

In this paper the authors have re-engineered the Spinach RNA aptamer into a conditionally fluorescent molecular beacon (Spinach.ST). Spinach.ST was designed to cotranscriptionally fold into a nonfluorescent structure that undergoes a conformational switch to the fluorescent state only upon hybridization with specific DNA or RNA oligonucleotides via toehold-mediated strand displacement. In situ-transcribed Spinach.ST was used for real-time quantitation and distinction of single-nucleotide mismatches in amplicons produced during nucleic acid sequence-based amplification (NASBA).
Keywords: nucleic acid engineering, aptamer, fluorescent RNA, strand displacement, aptamer beacon, molecular beacon, nucleic acid diagnostics
Abstract
We have re-engineered the fluorescent RNA aptamer Spinach to be activated in a sequence-dependent manner. The original Spinach aptamer was extended at its 5′- and 3′-ends to create Spinach.ST, which is predicted to fold into an inactive conformation and thus prevent association with the small molecule fluorophore DFHBI. Hybridization of a specific trigger oligonucleotide to a designed toehold leads to toehold-initiated strand displacement and refolds Spinach into the active, fluorophore-binding conformation. Spinach.ST not only specifically detects its target oligonucleotide but can discriminate readily against single-nucleotide mismatches. RNA amplicons produced during nucleic acid sequence-based amplification (NASBA) of DNA or RNA targets could be specifically detected and reported in real-time by conformational activation of Spinach.ST generated by in vitro transcription. In order to adapt any target sequence to detection by a Spinach reporter we used a primer design technique that brings together otherwise distal toehold sequences via hairpin formation. The same techniques could potentially be used to adapt common Spinach reporters to non-nucleic acid analytes, rather than by making fusions between aptamers and Spinach.
INTRODUCTION
RNA aptamers (Ellington and Szostak 1990; Tuerk and Gold 1990) have been described that bind and enhance the fluorescence of small molecule dyes such as the Hoechst 33258 derivative 2,6-Di-tert-butyl-4-[5-(4-methylpiperazin-1-yl)-1H,1′H-2,5′-bibenzo[d]imidazol-2′-yl]phenol (Sando et al. 2008), triphenylmethane dyes including malachite green, patent blue VF, and patent blue violet (Holeman et al. 1998; Grate and Wilson 1999; Babendure et al. 2003), cyanine dyes such as dimethylindole red (Constantin et al. 2008) and thiazole orange conjugates (Pei et al. 2009), and photoinduced N-(p-methoxyphenyl)piperazine derivatives of 2′,7′-dichlorofluorescein (Sparano and Koide 2005, 2007). Some of these aptamers have been adapted to function as biosensors whose fluorescence enhancement is triggered by analytes such as nucleotides, theophylline (Stojanovic and Kolpashchikov 2004; Furutani et al. 2010), and nucleic acid sequences (Kolpashchikov 2005; Afonin et al. 2008). For instance, a binary probe was constructed by splitting the malachite green aptamer such that aptamer structure, and hence fluorophore-binding and enhancement, were only restored upon hybridization to a target nucleic acid (Kolpashchikov 2005). Label-free, sequence-specific detection of prefolded RNA molecules has also been facilitated by using RNA-binding motifs that are covalently coupled to the malachite green aptamer (Afonin et al. 2008).
In order to develop RNA sensors that might have the same general utility as protein sensors, selections were carried out against fluorophores that resembled those found in green fluorescent protein (GFP) (Paige et al. 2011; Strack et al. 2013). In particular, a RNA aptamer (Spinach) that binds to the fluorophore DFHBI ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one) has been shown to lead to a large increase in green fluorescence emission. Spinach has been adapted to act as a genetically encoded sensor for RNA transcription in cells (Pothoulakis et al. 2013), as an imaging agent, as a reagent for intracellular detection of small molecule analytes such as adenosine 5′-diphosphate (ADP) and S-adenosylmethionine (SAM) (Paige et al. 2012), and as a sensor for proteins such as streptavidin, thrombin, and the MS2 coat protein (Song et al. 2013).
In order to further expand the utility of this versatile molecule we have engineered Spinach variants that can only be activated in the presence of specific target sequences. To engineer Spinach we programmed a conformational switch that should be triggered by nucleic acid strand displacement reactions (Zhang and Seelig 2011b). An inactive Spinach conformer was generated that also contained a small single-stranded domain (termed a toehold). Binding of an antisense sequence to the toehold allowed branch migration that destabilized the inactive conformer and promoted refolding into an active, fluorescent conformer. Similar programmable DNA strand displacement mechanisms underlie multiple nucleic acid assays and devices, including molecular logic circuits and motors, catalytic amplifiers, and reconfigurable self-assembled nanostructures (Yurke et al. 2000; Seelig et al. 2006; Yin et al. 2008; Andersen et al. 2009; Chen and Ellington 2010; Han et al. 2010; Li et al. 2011; Qian and Winfree 2011a; Qian et al. 2011; Zhang and Seelig 2011b).
The resultant sequence-dependent Spinach is the aptamer-based equivalent of a molecular beacon but no longer relies on the chemical conjugation of dye and quencher moieties for sequence detection. Such a molecular beacon should prove to be generally useful for directly generating target sequence biosensors by transcription both in vitro and in vivo. In particular, strand displacement nucleic acid circuits have proven to be useful tools for the sequence-specific detection of amplicons, especially those generated by isothermal enzymatic amplification reactions such as rolling circle amplification (RCA), strand displacement amplification (SDA), and loop-mediated isothermal amplification (LAMP) (Jiang et al. 2013). In the current work we demonstrate that sequence-dependent Spinach can be transcribed in parallel with and function as a biosensor for an isothermal amplification reaction, NASBA.
More generally, nucleic acid circuits have unprecedented programmability and modular composability that can be applied to logical decision-making and noise cancellation in diagnostics (Li et al. 2011, 2012; Allen et al. 2012). This has proven true not only for DNA-based circuits, but also for circuits whose input or components are made of RNA, both in vitro and in vivo (Isaacs et al. 2004; Kim et al. 2006; Choi et al. 2010; Kim and Winfree 2011; Lucks et al. 2011; Mutalik et al. 2012; Bhadra and Ellington 2014). Of particular interest, the hybridization chain reaction and RNA hairpins have been used for in situ mRNA detection (Choi et al. 2010). The sequence-activated Spinach molecular beacon should occupy a unique niche that bridges the programmable kinetic and thermodynamic properties of nucleic acid circuits with reporter molecules that can potentially be generated in vitro, in situ, or even in vivo.
RESULTS AND DISCUSSION
Design of sequence-modulated variants of Spinach
In order to generate a sequence-responsive reporter, a new version of the Spinach aptamer, Spinach.ST, was engineered by extending its 5′- and 3′-ends with nucleotide sequences that should fold back upon and base-pair with Spinach, thus preventing it from folding into an active conformation that would allow DFHBI binding (Fig. 1). If the construct folds as desired, a “trigger” oligonucleotide that encodes sequences complementary to the 3′-end extension of Spinach.ST should initiate a toehold-mediated strand displacement reaction that will ultimately restore the native conformation of the aptamer, and thus fluorophore binding and fluorescence. The designed Spinach.ST molecular beacon was embedded within a tRNA scaffold to stabilize the RNA (Iioka et al. 2011), including for in vivo applications (Ponchon and Dardel 2007; Paige et al. 2011).
FIGURE 1.
