

Abstract
Epidemiological evidence suggests that obesity is associated with inflammation of the respiratory tract and the pathogenesis of asthma. The purpose of this study was to examine the role of phospholipase D1 (PLD1) in leptin-induced expression of the proinflammatory cytokine, tumor necrosis factor (TNF)-α, and to suggest a molecular link between obesity and respiratory tract inflammation. We investigated whether leptin, a typical adipocytokine, plays a role in the expression of TNF-α through increased PLD1 activity in Raw 264.7. Leptin enhanced the activity of PLD1 through activation of PLCγ and Src, while PLD1 siRNA decreased the leptin-induced expression and production of TNF-α. Leptin-induced PLD activation was also inhibited by a PLCγ inhibitor (PAO) and Src kinase inhibitor (PP2), indicating that PLCγ and Src kinase are upstream activators of PLD1. Down-regulation of PLD1 also completely blocked activation of p70S6K, an activator of JNK. Leptin-induced expression of TNF-α was also prevented by inhibition of p70S6K and JNK. Taken together, these results indicate that PLD1 acts as an important regulator of leptin-induced expression of TNF-α by participating in the PLCγ/Src/PLD1/PA/p70S6K/JNK pathway.
Introduction
Leptin, a hormone/cytokine secreted by adipocytes, is an important regulator of metabolic, reproductive, and immune functions [1], [2]. Increased levels of leptin may play a role in energy homeostasis [3], neuroendocrine function [4], angiogenesis [5], and production of inflammatory mediators [6], [7]. Indeed, leptin can activate dendritic cells (DC), monocytes, and macrophages and stimulate them to express and release Th1-type cytokines [8]. Leptin also regulates pro-inflammatory immune responses in the white adipose tissue of obese mice, rats [9], and humans [10]. Despite the evidence for a role of leptin in the immune response, the intracellular signaling mechanisms involved, including those affecting of cytokine secretion, are not well understood.
Phospholipase D (PLD) catalyzes the hydrolysis of phosphatidylcholine to phosphatidic acid (PA) and choline [11]. PA, one of the enzymatic products of PLD, can be metabolically converted to diacylglycerol (DAG) and lyso-PA (LPA), both of which have second messenger roles that could contribute to the effects of PLD. Two mammalian PLD isoforms, PLD1 and PLD2, have been cloned and found to share 55% sequence homology. PLD is responsible for signaling in a variety of cellular processes, including cell proliferation [12], differentiation, cell survival, apoptosis [13], and the immune response [14]. Phospholipase C (PLC) also plays an important regulatory role in cellular immune responses [15], [16]. Activated PLCγ hydrolyses phosphatidylinositol 4,5-bisphosphate (PIP2) to DAG and inositol 1,4,5-trisphosphate (IP3), resulting in activation of protein kinase C (PKC) [17]. Several studies have shown that PLCγ is responsible for the activity of PLD in intracellular signaling events [18], [19].
mTOR is a serine/threonine protein kinase thought to be involved in inflammatory and thrombotic processes as a regulator of signal-dependent translation in various cell types [20], [21]. The best-known downstream effectors of mTOR include the ribosomal subunit S6 kinase (S6K) and the eukaryotic translation initiation factor 4E binding protein 1 (4EBP1), two regulators of mitogen-stimulated translation initiation [22]. A recent study demonstrated a critical relationship between PLD and PA in the mTOR pathway [23]. Moreover, mTOR is involved in leptin-induced inflammatory cytokine production in macrophages [24]. Leptin can induce c-jun N-terminal protein kinase (JNK) through PLC and, subsequently, PKC activation [25]. Additionally, in Kupffer cells, leptin stimulates TNF-α production via the JNK and p38 MAPK pathways [26].
In the present study, we investigated the relationship between leptin-induced TNF-α production and PLD signaling, and demonstrated that PLD1 is activated by leptin via PLCγ/Src kinase and is involved in activation of the p70S6K/JNK pathway that leads to increased TNF-α expression/production in Raw 264.7 cells.
Materials and Methods
Cell culture
Raw 264.7 murine macrophage cells were purchased from the American Type Culture Collection (ATCC) and cultured in Dulbecco's Modified Eagle's Medium (DMEM, low glucose) with 10% fetal bovine serum, 100 units/mL penicillin and 100 ng/mL streptomycin (Invitrogen) at 37°C with 5% CO2 in humidified air.
