitland, Ravi Salgia
Data analysis and interpretation: Federico Innocenti, Richard L. Schilsky, Jacqueline Ramírez, Samir Undevia, Kehua Wu, Robert Marsh, Theodore Karrison, Michael L. Maitland, Ravi Salgia, Mark J. Ratain
Manuscript writing: All authors
Final approval of manuscript: All authors
REFERENCES
- 1.Meyerhardt JA, Mayer RJ. Systemic therapy for colorectal cancer. N Engl J Med. 2005;352:476–487. doi: 10.1056/NEJMra040958. [DOI] [PubMed] [Google Scholar]
- 2.Innocenti F, Ratain MJ. Pharmacogenetics of irinotecan: Clinical perspectives on the utility of genotyping. Pharmacogenomics. 2006;7:1211–1221. doi: 10.2217/14622416.7.8.1211. [DOI] [PubMed] [Google Scholar]
- 3.Ramchandani RP, Wang Y, Booth BP, et al. The role of SN-38 exposure, UGT1A1+28 polymorphism, and baseline bilirubin level in predicting severe irinotecan toxicity. J Clin Pharmacol. 2007;47:78–86. doi: 10.1177/0091270006295060. [DOI] [PubMed] [Google Scholar]
- 4.Innocenti F, Undevia SD, Iyer L, et al. Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J Clin Oncol. 2004;22:1382–1388. doi: 10.1200/JCO.2004.07.173. [DOI] [PubMed] [Google Scholar]
- 5.Minami H, Sai K, Saeki M, et al. Irinotecan pharmacokinetics/pharmacodynamics and UGT1A genetic polymorphisms in Japanese: Roles of UGT1A1+6 and +28. Pharmacogenet Genomics. 2007;17:497–504. doi: 10.1097/FPC.0b013e328014341f. [DOI] [PubMed] [Google Scholar]
- 6.Han JY, Lim HS, Shin ES, et al. Comprehensive analysis of UGT1A polymorphisms predictive for pharmacokinetics and treatment outcome in patients with non-small-cell lung cancer treated with irinotecan and cisplatin. J Clin Oncol. 2006;24:2237–2244. doi: 10.1200/JCO.2005.03.0239. [DOI] [PubMed] [Google Scholar]
- 7.Toffoli G, Cecchin E, Corona G, et al. The role of UGT1A1+28 polymorphism in the pharmacodynamics and pharmacokinetics of irinotecan in patients with metastatic colorectal cancer. J Clin Oncol. 2006;24:3061–3068. doi: 10.1200/JCO.2005.05.5400. [DOI] [PubMed] [Google Scholar]
- 8.Hoskins JM, Goldberg RM, Qu P, et al. UGT1A1+28 genotype and irinotecan-induced neutropenia: Dose matters. J Natl Cancer Inst. 2007;99:1290–1295. doi: 10.1093/jnci/djm115. [DOI] [PubMed] [Google Scholar]
- 9.Pfizer. Camptosar Package Insert. New York, NY: Pfizer; 2012. [Google Scholar]
- 10.Egorin MJ. Horseshoes, hand grenades, and body-surface area-based dosing: Aiming for a target. J Clin Oncol. 2003;21:182–183. doi: 10.1200/JCO.2003.10.084. [DOI] [PubMed] [Google Scholar]
- 11.Mathijssen RH, Verweij J, de Jonge MJ, et al. Impact of body-size measures on irinotecan clearance: Alternative dosing recommendations. J Clin Oncol. 2002;20:81–87. doi: 10.1200/JCO.2002.20.1.81. [DOI] [PubMed] [Google Scholar]
- 12.Iyer L, Das S, Janisch L, et al. UGT1A1+28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J. 2002;2:43–47. doi: 10.1038/sj.tpj.6500072. [DOI] [PubMed] [Google Scholar]
- 13.Iyer L, Hall D, Das S, et al. Phenotype-genotype correlation of in vitro SN-38 (active metabolite of irinotecan) and bilirubin glucuronidation in human liver tissue with UGT1A1 promoter polymorphism. Clin Pharmacol Ther. 1999;65:576–582. doi: 10.1016/S0009-9236(99)70078-0. [DOI] [PubMed] [Google Scholar]