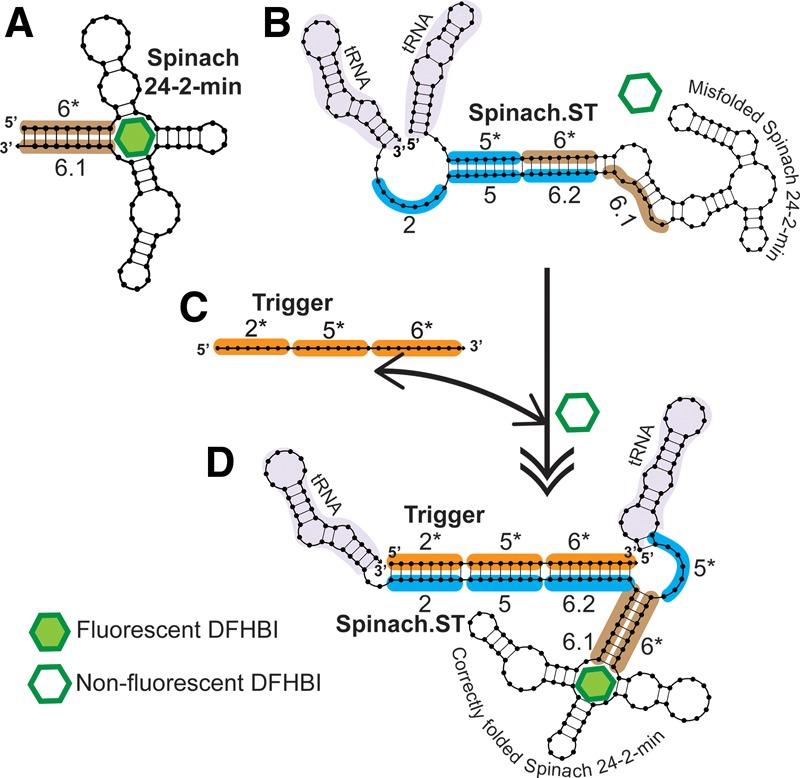
Re-engineering the RNA aptamer Spinach to be a sequence-dependent molecular beacon. All sequence blocks are designated as numbered domains (* indicates complementary domains). (A) The 9-bp stem (highlighted in brown) of the folded, 80-nt minimized Spinach aptamer (24-2-min) was designated domain 6* at its 5′-end and domain 6.1 at its 3′-end. (B) The 5′-end was extended by adding an 8-nt domain 5* (highlighted in blue) while the 3′-end was extended by adding a duplicate of domain 6.1 (termed 6.2) and two 8-nt domains, designated 5 and 2 (highlighted in blue), respectively. Sequences derived from Saccharomyces cerevisiae tRNAtrp (highlighted in light purple) flanked the engineered Spinach. RNA folding should result in domains 5 and 6.2 being contiguously paired, trapping the aptamer in a nonfluorescent conformation in which domain 2 remains unpaired. (C,D) Domain 2 serves as a toehold for strand displacement by a trigger sequence containing domains 2*–5*–6* (highlighted in orange), leading to refolding of the aptamer into its active conformation. Associations between trigger and Spinach.ST are indicated with a double-sided arrow, while the conformational change that Spinach.ST undergoes is indicated by a double-tipped arrow. All nucleic acid structures were generated using NUPACK (Dirks and Pierce 2003, 2004; Dirks et al. 2007; Zadeh et al. 2011).
The folded 80-nt minimized Spinach aptamer (24-2-min) contains a 9-bp stem that was designated domain 6* at the 5′-end and domain 6.1 at the 3′-end (Fig. 1). The 5′-end was then extended by adding an 8-nt domain 5* immediately upstream of domain 6*. The 3′-end was similarly extended by adding a duplicate 9-nt domain 6 (termed 6.2), followed by two 8-nt domains designated 5 and 2, respectively. RNA folding should result in domains 5 and 6.2 being contiguously paired, trapping the aptamer in a nonfluorescent conformation in which domain 2 remains unpaired. While it was possible that the aptamer would initially fold such that domain 6.1 is paired with domain 6*, we ruled this out by modeling the cotranscriptional folding of the sequence-dependent Spinach using Kinefold, a web-based server for RNA/DNA folding path and structure prediction (Xayaphoummine et al. 2005) (http://kinefold.curie.fr/). The program predicted that initial base-pairing during transcription would occur between domain 6* and the first domain 6.1, but then predicted rearrangement into the nonfluorescent trapped conformation as transcription proceeded. Thus, we anticipated that no post-transcriptional thermal equilibration would be required to achieve the inactive Spinach conformation. In the inactive conformation domain 2 serves as a toehold for strand displacement by a trigger sequence that is a contiguous arrangement of domains 2*–5*–6*. Binding of trigger toehold 2* to the toehold domain 2 of Spinach.ST should initiate branch migration through domain 5 and the duplicate domain 6, regenerating the Spinach basal stem and active conformation and allowing it to complex with DFHBI (Fig. 1).
To demonstrate the generality of this design principle three versions of Spinach molecular beacons (Spinach.ST1, Spinach.ST2, and Spinach.ST3) were engineered to undergo conformational activation in response to Trigger sequences 1, 2, or 3, respectively. PCR-generated Spinach.ST transcription templates were transcribed for 2 h at 42°C using T7 RNA polymerase. Transcripts were filtered through Sephadex-G25 (in order to preserve the kinetic trap) and then incubated at 37°C in 1X TNaK buffer containing 70 μM DFHBI and 0.5 μM of various trigger DNA oligonucleotides. Although strand displacement of RNA by DNA is generally energetically unfavorable (since RNA:RNA interactions are typically stronger than DNA:RNA interactions) (Lesnik and Freier 1995), we anticipated that folding intermediates during strand displacement of Spinach.ST would be stabilized by binding of DFHBI to the active aptamer conformation. Sequence-dependent activation of Spinach.ST beacons was monitored in real-time by measuring fluorescence accumulation over 4–8 h using a TECAN Safire plate reader.
Our results demonstrated that each Spinach.ST variant could specifically identify its cognate trigger sequence and undergo conformational rearrangement into RNA–fluorophore complexes with significant fluorescence (Fig. 2A). In contrast, nonspecific trigger sequences failed to significantly activate the Spinach.ST molecular beacons. The observed fluorescence intensities increased with increasing trigger concentration (Supplemental Fig. S6) and showed a four- to 70-fold increment over the fluorescence of DFHBI alone. This level of activation is similar to the approximately four- to 32-fold fluorescence increase previously reported for Spinach-based biosensors when incubated with 103- to 104-fold excess of small molecule ligands such as adenosine, ADP, SAM, guanine, guanosine 5′-triphosphate (GTP), cyclic di-GMP, and cyclic AMP-GMP (Paige et al. 2012; Kellenberger et al. 2013). While the fluorescence quantum yield of the unmodified Spinach–DFHBI complex is reported to be over 1000-fold greater than DFHBI alone, Spinach biosensors typically demonstrate reduced fluorescence (Kellenberger et al. 2013), likely due to the requirements for aptamer conformational rearrangement. Similar reductions in efficiency may be expected for conformational rearrangements induced by sequences, rather than small ligands. Spinach.ST might also be expected to demonstrate reduced fluorescence due to the addition of flanking sequences (Strack et al. 2013).
FIGURE 2.

Sequence-dependent activation of Spinach.ST molecular beacons. (A) The molecular beacons Spinach.ST1, Spinach.ST2, and Spinach.ST3 were designed to be specific for three different target nucleic acid sequences. They are activated by only their complementary trigger oligonucleotides Trigger 1, Trigger 2, and Trigger 3, respectively. Spinach.ST RNAs were synthesized by T7 RNA polymerase-mediated transcription of 500 ng of PCR-generated duplex DNA transcription templates. Spinach.ST transcripts were incubated with 0.5 μM trigger DNA oligonucleotides and Spinach.ST activation was measured as fluorescence accumulation over time at 37°C. Raw fluorescence values are shown in arbitrary units (a.u.) and data representative of replicate experiments are depicted. (B) Spinach.ST molecular beacons can detect single-nucleotide mismatches in their target oligonucleotides. Spinach.ST1 transcripts were incubated with 0.7 μM cognate trigger oligonucleotides that were either fully complementary (1T) or contained a single-base mismatch at various positions within the toehold domain 2* (1T-1M to 1T-5M). Trigger sequences with 2*–5*–6* domain organization are depicted in the 5′–3′ direction with mismatches highlighted in red. Spinach.ST1 activation was measured in real-time at 37°C. Incubation with a noncognate trigger oligonucleotide (2T) served as the negative control for Spinach.ST1 activation. Raw fluorescence values are shown in arbitrary units (a.u.) and data representative of replicate experiments are depicted.