Reagents
Fetal bovine serum, penicillin/streptomycin, and DMEM were purchased from Invitrogen. PAO, rapamycin, and SP600125 were obtained from Calbiochem, and [3H]-palmitic acid was from Perkin Elmer Life Sciences. 1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphate (PA) dissolved in chloroform was purchased from Avanti Polar Lipids (Alabaster, AL, USA). The following polyclonal antibodies from Cell Signaling (Beverly, MA, USA) were used: PLD1, PLCγ, p-PLCγ, Src kinase, p-Src kinase, mTOR, p-mTOR, p-p70S6K antibody, and p70S6K. GAPDH and PLCγ were from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Semi-quantitative and real-time PCR
Total cellular RNA was isolated using TRizol reagent (Invitrogen) according to the manufacturer's instructions. cDNA synthesis was performed using Superscript kit (Invitrogen). Each PCR procedure was carried out in optimal MgCl2 concentration, annealing temperature, and cycle number for the linear amplification range. PCR products were analyzed by 1% agarose gel electrophoresis. Real-time PCR was performed on a CFX96 Real time system using iQ SYBR green supermix (Bio-Rad). All gene expression values were normalized to those of GAPDH. The primer sequences were as follows: TNF-α sense (5′AGCCCACG-TCGTAGCAAACCACCAA3′) and antisense (5′AACACCCATTCCC-TTC-ACAGAGCAAT3′) (PCR product, 200 bp); PLD1 sense (5′ACTCTGTCCAAAGTTAACA TGT CACTG3′) and antisense (5′GGCTTTGTCTTGAGCAGCTCTCT3′) (PCR product, 245 bp); GAPDH sense (5′CATGAGAAGTATGACAACAGCCT3′) and antisense (5′AGTCCTTCCAC GATA-CCAAAGT3′) (PCR product, 300 bp).
Transient transfection with plasmid DNA in Raw 264.7 cells
Raw 264.7 cells were transiently transfected using 5 µg each of pEGFP-C1 (vector), EGFP-PLD1, EGFP-dominant negative PLD1, EGFP-PLD2, or EGFP-dominant negative PLD2 plasmid using Lipofectamine reagents (Invitrogen). 48 h after transfection the cells were serum-starved for 18 h and then treated with leptin.
Western blot analysis
Cells were first lysed in 20 mM Tris, pH 7.5, containing 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1% Triton X-100, 1 mM phenylmethylsulfonyl fluoride, and 1 mM Na3VO4. Protein samples of 10 to 20 µg were loaded onto SDS-polyacrylamide gels (10%), electrophoresed, and transferred to nitrocellulose membranes (Amersham Biosciences). After being blocked with 5% dried skim milk for 2 h, the membranes were incubated with primary antibodies, followed by horseradish peroxidase-conjugated secondary antibody (1∶2000, New England Biolabs, Beverly, MA), and specific bands were detected by ECL (Amersham Biosciences).
Determination of PLD activity
PLD activity was determined by the formation of phosphatidylbutanol (PBt), the product of PLD-mediated transphosphatidylation in the presence of butanol as described previously, with a slight modification [27]. The cells, which had been incubated for the indicated times in 6-well plates, were radioactively labeled with 2 µCi/mL of [3H]-palmitic acid in serum-free medium for 20 h, then pretreated with 0.3% (v/v) 1-butanol for 15 min before stimulation with leptin. Additionally, cells were preincubated with PAO or PP2 for 30 min after being labeled with [3H]-palmitic acid, and serum-starved for 18 h. After treatment with leptin for 30 min, they were quickly washed with ice-cold phosphate-buffered saline and suspended in ice-cold methanol. Lipids were extracted according to the Bligh and Dyer method [28], and PBt was separated by TLC using the acetate/isooctane/acetic acid/water (110∶50∶20∶100, v/v) solvent system. Regions corresponding to authentic PBt bands were identified with 0.002% (w/v) primulin in 80% (v/v) acetone, scraped, and counted using a liquid scintillation counter.
ELISA
Cell supernatants were collected after leptin treatment and the concentration of TNF-α was measured with commercial ELISA kits (Bender Medsystems, Vienna, Austria) according to the manufacturer's instructions. The concentration of each sample was determined from a standard curve, and ranged from 3.13 to 2000 pg/mL. The means of triplicate ELISA values for each of the doses of PLD1 knockdown and TNF-α protein expression were calculated by linear regression.
Statistical analysis
All experiments were repeated at least 3 times. Statistical comparisons were made using one-way Student's t-test or multi-factorial ANOVA. GraphPad Prism (version 6; GraphPad Software, San Diego, Calif., USA) was used for statistical analysis. Values of p<0.05 were considered statistically significant.
Results
PLD1 regulates leptin-induced TNF-α expression and production in Raw 264.7 cells
To determine whether leptin induces production of TNF-α, Raw 264.7 cells were treated with leptin for the indicated times. Treatment of the cells with leptin increased TNF-α expression (Figure 1A,B) and production(Figure 1C)). To determine the role of PLD in this increase, we measured the activity of PLD. As shown in Figure 2A, leptin increased PLD activity in treated cells nearly 2.2-fold within 15 min. Next, we investigated which isozymes of PLD in Raw 264.7 cells were involved in the observed effect. We found that knockdown of PLD1 by siRNA significantly reduced the expression and production of TNF-α, while PLD2 siRNA did not. This suggested that only PLD1 had an impact on the expression (Figure 2B,C) and production of TNF-α (Figure 2F). A similar effect on TNF-α was also induced by PA, the end product of PLD (Figure 2D,E), confirming that PLD1 plays an important role in the expression and production of TNF-α in response to leptin.