- 14.Therasse P, Eisenhauer EA, Verweij J. RECIST revisited: A review of validation studies on tumour assessment. Eur J Cancer. 2006;42:1031–1039. doi: 10.1016/j.ejca.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 15.Gillis NK, Patel JN, Innocenti F. Clinical implementation of germ line cancer pharmacogenetic variants during the next-generation sequencing era. Clin Pharmacol Ther. 2014;95:269–280. doi: 10.1038/clpt.2013.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cancer Genome Atlas Research Network. Weinstein JN, Collisson EA, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sleijfer S, Bogaerts J, Siu LL. Designing transformative clinical trials in the cancer genome era. J Clin Oncol. 2013;31:1834–1841. doi: 10.1200/JCO.2012.45.3639. [DOI] [PubMed] [Google Scholar]
- 18.Innocenti F, Schilsky RL. Translating the cancer genome into clinically useful tools and strategies. Dis Model Mech. 2009;2:426–429. doi: 10.1242/dmm.004119. [DOI] [PubMed] [Google Scholar]
- 19.Marcuello E, Páez D, Paré L, et al. A genotype-directed phase I-IV dose-finding study of irinotecan in combination with fluorouracil/leucovorin as first-line treatment in advanced colorectal cancer. Br J Cancer. 2011;105:53–57. doi: 10.1038/bjc.2011.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hall D, Ybazeta G, Destro-Bisol G, et al. Variability at the uridine diphosphate glucuronosyltransferase 1A1 promoter in human populations and primates. Pharmacogenetics. 1999;9:591–599. [PubMed] [Google Scholar]
- 21.Merrouche Y, Extra JM, Abigerges D, et al. High dose-intensity of irinotecan administered every 3 weeks in advanced cancer patients: A feasibility study. J Clin Oncol. 1997;15:1080–1086. doi: 10.1200/JCO.1997.15.3.1080. [DOI] [PubMed] [Google Scholar]
- 22.Toffoli G, Cecchin E, Gasparini G, et al. Genotype-driven phase I study of irinotecan administered in combination with fluorouracil/leucovorin in patients with metastatic colorectal cancer. J Clin Oncol. 2010;28:866–871. doi: 10.1200/JCO.2009.23.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang D, Zhang D, Cui D, et al. Characterization of the UDP glucuronosyltransferase activity of human liver microsomes genotyped for the UGT1A1+28 polymorphism. Drug Metab Dispos. 2007;35:2270–2280. doi: 10.1124/dmd.107.017806. [DOI] [PubMed] [Google Scholar]
- 24.Ramírez J, Mirkov S, Zhang W, et al. Hepatocyte nuclear factor-1 alpha is associated with UGT1A1, UGT1A9 and UGT2B7 mRNA expression in human liver. Pharmacogenomics J. 2008;8:152–161. doi: 10.1038/sj.tpj.6500454. [DOI] [PubMed] [Google Scholar]
- 25.Innocenti F, Grimsley C, Das S, et al. Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics. 2002;12:725–733. doi: 10.1097/00008571-200212000-00006. [DOI] [PubMed] [Google Scholar]
- 26.Chabot GG, Abigerges D, Catimel G, et al. Population pharmacokinetics and pharmacodynamics of irinotecan (CPT-11) and active metabolite SN-38 during phase I trials. Ann Oncol. 1995;6:141–151. doi: 10.1093/oxfordjournals.annonc.a059109. [DOI] [PubMed] [Google Scholar]
- 27.Kmietowicz Z. Task force hopes to deliver affordable cancer drugs to developing countries. BMJ. 2009;339:4506b. doi: 10.1136/bmj.b4506. [DOI] [PubMed] [Google Scholar]
- 28.Farmer P, Frenk J, Knaul FM, et al. Expansion of cancer care and control in countries of low and middle income: A call to action. Lancet. 2010;376:1186–1193. doi: 10.1016/S0140-6736(10)61152-X. [DOI] [PubMed] [Google Scholar]