The Spinach.ST1 and Spinach.ST2 molecular beacons require ∼30–60 min to develop maximal fluorescence in response to trigger oligonucleotides (Fig. 2; Supplemental Fig. S6). The slow kinetics of Spinach.ST signal accumulation might be due to one or a combination of potential reasons, such as the stochastic process of strand displacement, slow conversion of the trigger-bound Spinach.ST to a fluorescent conformation, or the overall low fluorescent quantum yield of Spinach.ST–DFHBI complexes. While faster (10–12-min) activation times have been reported for Spinach-based ADP and SAM biosensors incubated with a 1000-fold excess of analyte (Paige et al. 2012), a set of Spinach biosensors for cyclic di-GMP and cyclic AMP-GMP required 25–40 min at 37°C and 80 min at 25°C for full activation upon incubation with ligand in 1000-fold excess (Kellenberger et al. 2013). In fact, another version of the modular Spinach-based cyclic di-GMP biosensor required 12-h incubation with analyte in 10-fold excess for signal generation (Nakayama et al. 2012).
While Spinach.ST initial activation rates might be improved in the presence of excess trigger oligonucleotides (Supplemental Fig. S6) or by operating at a higher temperature, design improvements might also accelerate responsivity. Spinach.ST activation involves two events—toehold-mediated strand displacement by the trigger and conformational rearrangement. Given that strand displacement kinetics is principally influenced by toehold strength (Zhang and Winfree 2009; Srinivas et al. 2013), the length and sequence of toeholds can be further optimized. Destabilization of the nonbinding conformation would also enable faster activation, but could increase nonspecific signals (Hall et al. 2009).
As an example of how these considerations may play out, the Spinach.ST3 beacon was found to require a longer response time than the other two designed beacons (Fig. 2A). Spinach.ST3 is identical to Spinach.ST1 except in having an additional 8-nt domain 4 (and complement) embedded between domains 2 and 5 (Supplemental Table S1). The increased length of the branch migration domain might be partly responsible for the slower activation of Spinach.ST3. Consistent with this interpretation, Trigger 3 fails to activate Spinach.ST1, potentially indicating that even with equivalent toehold binding the eight unpaired nucleotides in domain 4* in the inactive conformation forestalled further branch migration.
The Spinach.ST molecular beacon system is functional in other physiologically relevant buffers such as phosphate buffered saline (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 at pH 7.2) with or without 3 mM MgCl2 and also in the IC buffer (20 mM HEPES, 140 mM KCl, 10 mM NaCl, 2 mM MgCl2, 5 mM KH2PO4 at pH 7.2), which mimics intracellular salt concentrations (Supplemental Fig. S5; Lodish and Darnell 1995). Spinach molecular beacons can distinguish specific trigger oligonucleotides in the 1X HEPES buffer (40 mM HEPES at pH 7.4,125 mM KCl, 5 mM MgCl2, 5% DMSO) originally used for Spinach aptamer selection (Paige et al. 2011). However, nonspecific activation of Spinach.ST is unacceptably high in this buffer (Supplemental Fig. S5).
The TNaK buffer was originally developed to execute nucleic acid circuits in physiological samples (Li et al. 2011). Magnesium ions were intentionally omitted to mitigate the enzymatic activity of contaminating nucleases (Li et al. 2011). We have previously used this buffer to operate a catalytic hairpin assembly circuit composed of cotranscriptionally generated RNA (Bhadra and Ellington 2014). In the present work we chose to analyze Spinach molecular beacons in the TNaK buffer so that we could directly interface them with nucleic acid amplicons and RNA circuit outputs.
Similar to DNA triggers, RNA sequences have also been observed to trigger Spinach.ST activation with extraordinary specificity (Supplemental Fig. S1). In addition, we split the Spinach.ST trigger to demonstrate the concerted role of the toehold and branch migration domains in effecting a conformational change in Spinach.ST. The Spinach.ST2 trigger was dispersed between two RNA transcripts such that the toehold domain 2* along with the first two nucleotides of domain 5* were placed at the 5′-end of the transcript followed by a 28-nucleotide dimerization domain D that lacked complementarity to Spinach.ST2 (trigger transcript 1). The second transcript contained the complementary domain D* followed by the remaining branch migration domains 5* and 6* (trigger transcript 2; Supplemental Fig. S1A).
The RNA triggers 1 and 2 and Spinach.ST2 were separately transcribed and purified on Sephadex G25. Spinach.ST2 activation and signal enhancement occurred only in the presence of both triggers 1 and 2, while either trigger transcript alone failed to facilitate the conformational switch of Spinach.ST2 (Supplemental Fig. S1B).
Sequence-modulated Spinach as a real-time molecular transducer
While Spinach.ST molecular beacons can specifically detect both DNA and RNA targets, it would be especially useful to demonstrate cotranscriptional activation of Spinach.ST. Such a demonstration could pave the way toward applications such as in vivo RNA sensing and in vitro real-time diagnostics. The transcription templates for Spinach.ST1 and Trigger 1 were cotranscribed using T7 RNA polymerase in the presence of 40 μM DFHBI. Fluorescence accumulation due to cotranscriptional Spinach.ST1 activation was then monitored in real-time at 39°C (Supplemental Fig. S2). The Spinach.ST1 aptamer RNA transcribed alone did not demonstrate an increase in DFHBI fluorescence; however, target RNA cotranscribed with Spinach.ST in the presence of DFHBI led to real-time accumulation of fluorescence, indicating sequence-mediated aptamer activation.
Since Spinach.ST beacons can undergo cotranscriptional activation in the presence of a complementary trigger RNA (Supplemental Fig. S2), we hypothesized that it should be possible to develop Spinach.ST molecular beacons into real-time cotranscriptional signal transducers of enzymatic amplification reactions involving RNA (rather than DNA) amplicons, such as nucleic acid sequence-based amplification (NASBA) (Supplemental Fig. S3A). In NASBA (Compton 1991) RNA templates are reverse transcribed into double-stranded DNA using forward and reverse primers, one of which carries the T7 RNA polymerase promoter sequence. The resulting double-stranded DNA can then be transcribed to generate additional RNA copies, which can in turn continuously be converted into additional double-stranded DNA templates. Although NASBA is a powerful amplification technique, like many continuous amplification techniques it is limited by nonspecific background amplification, thus making it an excellent candidate for sequence-specific signal transduction using Spinach.ST.