Figure 1. Effect of leptin on TNF-α expression in Raw 264.7 cells.
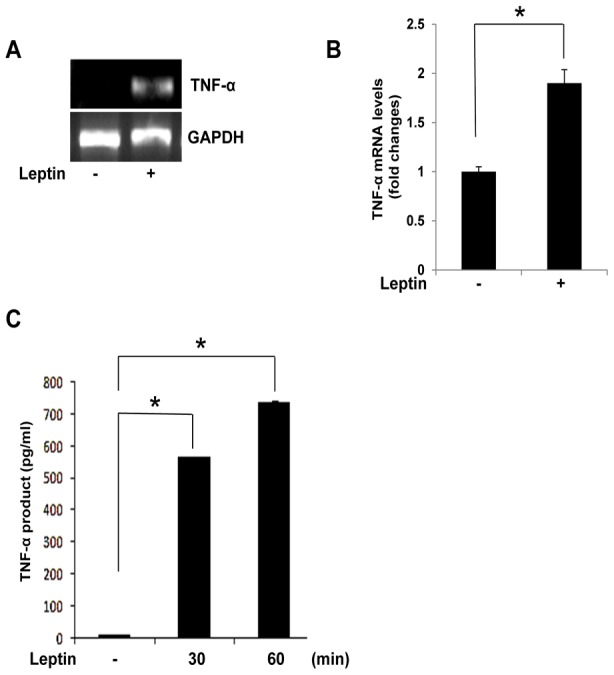
(A,B) Raw 264.7 cells were treated with leptin (20 nM) for 30 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs control. (C) For ELISA, cells in 96-well culture plates were treated with leptin (20 nM) for the indicated times. Results are the mean ± S.E. for each group of samples. Data are means ± S.E. of eight values. *p<0.05 vs control.
Figure 2. Effect of PLD knockdown on leptin-induced TNF-α expression in Raw 264.7 cells.

(A) Raw264.7 cells were labeled with 2 µCi/mL [3H]-palmitic acid and stimulated with leptin (20 nM) for the indicated times. PLD activity was determined by estimating the amount of [3H]-PBt in the presence of 1-butanol. Results are the mean ± S.D. of three independent experiments. *p<0.05 vs control. (B,C) Raw264.7 cells were transiently transfected with 200 nM PLD1 siRNA or PLD2 siRNA for 48 h and were stimulated with leptin (20 nM) for 30 min. Cells lysates were subjected to Western blotting. The cells were harvested and total RNA was isolated using TRIzol reagent, and mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs leptin-treated control. (D,E) Cells were treated with leptin (20 nM) or PA (10 µM) for 30 min and analyzed. mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs control. (F) Cells on 96-well culture plates were transfected with 200 nM PLD1 siRNA or PLD2 siRNA for 48 h and then stimulated with leptin (20 nM) for 1 h. Cells were also treated with PA (10 µM) for 1 h. Results are the mean ± S.E. amounts of TNF-α measured by ELISA for each group of samples. Data are means ± S.E. of eight values. *p<0.05 vs leptin-treated control.
Leptin activates PLCγ/Src kinase, resulting in increased PLD activity and TNF-α expression and production
To investigate the mechanism by which leptin activates PLD1, we examined PLCγ signaling, as PLCγ activation was previously shown to increase PLD activity [18]. As shown in Figure 3A, leptin induced the phosphorylation of both PLCγ and Src kinase. Therefore, we asked whether phosphorylation of Src was dependent on the presence of PLCγ. PAO, a PLCγ inhibitor, completely blocked the phosphorylation of Src kinase induced by leptin (Figure 3B). Next, to determine whether PLCγ/Src kinase affected PLD activation in response to leptin, cells were pretreated with a specific PLCγ inhibitor (PAO) or a Src kinase inhibitor (PP2). As shown in Figure 3C, pretreatment with PAO or PP2 completely inhibited PLD activation. Furthermore, when the activity of PLCγ and Src kinase was blocked, significant inhibition of TNF-α expression (Figure 3D,E) and its production (Figure 3F) were observed, demonstrating that the PLCγ/Src kinase pathway is located upstream of PLD1. Taken together, these results establish that PLCγ/Src kinase activation regulates the leptin-induced PLD activation that leads to TNF-α expression in Raw 264.7 cells.
Figure 3. Effect of the PLCγ inhibitor (PAO) and Src kinase inhibitor (PP2) on leptin-induced TNF-α expression in Raw 264.7 cells.