As a proof-of-principle we first used a NASBA template (SNV3TO) that contained a reverse complement (designated as domains 6–5–2 in Supplemental Fig. S3A) to the previously described Trigger sequence and flanking primer-binding sites. Single-stranded DNA or RNA templates transcribed from PCR-generated DNA were then amplified by NASBA (using the FP1:RP1pT7 primer pair) in the presence of the Spinach.ST1 reporter template and 40 μM DFHBI. During NASBA the cotranscribed Spinach.ST1 reporter RNA was activated, as determined by real-time fluorescence analysis (Supplemental Figs. S3B, S4A). Within 1 h of amplification 1–10 nM ssDNA targets yielded signals that were two- and sevenfold signal above background, respectively. As little as 10 pM of target could be detected (at twofold signal-to-noise) following 8 h of amplification. Although comparable amounts of Spinach.ST1 were transcribed in all reactions, NASBA amplicons containing a noncomplementary trigger sequence (generated using the pT7FP2:RP2 primer pair) failed to activate Spinach.ST1 (Supplemental Figs. S3C,D, S4B). Even after 8 h of amplification no significant signal above background was observed even when reactions were seeded with 10 nM template. Amplification starting from RNA templates proved to be slower, with only the 10-nM template concentration being detectable within 1 h of amplification at a twofold signal-to-noise ratio. This is not surprising, given that the internal structure of RNA templates typically limits the efficiency of reverse transcription. That said, by 5 h some 1-nM target RNA could be reliably distinguished at a fourfold signal above noise, while a 10 pM template generated a 1.4-fold increase in signal. RNA amplification with the pT7FP2:RP2 primer pair (that generates noncognate amplicons) failed to generate a fluorescent signal above background, further demonstrating the specificity of the transduction reaction.
Engineering the Spinach reporter for any amplicon
Although Spinach.ST can be designed to accept many different sequences via Trigger domains 2* and 5*, domain 6* remains invariant as it is a part of the aptamer basal stem (Fig. 1). Since most target sequences of interest will not have a 9-nt long stretch that is perfectly complementary to aptamer domain 6*, the real-time Spinach.ST-NASBA system was re-engineered to be adaptable to any sequence target. The Trigger was split such that only domains 2* and 5* would originate from the target (Fig. 3A) while a 9-nt complementary domain 6 was appended to the 5′-end of the NASBA forward primer (NV4FP). Thus, as NASBA proceeded the RNA products should contain the requisite Trigger domain 6* (Fig. 3A), although it will be separated from target-originated Trigger domains 2* and 5* by a short target-specific sequence (designated as PBS-F*).
FIGURE 3.

Application of Spinach.ST to the detection of NASBA amplicons. (A) Schematic of primer design for real-time, sequence-specific NASBA detection using Spinach.ST2. Sequence blocks are depicted as numbered or lettered domains (* indicates complementarity). NV4FP and NV4RP are the forward and reverse primers used for NASBA. PBS-F and PBS-R* domains hybridize with the target nucleic acid and initiate polymerization. Hybridization is depicted using vertical slashes, while the potential hybridization of the PBS-F domain within the NV4FP primer to a complementary region is indicated by colons. Dashed regions in the target, double-stranded DNA transcription template, and RNA transcript denote target sequences not involved in priming or in Spinach.ST activation. The Spinach.ST trigger in the NASBA RNA amplicons is highlighted in gray. Spinach.ST structures were generated using NUPACK. (B,C) Sequence-specific fluorescent detection of single-stranded DNA (B) or RNA (C) templates by real-time Spinach.ST-NASBA. Varying concentrations of NV4 template DNA or RNA were amplified at 37°C by NASBA using T7 RNA polymerase and MMLV RT in the presence of cotranscribed Spinach.ST2 and 40 µM DFHBI. Raw fluorescence values are shown in arbitrary units (a.u.) and data representative of replicate experiments are depicted. (D) Denaturing 10% polyacrylamide gel analysis of NASBA amplification reactions of single-stranded DNA (lanes 1–5) or RNA (lanes 6–10) templates; cotranscription of Spinach.ST2 is also shown. Template concentrations are in nanomolar (nM) amounts. Two microliters of each NASBA reaction were analyzed. Single-stranded DNA oligonucleotides were used as size markers (lane 11).
As seen in Figure 2A the presence of as few as eight intervening, nonpairing nucleotides in the Trigger 3 sequence rendered it unable to activate Spinach.ST1. Therefore, to efficiently bridge the PBS-F* gap we structured the primer so that the resultant RNA would create a hairpin that brought domains 5* and 6* into apposition (Fig. 3A). A 20-nt long sequence designated as domain D* was designed to be complementary to a portion of the NV4 template (designated domain D), and was inserted between domain 6 and the primer binding site (PBS-F) of the NV4FP primer. Upon amplification, domain D* will pair with the domain D segment generated from the template, forming a hairpin and bringing domains 5* and 6* close to one another for activation of Spinach. It has previously been shown that “associative toehold activation,” wherein the toehold and branch migration domains become connected via the hybridization of auxiliary domains, allows strand displacement across a three-way junction (Chen 2012). We have now for the first time successfully applied a similar concept to the cotranscriptional formation of RNA associative trigger sequences for the real-time sequence-specific detection of enzymatically generated RNA amplicons.
Finally, a T7 RNA polymerase promoter was placed at the 5′-end of the reverse primer (NV4RP) adjacent to the primer binding site PBS-R*. The resultant NASBA-generated RNA amplicons should have the following domain structure: 5′-PBS-R*–2*–5*–D*–PBS-F*–D–6*-3′. As stated above, hybridization of the complementary domains D* and D into a stem with PBS-F* loop should juxtapose domains 2*, 5*, and 6* in the NASBA-amplified RNA, leading to the activation of Spinach.ST (Fig. 3A).
NASBA reactions were set up with single-stranded DNA (Fig. 3B) or RNA templates (Fig. 3C) in the presence of the Spinach.ST2 transcription template and 40 μM DFHBI. Although similar amounts of Spinach.ST2 reporter RNA were transcribed in all reactions (Fig. 3D), fluorescence nonetheless increased in a sequence-specific manner with increasing accumulation of the NASBA-generated RNA product.
We believe that the trigger RNA sequences in the NASBA-generated RNA amplicons did not show significant reverse transcriptase (RT) priming ability because (1) the trigger was embedded within the SNV3TO RNA amplicons such that the target-specific PBS-F* domain (located at the 3′-end of the amplicon) had no significant complementarity to Spinach.ST1; and (2) MMLV RT is known to be strongly inhibited by RNA secondary structures such as those present in Spinach.ST (Harrison et al. 1998).
Allele discrimination by sequence-modulated Spinach
It has previously been shown that toehold design can significantly modulate the kinetics of strand displacement (Zhang and Winfree 2009; Zhang et al. 2012), and we hoped to further optimize the performance of the Spinach molecular beacons to distinguish even single-nucleotide mismatches within the trigger sequences. Nucleotide positions 1 through 5 of the Trigger 1 toehold domain 2* were individually mutated (1T-1M through 1T-5M). We hypothesized that some mismatches would likely render the toehold interaction too weak to support strand displacement, and that this knowledge could then be used to design circuits that were particularly sensitive to mismatches. To test this hypothesis some 0.7 μM mismatched trigger oligonucleotides were incubated with Sephadex-filtered Spinach.ST1 transcripts. We observed that trigger oligonucleotides containing mismatches in either the first or second nucleotide positions of the toehold activated Spinach.ST1 to the same degree as the fully complementary Trigger 1 and gave initial rates of ∼2 a.u./min for the first 13–40 min of the reaction (Fig. 2B). In contrast, mismatches placed at nucleotide positions 3, 4, or 5 almost completely destroyed the ability of the trigger oligonucleotides to activate Spinach.ST1, resulting in near zero initial rates that were similar to those observed with an entirely nonspecific Trigger 2 (Fig. 2B). Our results clearly demonstrate that Spinach beacons can distinguish even single-nucleotide polymorphisms (SNP) within the target sequences.