(A) Raw 264.7 cells were treated with leptin (20 nM) for 5 min. Cells were harvested, and cell extracts were subjected to immunoblotting for PLCγ and Src kinase, respectively. (B) Cells were pretreated with PAO (20 µM) for 30 min and then stimulated with leptin (20 nM) for 5 min. The cell lysates were then analyzed by Western blotting using total Src and p-Src antibodies. (C) Cells were labeled with 2 µCi/ml [3H]-palmitic acid and then pretreated with PAO or PP2 (20 µM) for 30 min before stimulation with leptin (20 nM) for 15 min. PLD activity was determined by estimating the formation of [3H]-PBt in the presence of 1-butanol. Results are the mean ± S.D. of three independent experiments. *p<0.05 vs leptin-treated control (D,E) Cells were pretreated with PAO or PP2 (20 µM) for 30 min and then stimulated with leptin (20 nM) for 30 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs leptin-treated control. (F) Cells in 96-well culture plates were pretreated with PAO or PP2 (20 µM) for 30 min and then stimulated with leptin (20 nM) for 1 h. Results are the mean ± S.E. amounts of TNF-α measured by ELISA for each group of samples. Data are means ± S.E. of eight values. *p<0.05 vs leptin-treated control.
mTOR/JNK signaling is required for leptin-induced TNF-α expression and production
The mTOR pathway integrates insulin and nutrient signaling in many cells [29], [30]. We observed that leptin stimulated the phosphorylation of p70S6K (Figure 4A). To determine whether the PLCγ/Src kinase pathway was required for this effect, cells were pretreated with the specific PLCγ inhibitor (PAO) or the Src kinase inhibitor (PP2), respectively. As shown in Figure 4B, these treatments completely inhibited he p70S6K phosphorylation. We also studied the role of PLD1 in this effect using siRNA transfection; this completely abolished p70S6K phosphorylation in response to leptin (Figure 4C). To confirm the role of PLD1, we treated cells with PA and found that it also increased p70S6K phosphorylation, even in the absence of leptin (Figure 4D). As a final step, we investigated whether TNF-α expression and production were regulated by mTOR. Pretreatment of cells with rapamycin, a specific inhibitor of mTOR, completely blocked TNF-α expression (Figure 4E,F) and production in response to leptin (Figure 4G).
Figure 4. Involvement of p70S6K in leptin-induced TNF-α expression in Raw 264.7 cells.

(A) Cells were treated with leptin (20 nM) for 5 min and then harvested, and the amounts of total p70S6K and p-p70S6K were determined by Western blotting. (B) Cells were pretreated with PAO or PP2 (20 µM) for 30 min and then treated with leptin as above. (C) Cells were transiently transfected with 200 nM PLD1 siRNA for 48 h and stimulated with leptin (20 nM) for 30 min. Cell lysates were analyzed as above. (D) Cells were treated with PA (10 µM) for 1 h, and cell lysates were analyzed as above. (E,F) Cells were pretreated with rapamycin (50 µM) for 30 min and then stimulated with leptin (20 nM) for 30 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs leptin-treated control (G) Cells in 96-well culture plates were pretreated with rapamycin (100 nM) for 30 min and then stimulated with leptin (20 nM) for 1 h. Results are the mean ± S.E. amounts of TNF-α measured by ELISA for each group of samples. Data are means ± S.E. of eight values. *p<0.05 vs leptin-treated control.
One of the targets of leptin is MAPK [26], And we confirmed that leptin stimulated the phosphorylation of MAPKs: treatment with leptin increased the phosphorylation of JNK, p38MAPK, and ERK (Figure 5A). Interestingly, rapamycin only blocked the phosphorylation of JNK not the other MAPKs, suggesting that only JNK signaling was mTOR-dependent (Figure 5B). Finally, we investigated the effect of JNK activation on the TNF-α expression and production in leptin-treated Raw264.7 cells. The JNK inhibitor, SP600125, significantly inhibited the expression (Figure 5C,D) and production (Figure 5E) of TNF-α, which was induced by leptin. These results demonstrate that both the expression and production of TNF-α in response to leptin are regulated by the mTOR/JNK pathway in Raw 264.7 cells.
Figure 5. Effect of JNK phosphorylation on leptin-induced TNF-α expression in Raw 264.7 cells.

(A) Raw 264.7 cells treated with leptin (20 nM) for 5 min were harvested, and cell extracts were subjected to immunoblotting for active and total MAPKs. (B) Cells were pretreated with rapamycin (100 nM) for 30 min and then stimulated with leptin (20 nM) for 5 min. Phosphorylated JNK was determined by Western blotting. (C,D) Cells were pretreated with SP600125 (50 µM) for 30 min and then stimulated with leptin (20 nM) for 30 min. Total RNA was isolated using TRIzol reagent, and mRNA levels were determined by semi-quantitative and real-time RT-PCR with primers for TNF-α or GAPDH. *p<0.05 vs leptin -treated control. (E) Cells in 96-well culture plates were pretreated with SP600125 (50 µM) for 30 min and then stimulated with leptin (20 nM) for 1 h. Results are the mean ± S.E. amounts of TNF-α measured by ELISA for each group of samples. Data are means ± S.E. of eight values. *p<0.05 vs leptin -treated control.
Discussion
The effect of leptin on cells of the immune system such as macrophages and B cells is typically achieved via stimulation of Th1 cytokine expression [31], [32]. Regulation of leptin-induced signaling pathways is a key step in immune-related diseases. Interestingly leptin potentiates the production of proinflammatory cytokines (including TNF-α and IL-6) in macrophages in response not only to LPS but also to ozone exposure [33], [34]. However, the leptin-induced intracellular signaling pathways leading to cytokine expression are not yet fully understood. Here, we investigated the molecular mechanism of PLD1-mediated TNF-α expression and production in response to leptin. Many researchers used a supraphysiological concentration of leptin to find out mechanisms for leptin-induced proinflammatory cytokines in several cell models [6], [7], [24], [35].
In the present study, leptin increased PLD1 activity, reaching a maximum value at 15 min (Figure 1), without a concurrent change in PLD1 and PLD2 expression (data not shown), which suggests that PLD1 activation was caused by leptin-regulated signaling. A PLD-knockdown experiment using PLD1 and PLD2 siRNAs showed that leptin-induced TNF-α expression and production were coupled to PLD1 activation in Raw 264.7 cells, these studies suggest a possible functional role of PLD1 in cytokine expression and production. PLD is a key enzyme that transduces direct and receptor-mediated signaling from several molecules, in particular Ras, Src kinase, and PLCγ [14], [19], [36]. PLC activation is required for the signal transduction events leading to inflammatory responses [16], [37]. In LPS-stimulated macrophages and human endothelial cells, proinflammatory cytokine secretion and expression are dependent on Src kinase [38], [39], [40]. Therefore, the role of Src kinase in immune response is rapidly emerging. Prior to this report, however, the relationship between leptin and PLCγ/Src kinase, which regulate PLD1 activation in the immune response, had not been examined. In the present study, we showed that leptin stimulated the activation of PLCγ and Src kinase. As expected, PAO and PP2 attenuated not only PLD1 activation but also TNF-α production, thus supporting the idea that the PLCγ/Src kinase pathway regulates PLD1 activation and TNF-α production induced by leptin in Raw 264.7 cells.
Accumulating evidence shows that the mTOR signaling pathway regulates inflammatory mechanisms in a number of cell types [41], [42]. Activation of mTOR has been associated with cytokine and hormone receptor signaling that involving interferon and insulin [43], [44]. Indeed, in macrophages, leptin was shown to induce time-dependent phosphorylation of 4EBP1 and p70S6K via a rapamycin-sensitive mechanism [24]. Yet, the pathway responsible for p70S6K activation in TNF-α production is not fully understood. Therefore, we hypothesized that leptin might modulate TNF-α production via the mTOR pathway. In the present study, we found that phosphorylation of p70S6K was increased by leptin, and that rapamycin could suppress TNF-α expression and production. These results demonstrate that mTOR is critically important in leptin signaling for macrophage activation leading to cytokine production.
The MAPK family plays important roles in the expression of proinflammatory cytokines in many cell types [45], [46]. Among these kinases, JNK is another candidate for regulation by PLD [47], and is involved in inflammation [48], [49]. Leptin is reported to induce JNK phosphorylation through PLC and, subsequently, PKC activation [25]. Leptin also stimulates production of TNF-α via the p38 MAPK and JNK pathways in LPS-treated cells [26]. Furthermore, it activates the MAPK pathways to mediate anti-apoptotic effects in mononuclear cells [50]. Thus the MAPK pathways appear to be essential in a wide range of immune responses. In the present study, phosphorylation of JNK was increased by leptin, and decreased when the cells were pretreated with rapamycin. This led us to think that leptin-induced JNK activation was involved in the mTOR pathway. As expected, a JNK inhibitor, SP600125, attenuated the leptin-induced expression and production of TNF-α, thus raising the possibility that JNK regulates TNF-α expression and production induced by leptin in Raw 264.7cells.
In conclusion, the new evidence found in our study suggests that leptin-induced PLD1 activation occurs via PLCγ/Src, and that activation of PLD1 is essential for TNF-α expression and production via the mTOR/JNK pathway in Raw 264.7 cells (Figure 6).
Figure 6. PLD1 activation is crucial for leptin-induced TNF-α expression and production in Raw264.7 cells.