We then sought to demonstrate the ability of Spinach.ST for real-time SNP distinction during NASBA. A SNP-containing allelic variant of the NV4 NASBA template (NV4.4M) was synthesized that harbored a single G to C point mutation at nucleotide position 5 (counted from the 5′-end) of the 8-nt-long toehold complementary domain 2 (Fig. 4A). The complementary RNA amplicons generated during NASBA-mediated amplification of NV4.4M should harbor a single C to G point mutation at nucleotide position 4 (counted from the 5′-end) of the 8-nt-long toehold domain 2*, compared with the original NV4 amplicons. The single mismatch between the NV4.4M RNA toehold domain 2* and the Spinach.ST2 toehold domain 2 was expected to significantly diminish Spinach.ST2 activation when compared with the activation achieved by the NV4 transcript. To further test this hypothesis, a Spinach.ST2 variant (Spinach.ST2.4M) that contained a complementary mutation in its toehold domain 2 and that restored complete base-pairing to the NV4.4M RNA toehold domain 2* was constructed. Spinach.ST2.4M was expected to efficiently detect NV4.4M RNA while showing minimal activation by NV4 RNA.
FIGURE 4.

Discrimination of alleles in real-time by NASBA and sequence-modulated Spinach. (A) Schematic depicting the target alleles NV4 and the SNP-containing NV4.4M. Only the domain 2 and 2* sequences are shown for clarity. The single-nucleotide difference between the two alleles (highlighted in a large bold font) is located within the target region termed domain 2. Both NV4 and NV4.4M alleles were amplified using the common primers NV4FP and NV4RP. NASBA-generated NV4 and NV4.4M RNA amplicons form associative Spinach.ST triggers that have a single base difference between them (highlighted in bold font) in their 2* domains. The sequence-modulated molecular beacons Spinach.ST2 and the SNP-complementary Spinach.ST2.4M have corresponding single-nucleotide differences (highlighted in a large bold font) in their toehold domain 2. RNA amplicons bearing fully complementary associative triggers should activate Spinach.ST molecular beacons by facilitating toehold-mediated strand displacement. Activated Spinach.ST molecules increase the fluorescence of DFHBI (depicted as a green hexagon). Activation of Spinach.ST molecules by RNA amplicons bearing a single-nucleotide mismatch in their toehold domain should be significantly impaired (denoted as dashed double-tipped arrows interrupted with an X). (B,C) Detection of NASBA amplicons by allele-specific, sequence-modulated Spinach.ST. NASBA reactions were used to amplify either no template (black line) or 20 nM of ssDNA targets NV4 (red line) or NV4.4M (purple line) in the presence of 40 µM DFHBI and 138 ng of dsDNA transcription templates for either Spinach.ST2 (B) or Spinach.ST2.4M (C). Raw fluorescence values are shown in arbitrary units (a.u.) and data representative of replicate experiments are depicted.
To experimentally validate our design for Spinach.ST-based real-time allelic distinction either no template or 20 nM of the NV4 or NV4.4M ssDNA templates were amplified by NASBA using the NV4FP and NV4RP primer pair (Fig. 4B,C). NASBA amplicons were detected in real-time by including 40 µM DFHBI and either Spinach.ST2 or Spinach.ST2.4M dsDNA transcription templates in duplicate amplification reactions. Both Spinach.ST2 and Spinach.ST2.4M detected their matched NV4 and NV4.4M RNA amplicons, respectively, with a seven- to ninefold signal-to-noise ratio. However, in both cases activation by the noncognate SNP-containing RNA was significantly impaired, resulting in less than twofold signal-over-noise. Accurate detection of amplicons could likely be improved through the introduction of “thresholding” gates that also operate via strand displacement (Qian and Winfree 2011b; Qian et al. 2011; Zhang and Seelig 2011a).
We have successfully engineered the fluorescent RNA aptamer Spinach into a sequence-dependent molecular beacon that is first cotranscriptionally trapped into a nonfluorescent conformation and is then switched into an active fluorescent state upon sequence-specific, toehold-mediated strand displacement. These experimental results are especially interesting as they accord with modeling results for cotranscriptional entrapment of the Spinach.ST beacon into a nonfluorescent conformation, suggesting that further engineering of Spinach reporters using design algorithms may be possible. We have demonstrated the application of Spinach.ST as a signal transducer for detecting DNA and RNA sequences in vitro. Even in the complex milieu of transcription or NASBA reactions Spinach.ST beacons cotranscriptionally folded into stable inactive conformations that were only released in the presence of cognate Trigger RNA sequences. The sensor was able to readily distinguish single-nucleotide mismatches within its sequence targets.
Sequence-modulated Spinach should prove especially useful in point-of-care assays because it can be generated by transcription in parallel during amplification assays. As an example, we developed a one-pot in vitro isothermal NASBA reaction wherein the label-free Spinach.ST reporters were enzymatically generated in situ and acted as real-time fluorescent reporters of target nucleic acid amplicons. The detection of realistic target amplicons was readily accomplished by designing NASBA primers to insert a Spinach.ST Trigger sequence within the RNA amplicons. The gap between target-generated and primer-originated domains in the associative Triggers was bridged by sequestration of intervening sequences into a stem–loop structure. Strand displacement across this three-way RNA junction not only activates the Spinach.ST reporter but also allows for SNPs located in the middle of the toehold domain to be readily distinguished. This design advance ultimately simplifies assay development by allowing ready adaptation of Spinach.ST reporter for detection of different targets by simply changing the domains 2, 5, and 5*. Future assay optimizations will also focus on increasing the speed of detection.
In fact, an invariant Spinach.ST molecular beacon could be used as a universal reporter for a wide variety of amplicons by the simple expedient of redesigning NASBA primers such that the trigger complementary branch migration domains are appended to the forward primer (5′-6–5–D1*–PBS-F-3′), while the toehold domain is situated in the reverse primer (5′-pT7–2*–D2–PBS-R*-3′). Primer domains D1* and D2 are derived from two adjoining target domains D1 and D2. The resulting NASBA RNA amplicons will display the domain organization: 5′-2*–D2–PBS-R*–D2*–D1*–PBS-F*–D1–5*–6*-3′. Associative trigger assembly can then be mediated by double stem–loop formation upon hybridization of the adjoining target domains D1* and D2* to their respective complementary domains. In a second, perhaps structurally less complex, approach universal Spinach.ST trigger branch migration and toehold domains may be split between 3′-end-blocked hybridization probe 1 (5′-2*–D2-3′) and probe 2 (5′-D1–5*–6*-3′). During NASBA with target-specific primers hybridization of D1 and D2 to adjacent complementary sequences on the NASBA amplicons would juxtapose the Spinach.ST trigger domains, leading to activation of universal Spinach.ST.
The demonstrated robustness of the transcribed Spinach.ST reporter bodes well for potentially expanding its applications in vivo as a sensor for endogenously expressed RNA sequences, just as it has been adapted to sensing other analytes (Paige et al. 2012; Song et al. 2013). Indeed, while previously Spinach has been fused to aptamers (Paige et al. 2012; Strack et al. 2014), the method we propose in which the Spinach reporter is trans to the transducer, rather than cis, may also prove useful for the detection of non-nucleic acid analytes. Structure-switching aptamers can be designed to undergo analyte-induced intra- or intermolecular rearrangement (Nutiu and Li 2003; Cho et al. 2009). Such strategies have been widely used in developing aptamer-based optical (Li and Ho 2008) and electrochemical sensors (Lubin and Plaxco 2010) for analytes such as nucleotides, cocaine, and thrombin (Liu and Lu 2006; Tang et al. 2008). Similar structure-switching aptamers could be designed to display sequestered Spinach.ST trigger sequences or toeholds in response to non-nucleic acid ligand recognition, and the triggers would in turn activate standalone Spinach.ST molecular beacons.