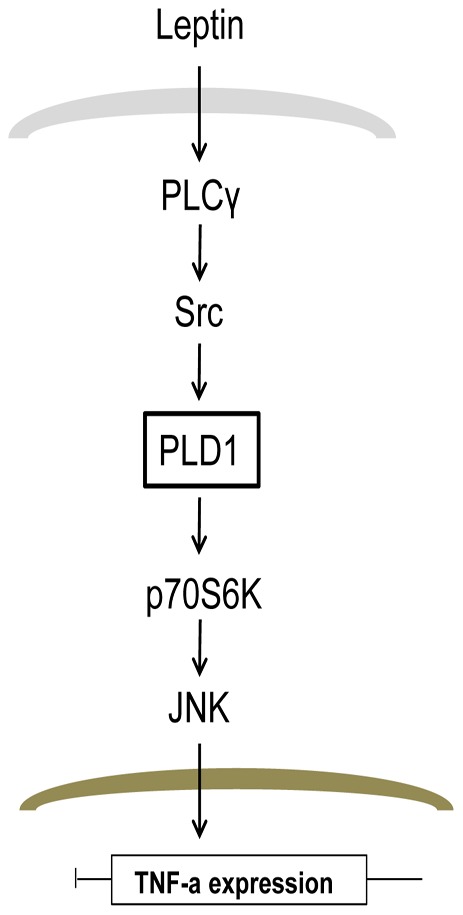
Our novel finding in this experiment is that leptin-induced TNF-α expression is controlled by PLD1 activation.
Funding Statement
This study was supported by the Korea Science and Engineering Foundation (NRF-2012R1A5A2A34671243) through the Medical Research Center at Hanyang University College of Medicine, Republic of Korea, and the National Research Foundation of Korea (NRF), funded by the Mid-career Researcher Program (NRF-2010-0026844). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Friedman JM, Halaas JL (1998) Leptin and the regulation of body weight in mammals. Nature 395: 763–770. [DOI] [PubMed] [Google Scholar]
- 2. Spiegelman BM, Flier JS (2001) Obesity and the regulation of energy balance. Cell 104: 531–543. [DOI] [PubMed] [Google Scholar]
- 3. Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW (2006) Central nervous system control of food intake and body weight. Nature 443: 289–295. [DOI] [PubMed] [Google Scholar]
- 4. Haynes WG, Morgan DA, Walsh SA, Mark AL, Sivitz WI (1997) Receptor-mediated regional sympathetic nerve activation by leptin. J Clin Invest 100: 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, et al. (1998) Biological action of leptin as an angiogenic factor. Science 281: 1683–1686. [DOI] [PubMed] [Google Scholar]
- 6. Yamagishi SI, Edelstein D, Du XL, Kaneda Y, Guzman M, et al. (2001) Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein-1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem 276: 25096–25100. [DOI] [PubMed] [Google Scholar]
- 7. Mancuso P, Canetti C, Gottschalk A, Tithof PK, Peters-Golden M (2004) Leptin augments alveolar macrophage leukotriene synthesis by increasing phospholipase activity and enhancing group IVC iPLA2 (cPLA2gamma) protein expression. Am J Physiol Lung Cell Mol Physiol 287: L497–502. [DOI] [PubMed] [Google Scholar]
- 8. Mattioli B, Straface E, Quaranta MG, Giordani L, Viora M (2005) Leptin promotes differentiation and survival of human dendritic cells and licenses them for Th1 priming. J Immunol 174: 6820–6828. [DOI] [PubMed] [Google Scholar]
- 9. Yamakawa T, Tanaka S, Yamakawa Y, Kiuchi Y, Isoda F, et al. (1995) Augmented production of tumor necrosis factor-alpha in obese mice. Clin Immunol Immunopathol 75: 51–56. [DOI] [PubMed] [Google Scholar]
- 10. Kern PA, Saghizadeh M, Ong JM, Bosch RJ, Deem R, et al. (1995) The expression of tumor necrosis factor in human adipose tissue. Regulation by obesity, weight loss, and relationship to lipoprotein lipase. J Clin Invest 95: 2111–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Billah MM, Anthes JC (1990) The regulation and cellular functions of phosphatidylcholine hydrolysis. Biochem J 269: 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jones D, Morgan C, Cockcroft S (1999) Phospholipase D and membrane traffic. Potential roles in regulated exocytosis, membrane delivery and vesicle budding. Biochim Biophys Acta 1439: 229–244. [DOI] [PubMed] [Google Scholar]
- 13. Nakashima S, Nozawa Y (1999) Possible role of phospholipase D in cellular differentiation and apoptosis. Chem Phys Lipids 98: 153–164. [DOI] [PubMed] [Google Scholar]
- 14. Park SY, Cho JH, Oh DY, Park JW, Ahn MJ, et al. (2009) House dust mite allergen Der f 2-induced phospholipase D1 activation is critical for the production of interleukin-13 through activating transcription factor-2 activation in human bronchial epithelial cells. J Biol Chem 284: 20099–20110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee CH, Shieh DC, Tzeng CY, Chen CP, Wang SP, et al. (2008) Bradykinin-induced IL-6 expression through bradykinin B2 receptor, phospholipase C, protein kinase Cdelta and NF-kappaB pathway in human synovial fibroblasts. Mol Immunol 45: 3693–3702. [DOI] [PubMed] [Google Scholar]