The ability of transcriptionally generated Spinach.ST to readily transform upstream events into conformational state changes and signals without requiring covalent coupling or fusion to other sequences should make it a particularly useful tool for cell-based sensing. Since Spinach.ST signal accumulation occurs on a timescale of several minutes to hours, real-time detection of nascent transcripts (with a timescale of ∼1 sec) is perhaps unrealistic. In fact, the hybridization rate for even simple, short oligonucleotide probes is typically several orders of magnitude slower than diffusion under physiological conditions, thus precluding real-time RNA detection via methods such as total internal reflection fluorescence (TIRF) microscopy (Zhang et al. 2014). Nonetheless, since mRNA half-lives range from a few minutes in bacteria, to minutes to hours in yeast and mammalian cells (Bernstein et al. 2002; Wang et al. 2002; Yang et al. 2003), we believe that the Spinach.ST system may prove to be a valuable tool for probing endogenous RNA in live cells, especially when combined with sensitive microscopic detection methods. Perhaps the biggest impetus for exploring such an application lies in the fact that copious amounts of transcripts containing quenched reporters could be generated within cells, then activated by other transcripts based on spatial or temporal localization. In contrast, the delivery of nucleic acid reporters such as fluorophore-labeled antisense oligonucleotides and molecular beacons by conventional transfection methods requires at least 30 min to 4 h and is often inefficient (Nitin et al. 2004). Ultimately, we envision being able to program rational operating systems for cells based on hybridization between distinct nucleic acid regulators. Toward this goal we have also successfully used the Spinach.ST reporter to detect RNA conformational changes occurring during in vitro-catalyzed RNA hairpin assembly reactions, thus creating input/output circuits made only of RNA (Bhadra and Ellington 2014).
MATERIALS AND METHODS
Reagents, oligonucleotides, and transcription templates
Unless otherwise indicated all molecular biology grade chemical reagents were purchased from Sigma-Aldrich. Acrylamide was purchased from Bio-Rad and DFHBI ((Z)-4-(3,5-difluoro-4-hydroxybenzylidene)-1,2-dimethyl-1H-imidazol-5(4H)-one) was a generous gift from Dr. Zhan Zhang (The University of Texas at Austin).
All oligonucleotides were obtained from Integrated DNA Technologies (IDT). Oligonucleotides were resuspended at 100 μM concentration in TE (10:0.1) buffer (10 mM Tris-HCl at pH 7.5, 0.1 mM EDTA at pH 8.0) and stored at −20°C. The concentration of all DNA and RNA suspensions was measured by UV spectrophotometry using a Nanodrop 1000 spectrophotometer (Thermo Scientific). All transcription templates were built from DNA oligonucleotides obtained from IDT. Short transcription templates (≤60 bp) were prepared by annealing two completely complementary oligonucleotides mixed in equimolar concentration in TE (10:0.1) buffer containing 50 mM NaCl. Oligonucleotides were denatured for 5 min at 95°C prior to annealing by slow cooling (0.1°C/sec) to 25°C. Annealed oligonucleotides were quantitated and used directly for in vitro transcription reactions, sometimes after storage at −20°C. Longer transcription templates were sequentially assembled from sets of shorter overlapping oligonucleotides by oligonucleotide annealing, primer extension, and PCR reactions. Site-directed mutagenesis was performed by overlap PCR with mutagenic primers. All enzymatic amplification reactions were performed using high-fidelity Phusion DNA polymerase (New England Biolabs) or Taq DNA polymerase (NEB) according to the manufacturer's protocols. Templates generated by PCR using the proof-reading Phusion DNA polymerase were subjected to an A-tailing reaction to facilitate subsequent TA cloning. All DNA fragments were either purified using Wizard SV gel and PCR clean-up columns (Promega) or subjected to agarose gel purification prior to TOPO-TA cloning into pCR2.1TOPO plasmid (Life Technologies), again according to the manufacturer's instructions. Cloned plasmids were selected and maintained in the E. coli Top10 strain. All transcription templates were verified by sequencing at the Institute of Cellular and Molecular Biology Core DNA sequencing facility.
For in vitro run off transcription reactions, transcription templates that had been cloned into the pCR2.1-TOPO vector were amplified (from previously sequence-verified plasmids) by PCR using Phusion DNA polymerase. Spinach.ST transcription templates were amplified using primers specific to the flanking plasmid sequence at the 5′-end and primers (sphT.U.R) specific to the 3′-end sequence of Spinach.ST1. PCR products were analyzed by agarose gel electrophoresis and then purified using the Wizard SV gel and PCR Clean-up system (Promega, Madison).
Circuit design
All RNA structures, circuit designs, and intra- and intermolecular interactions were analyzed using the NUPACK server (Dirks and Pierce 2003, 2004; Dirks et al. 2007; Zadeh et al. 2011). RNA analysis at different temperatures was carried out using Serra and Turner (Serra and Turner 1995) RNA energy parameters with some Dangle treatment. Cotranscriptional RNA folding was modeled using the Kinefold web server (Xayaphoummine et al. 2005). Kinefold performs stochastic folding simulations of nucleic acids on second to minute time scales and can thereby simulate renaturation or cotranscriptional folding paths at the level of individual helices. The engineered Spinach.ST sequences used in this study were as follows:
Spinach.ST1, TAATACGACTCACTATAGGAAGCGGTGGCTCAATGGTAGAGCTTTCGACCTTGTCAGACGCGACCGAAATGGTGAAGGACGGGTCCAGTGCTTCGGCACTGTTGAGTAGAGTGTGAGCTCCGTAACTGGTCGCGTCGGTCGCGTCTGACAAGGGCTAGGTTTCGAAGGGTTGCAGGTTCAATTCCTGTCCGTTTC.
Spinach.ST2, TAATACGACTCACTATAGGAAGCGGTGGCTCAATGGTAGAGCTTTCGACGACATCTGACGCGACCGAAATGGTGAAGGACGGGTCCAGTGCTTCGGCACTGTTGAGTAGAGTGTGAGCTCCGTAACTGGTCGCGTCGGTCGCGTCAGATGTCGGTCAGGTCTCGAAGGGTTGCAGGTTCAATTCCTGTCCGTTTC.
Spinach.ST3, TAATACGACTCACTATAGGAAGCGGTGGCTCAATGGTAGAGCTTTCGACGACATCTCCTTGTCAGACGCGACCGAAATGGTGAAGGACGGGTCCAGTGCTTCGGCACTGTTGAGTAGAGTGTGAGCTCCGTAACTGGTCGCGTCGGTCGCGTCTGACAAGGAGATGTCGGCTAGGTTTCGAAGGGTTGCAGGTTCAATTCCTGTCCGTTTC. The 17-mer T7 RNA polymerase promoter is highlighted in bold, while the tRNA-derived sequences are underlined.
In vitro transcription and measurement of Spinach fluorescence
Some 100–500 ng of double-stranded DNA transcription templates were transcribed using 100 units of T7 RNA polymerase (NEB) in 50-μL reactions containing 40 mM Tris-HCl (pH 7.9), 30 mM MgCl2, 10 mM DTT, 2 mM spermidine, a 4-mM ribonucleotide (rNTP) mix, and 20 units of the recombinant ribonuclease inhibitor RNaseOUT (Life Technologies). Transcription reactions were incubated at 42°C for 120 min. Upon completion of transcription reactions were filtered through Sephadex G25 using the Illustra MicroSpin G-25 columns (GE Healthcare). Some 3–6 µL aliquots of filtered transcripts were incubated with 0.5–8 µM trigger DNA oligonucleotides or 1–3 µL of RNA transcripts 1 and/or 2 in 1X TNaK buffer (20 mM Tris-HCl at pH 7.5, 140 mM NaCl, 5 mM KCl) containing 70 μM DFHBI and 20 units of RNaseOUT. In some experiments 1X TNaK was replaced with one of the following buffers: 1X PBS (phosphate buffered saline: 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 at pH 7.2, with or without 3 mM MgCl2), 1X IC buffer (intracellular buffer: 20 mM HEPES, 140 mM KCl, 10 mM NaCl, 2 mM MgCl2, 5 mM KH2PO4 at pH 7.2), and 1X HEPES buffer (40 mM HEPES at pH 7.4, 125 mM KCl, 5 mM MgCl2, 5% DMSO). Spinach.ST activation was measured as fluorescence accumulation as a function of time at 37°C using the TECAN Safire plate reader.