- 16. Bandyopadhaya A, Das D, Chaudhuri K (2009) Involvement of intracellular signaling cascades in inflammatory responses in human intestinal epithelial cells following Vibrio cholerae infection. Mol Immunol 46: 1129–1139. [DOI] [PubMed] [Google Scholar]
- 17. Kang HK, Lee HY, Lee YN, Jo EJ, Kim JI, et al. (2004) Up-regulation of phospholipase Cgamma1 and phospholipase D during the differentiation of human monocytes to dendritic cells. Int Immunopharmacol 4: 911–920. [DOI] [PubMed] [Google Scholar]
- 18. Exton JH (1996) Regulation of phosphoinositide phospholipases by hormones, neurotransmitters, and other agonists linked to G proteins. Annu Rev Pharmacol Toxicol 36: 481–509. [DOI] [PubMed] [Google Scholar]
- 19. Oh DY, Park SY, Cho JH, Lee KS, Min do S, et al. (2007) Phospholipase D1 activation through Src and Ras is involved in basic fibroblast growth factor-induced neurite outgrowth of H19-7 cells. J Cell Biochem 101: 221–234. [DOI] [PubMed] [Google Scholar]
- 20. Kraiss LW, Weyrich AS, Alto NM, Dixon DA, Ennis TM, et al. (2000) Fluid flow activates a regulator of translation, p70/p85 S6 kinase, in human endothelial cells. Am J Physiol Heart Circ Physiol 278: H1537–1544. [DOI] [PubMed] [Google Scholar]
- 21. Weyrich AS, Denis MM, Schwertz H, Tolley ND, Foulks J, et al. (2007) mTOR-dependent synthesis of Bcl-3 controls the retraction of fibrin clots by activated human platelets. Blood 109: 1975–1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gingras AC, Raught B, Sonenberg N (1999) eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem 68: 913–963. [DOI] [PubMed] [Google Scholar]
- 23. Fang Y, Park IH, Wu AL, Du G, Huang P, et al. (2003) PLD1 regulates mTOR signaling and mediates Cdc42 activation of S6K1. Curr Biol 13: 2037–2044. [DOI] [PubMed] [Google Scholar]
- 24. Maya-Monteiro CM, Almeida PE, D'Avila H, Martins AS, Rezende AP, et al. (2008) Leptin induces macrophage lipid body formation by a phosphatidylinositol 3-kinase- and mammalian target of rapamycin-dependent mechanism. J Biol Chem 283: 2203–2210. [DOI] [PubMed] [Google Scholar]
- 25. Takekoshi K, Ishii K, Nanmoku T, Shibuya S, Kawakami Y, et al. (2001) Leptin stimulates catecholamine synthesis in a PKC-dependent manner in cultured porcine adrenal medullary chromaffin cells. Endocrinology 142: 4861–4871. [DOI] [PubMed] [Google Scholar]
- 26. Shen J, Sakaida I, Uchida K, Terai S, Okita K (2005) Leptin enhances TNF-alpha production via p38 and JNK MAPK in LPS-stimulated Kupffer cells. Life Sci 77: 1502–1515. [DOI] [PubMed] [Google Scholar]
- 27. Oh DY, Cho JH, Park SY, Kim YS, Yoon YJ, et al. (2008) A novel role of hippocalcin in bFGF-induced neurite outgrowth of H19-7 cells. J Neurosci Res 86: 1557–1565. [DOI] [PubMed] [Google Scholar]
- 28. Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37: 911–917. [DOI] [PubMed] [Google Scholar]
- 29. Sengupta S, Peterson TR, Sabatini DM (2010) Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 40: 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yecies JL, Manning BD (2011) Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res 71: 2815–2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shore SA, Schwartzman IN, Mellema MS, Flynt L, Imrich A, et al. (2005) Effect of leptin on allergic airway responses in mice. J Allergy Clin Immunol 115: 103–109. [DOI] [PubMed] [Google Scholar]
- 32. Shore SA (2010) Obesity, airway hyperresponsiveness, and inflammation. J Appl Physiol (1985) 108: 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kim SJ (2010) Leptin potentiates Prevotella intermedia lipopolysaccharide-induced production of TNF-alpha in monocyte-derived macrophages. J Periodontal Implant Sci 40: 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vaughan T, Li L (2010) Molecular mechanism underlying the inflammatory complication of leptin in macrophages. Mol Immunol 47: 2515–2518. [DOI] [PubMed] [Google Scholar]
- 35. Loffreda S, Yang SQ, Lin HZ, Karp CL, Brengman ML, et al. (1998) Leptin regulates proinflammatory immune responses. FASEB J 12: 57–65. [PubMed] [Google Scholar]
- 36. Lee JS, Kim JH, Jang IH, Kim HS, Han JM, et al. (2005) Phosphatidylinositol (3,4,5)-trisphosphate specifically interacts with the phox homology domain of phospholipase D1 and stimulates its activity. J Cell Sci 118: 4405–4413. [DOI] [PubMed] [Google Scholar]
- 37. Lin CI, Chen CN, Huang MT, Lee SJ, Lin CH, et al. (2008) Lysophosphatidic acid upregulates vascular endothelial growth factor-C and tube formation in human endothelial cells through LPA(1/3), COX-2, and NF-kappaB activation- and EGFR transactivation-dependent mechanisms. Cell Signal 20: 1804–1814. [DOI] [PubMed] [Google Scholar]
- 38. Anand AR, Cucchiarini M, Terwilliger EF, Ganju RK (2008) The tyrosine kinase Pyk2 mediates lipopolysaccharide-induced IL-8 expression in human endothelial cells. J Immunol 180: 5636–5644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Williams LM, Ridley AJ (2000) Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J Immunol 164: 2028–2036. [DOI] [PubMed] [Google Scholar]
- 40. Okenwa C, Kumar A, Rego D, Konarski Y, Nilchi L, et al. (2013) SHP-1-Pyk2-Src protein complex and p38 MAPK pathways independently regulate IL-10 production in lipopolysaccharide-stimulated macrophages. J Immunol 191: 2589–2603. [DOI] [PubMed] [Google Scholar]
- 41. Kim JH, Kim JE, Liu HY, Cao W, Chen J (2008) Regulation of interleukin-6-induced hepatic insulin resistance by mammalian target of rapamycin through the STAT3-SOCS3 pathway. J Biol Chem 283: 708–715. [DOI] [PubMed] [Google Scholar]
- 42. Park SY, Baik YH, Cho JH, Kim S, Lee KS, et al. (2008) Inhibition of lipopolysaccharide-induced nitric oxide synthesis by nicotine through S6K1-p42/44 MAPK pathway and STAT3 (Ser 727) phosphorylation in Raw 264.7 cells. Cytokine 44: 126–134. [DOI] [PubMed] [Google Scholar]
- 43. Lekmine F, Uddin S, Sassano A, Parmar S, Brachmann SM, et al. (2003) Activation of the p70 S6 kinase and phosphorylation of the 4E-BP1 repressor of mRNA translation by type I interferons. J Biol Chem 278: 27772–27780. [DOI] [PubMed] [Google Scholar]
- 44. Lekmine F, Sassano A, Uddin S, Smith J, Majchrzak B, et al. (2004) Interferon-gamma engages the p70 S6 kinase to regulate phosphorylation of the 40S S6 ribosomal protein. Exp Cell Res 295: 173–182. [DOI] [PubMed] [Google Scholar]
- 45. Hatziieremia S, Gray AI, Ferro VA, Paul A, Plevin R (2006) The effects of cardamonin on lipopolysaccharide-induced inflammatory protein production and MAP kinase and NFkappaB signalling pathways in monocytes/macrophages. Br J Pharmacol 149: 188–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Matsuda T, Omori K, Vuong T, Pascual M, Valiente L, et al. (2005) Inhibition of p38 pathway suppresses human islet production of pro-inflammatory cytokines and improves islet graft function. Am J Transplant 5: 484–493. [DOI] [PubMed] [Google Scholar]
- 47. Ura S, Nishina H, Gotoh Y, Katada T (2007) Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol Cell Biol 27: 5514–5522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Choi Y, Lee MK, Lim SY, Sung SH, Kim YC (2009) Inhibition of inducible NO synthase, cyclooxygenase-2 and interleukin-1beta by torilin is mediated by mitogen-activated protein kinases in microglial BV2 cells. Br J Pharmacol 156: 933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ajmone-Cat MA, De Simone R, Nicolini A, Minghetti L (2003) Effects of phosphatidylserine on p38 mitogen activated protein kinase, cyclic AMP responding element binding protein and nuclear factor-kappaB activation in resting and activated microglial cells. J Neurochem 84: 413–416. [DOI] [PubMed] [Google Scholar]
- 50. Sanchez-Margalet V, Martin-Romero C, Santos-Alvarez J, Goberna R, Najib S, et al. (2003) Role of leptin as an immunomodulator of blood mononuclear cells: mechanisms of action. Clin Exp Immunol 133: 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]