In vitro cotranscription reactions to measure sequence-dependent real-time trans-activation of Spinach.ST molecular beacons were performed in a 25-μL volume placed directly into 384-well flat bottom black plates (NUNC). The 500 ng of Spinach.ST1 and Trigger 1 transcription templates were transcribed using 100 units of T7 RNA polymerase in buffer containing 40 mM Tris-HCl (pH 7.9), 6 mM MgCl2, 10 mM DTT, 2 mM spermidine, 50 mM NaCl, 4.8 mM rNTP mix, 40 μM DFHBI, and 20 units of RNaseOUT. Reactions were incubated at 39°C in the TECAN Safire plate reader and real-time fluorescence measurements were recorded every 13 min.
NASBA with real-time sequence-dependent Spinach aptamer-based fluorimetric detection
NASBA reactions were set up with either ssDNA or gel-purified RNA inputs at a variety of concentrations. Single-stranded DNA inputs were purchased from IDT. The dsDNA templates for transcription were obtained by PCR with forward primers containing a T7 RNA polymerase promoter. Template molecules were diluted fresh into TE (10:0.1) buffer containing 1 μM oligo dT17. NASBA reactions that were to be reported by cotranscription of Spinach.ST were set up in 25-μL volumes in two steps. First, varying concentrations of template molecules were mixed with 24 mM MgCl2, 20 mM KCl, 50 mM NaCl, 100 nM each of forward and reverse primers, and 500 μM dNTP mix. This solution was heated at 65°C for 5 min followed by incubation on ice for 2 min. Subsequently, the remaining reaction components including 100–138 ng of Spinach.ST dsDNA transcription templates, 1X RNAPol reaction buffer (NEB: 40 mM Tris-HCl, 6 mM MgCl2, 10 mM DTT, 2 mM spermidine), 6 mM rNTP mix (NEB), 100 units of MMLV reverse transcriptase (NEB), 50 units of T7 RNA polymerase, 20 units of RNaseOUT, and 40 µM DFHBI were added. The reactions were directly transferred to 384-well flat bottom black plates that were incubated for 6–8 h in the TECAN Safire at 37°C and real-time fluorescence measurements were recorded every 13 min. At the completion of the amplification reaction 1–2 μL aliquots were analyzed by denaturing PAGE. Real-time NASBA reactions for allelic discrimination and using Spinach.ST2 were incubated at 42°C instead of 37°C.
Denaturing PAGE and RNA gel purification
The 10% polyacrylamide gels containing 7M urea were prepared using 40% acrylamide and bis-acrylamide solution, 19:1 (Bio-Rad) in 1X TBE buffer (89 mM Tris Base, 89 mM Boric acid, 2 mM EDTA at pH 8.0) containing 0.04% ammonium persulphate and 0.1% TEMED. An equal volume of 2X denaturing dye (7 M urea, 1X TBE, 0.1% bromophenol blue) was added to the RNA samples (volume made up to 6 μL with 0.1 mM EDTA), and then incubated at 65°C for 3 min, followed by cooling to room temperature prior to loading. A ssDNA ladder prepared by mixing 20-, 42-, 66-, and 99-nt-long oligonucleotides was included as a size marker. The gels were stained for 10 min with SYBR-Gold (Life Technologies) prior to visualization on the Storm Imager (GE Healthcare). For RNA purification, desired bands were excised from the gel and the RNA was eluted twice into TE (10:1) buffer (10 mM Tris-HCl at pH 7.5, 1 mM EDTA at pH 8.0) by incubation at 70°C and 1000 rpm for 20 min. Acrylamide traces were removed by filtering eluates through Ultrafree-MC centrifugal filter units (EMD Millipore), followed by precipitation with 2X volume of 100% ethanol in the presence of both 15 µg GlycoBlue (Life Technologies) and 0.3 M sodium acetate (pH 5.2). RNA pellets were washed once in 70% ethanol. Dried pellets of purified RNA samples were resuspended in 0.1 mM EDTA and stored at −80°C.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work was supported by the Welch Foundation (F1654), the National Institutes of Health in conjunction with the Boston University (1U54EB015403), and by the Gates Foundation (OPP1028808).
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.045047.114.
REFERENCES
- Afonin KA, Danilov EO, Novikova IV, Leontis NB 2008. TokenRNA: a new type of sequence-specific, label-free fluorescent biosensor for folded RNA molecules. Chembiochem 9: 1902–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen PB, Arshad SA, Li B, Chen X, Ellington AD 2012. DNA circuits as amplifiers for the detection of nucleic acids on a paperfluidic platform. Lab Chip 12: 2951–2958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen ES, Dong M, Nielsen MM, Jahn K, Subramani R, Mamdouh W, Golas MM, Sander B, Stark H, Oliveira CLP, et al. 2009. Self-assembly of a nanoscale DNA box with a controllable lid. Nature 459: 73–76 [DOI] [PubMed] [Google Scholar]
- Babendure JR, Adams SR, Tsien RY 2003. Aptamers switch on fluorescence of triphenylmethane dyes. J Am Chem Soc 125: 14716–14717 [DOI] [PubMed] [Google Scholar]
- Bernstein JA, Khodursky AB, Lin PH, Lin-Chao S, Cohen SN 2002. Global analysis of mRNA decay and abundance in Escherichia coli at single-gene resolution using two-color fluorescent DNA microarrays. Proc Natl Acad Sci 99: 9697–9702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhadra S, Ellington AD 2014. Design and application of cotranscriptional non-enzymatic RNA circuits and signal transducers. Nucleic Acids Res 42: e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X 2012. Expanding the rule set of DNA circuitry with associative toehold activation. J Am Chem Soc 134: 263–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ellington AD 2010. Shaping up nucleic acid computation. Curr Opin Biotechnol 21: 392–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Lee JW, Ellington AD 2009. Applications of aptamers as sensors. Annu Rev Anal Chem 2: 241–264 [DOI] [PubMed] [Google Scholar]
- Choi HM, Chang JY, Trinh le A, Padilla JE, Fraser SE, Pierce NA 2010. Programmable in situ amplification for multiplexed imaging of mRNA expression. Nat Biotechnol 28: 1208–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton J 1991. Nucleic acid sequence-based amplification. Nature 350: 91–92 [DOI] [PubMed] [Google Scholar]
- Constantin TP, Silva GL, Robertson KL, Hamilton TP, Fague K, Waggoner AS, Armitage BA 2008. Synthesis of new fluorogenic cyanine dyes and incorporation into RNA fluoromodules. Org Lett 10: 1561–1564 [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA 2003. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J Comput Chem 24: 1664–1677 [DOI] [PubMed] [Google Scholar]
- Dirks RM, Pierce NA 2004. An algorithm for computing nucleic acid base-pairing probabilities including pseudoknots. J Comput Chem 25: 1295–1304 [DOI] [PubMed] [Google Scholar]
- Dirks RM, Bois JS, Schaeffer JM, Winfree E, Pierce NA 2007. Thermodynamic analysis of interacting nucleic acid strands. SIAM Rev 49: 65–88 [Google Scholar]
- Ellington AD, Szostak JW 1990. In vitro selection of RNA molecules that bind specific ligands. Nature 346: 818–822 [DOI] [PubMed] [Google Scholar]
- Furutani C, Shinomiya K, Aoyama Y, Yamada K, Sando S 2010. Modular blue fluorescent RNA sensors for label-free detection of target molecules. Mol Biosyst 6: 1569–1571 [DOI] [PubMed] [Google Scholar]
- Grate D, Wilson C 1999. Laser-mediated, site-specific inactivation of RNA transcripts. Proc Natl Acad Sci 96: 6131–6136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall B, Cater S, Levy M, Ellington AD 2009. Kinetic optimization of a protein-responsive aptamer beacon. Biotechnol Bioeng 103: 1049–1059 [DOI] [PubMed] [Google Scholar]
- Han DR, Pal S, Liu Y, Yan H 2010. Folding and cutting DNA into reconfigurable topological nanostructures. Nat Nanotechnol 5: 712–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison GP, Mayo MS, Hunter E, Lever AML 1998. Pausing of reverse transcriptase on retroviral RNA templates is influenced by secondary structures both 5′ and 3′ of the catalytic site. Nucleic Acids Res 26: 3433–3442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holeman LA, Robinson SL, Szostak JW, Wilson C 1998. Isolation and characterization of fluorophore-binding RNA aptamers. Fold Des 3: 423–431 [DOI] [PubMed] [Google Scholar]
- Iioka H, Loiselle D, Haystead TA, Macara IG 2011. Efficient detection of RNA–protein interactions using tethered RNAs. Nucleic Acids Res 39: e53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacs FJ, Dwyer DJ, Ding CM, Pervouchine DD, Cantor CR, Collins JJ 2004. Engineered riboregulators enable post-transcriptional control of gene expression. Nat Biotechnol 22: 841–847 [DOI] [PubMed] [Google Scholar]
- Jiang YS, Li B, Milligan JN, Bhadra S, Ellington AD 2013. Real-time detection of isothermal amplification reactions with thermostable catalytic hairpin assembly. J Am Chem Soc 135: 7430–7433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger CA, Wilson SC, Sales-Lee J, Hammond MC 2013. RNA-based fluorescent biosensors for live cell imaging of second messengers cyclic di-GMP and cyclic AMP-GMP. J Am Chem Soc 135: 4906–4909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Winfree E 2011. Synthetic in vitro transcriptional oscillators. Mol Syst Biol 7: 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, White KS, Winfree E 2006. Construction of an in vitro bistable circuit from synthetic transcriptional switches. Mol Syst Biol 2: 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov DM 2005. Binary malachite green aptamer for fluorescent detection of nucleic acids. J Am Chem Soc 127: 12442–12443 [DOI] [PubMed] [Google Scholar]
- Lesnik EA, Freier SM 1995. Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: relationship with base composition and structure. Biochemistry 34: 10807–10815 [DOI] [PubMed] [Google Scholar]
- Li N, Ho CM 2008. Aptamer-based optical probes with separated molecular recognition and signal transduction modules. J Am Chem Soc 130: 2380–2381 [DOI] [PubMed] [Google Scholar]
- Li B, Ellington AD, Chen X 2011. Rational, modular adaptation of enzyme-free DNA circuits to multiple detection methods. Nucleic Acids Res 39: e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Chen X, Ellington AD 2012. Adapting enzyme-free DNA circuits to the detection of loop-mediated isothermal amplification reactions. Anal Chem 84: 8371–8377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JW, Lu Y 2006. Fast colorimetric sensing of adenosine and cocaine based on a general sensor design involving aptamers and nanoparticles. Angew Chem Int Ed 45: 90–94 [DOI] [PubMed] [Google Scholar]
- Lodish HF, Darnell JE 1995. Molecular cell biology. Scientific American Books, W.H. Freeman and Co., New York [Google Scholar]
- Lubin AA, Plaxco KW 2010. Folding-based electrochemical biosensors: the case for responsive nucleic acid architectures. Acc Chem Res 43: 496–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucks JB, Qi L, Mutalik VK, Wang D, Arkin AP 2011. Versatile RNA-sensing transcriptional regulators for engineering genetic networks. Proc Natl Acad Sci 108: 8617–8622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutalik VK, Qi L, Guimaraes JC, Lucks JB, Arkin AP 2012. Rationally designed families of orthogonal RNA regulators of translation. Nat Chem Biol 8: 447–454 [DOI] [PubMed] [Google Scholar]
- Nakayama S, Luo YL, Zhou J, Dayie TK, Sintim HO 2012. Nanomolar fluorescent detection of c-di-GMP using a modular aptamer strategy. Chem Commun 48: 9059–9061 [DOI] [PubMed] [Google Scholar]
- Nitin N, Santangelo PJ, Kim G, Nie SM, Bao G 2004. Peptide-linked molecular beacons for efficient delivery and rapid mRNA detection in living cells. Nucleic Acids Res 32: e58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutiu R, Li Y 2003. Structure-switching signaling aptamers. J Am Chem Soc 125: 4771–4778 [DOI] [PubMed] [Google Scholar]
- Paige JS, Wu KY, Jaffrey SR 2011. RNA mimics of green fluorescent protein. Science 333: 642–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paige JS, Nguyen-Duc T, Song W, Jaffrey SR 2012. Fluorescence imaging of cellular metabolites with RNA. Science 335: 1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei R, Rothman J, Xie YL, Stojanovic MN 2009. Light-up properties of complexes between thiazole orange-small molecule conjugates and aptamers. Nucleic Acids Res 37: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponchon L, Dardel F 2007. Recombinant RNA technology: the tRNA scaffold. Nat Methods 4: 571–576 [DOI] [PubMed] [Google Scholar]
- Pothoulakis G, Ceroni F, Reeve B, Ellis T 2013. The Spinach RNA aptamer as a characterization tool for synthetic biology. ACS Synth Biol 10.1021/sb400089c [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E 2011a. Scaling up digital circuit computation with DNA strand displacement cascades. Science 332: 1196–1201 [DOI] [PubMed] [Google Scholar]
- Qian L, Winfree E 2011b. A simple DNA gate motif for synthesizing large-scale circuits. J R Soc Interface 8: 1281–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian L, Winfree E, Bruck J 2011. Neural network computation with DNA strand displacement cascades. Nature 475: 368–372 [DOI] [PubMed] [Google Scholar]
- Sando S, Narita A, Hayami M, Aoyama Y 2008. Transcription monitoring using fused RNA with a dye-binding light-up aptamer as a tag: a blue fluorescent RNA. Chem Commun 33: 3858–3860 [DOI] [PubMed] [Google Scholar]
- Seelig G, Soloveichik D, Zhang DY, Winfree E 2006. Enzyme-free nucleic acid logic circuits. Science 314: 1585–1588 [DOI] [PubMed] [Google Scholar]
- Serra MJ, Turner DH 1995. Predicting thermodynamic properties of RNA. Methods Enzymol 259: 242–261 [DOI] [PubMed] [Google Scholar]
- Song W, Strack RL, Jaffrey SR 2013. Imaging bacterial protein expression using genetically encoded RNA sensors. Nat Methods 10: 873–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparano BA, Koide K 2005. A strategy for the development of small-molecule-based sensors that strongly fluoresce when bound to a specific RNA. J Am Chem Soc 127: 14954–14955 [DOI] [PubMed] [Google Scholar]
- Sparano BA, Koide K 2007. Fluorescent sensors for specific RNA: a general paradigm using chemistry and combinatorial biology. J Am Chem Soc 129: 4785–4794 [DOI] [PubMed] [Google Scholar]
- Srinivas N, Ouldridge TE, Sulc P, Schaeffer JM, Yurke B, Louis AA, Doye JP, Winfree E 2013. On the biophysics and kinetics of toehold-mediated DNA strand displacement. Nucleic Acids Res 41: 10641–10658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojanovic MN, Kolpashchikov DM 2004. Modular aptameric sensors. J Am Chem Soc 126: 9266–9270 [DOI] [PubMed] [Google Scholar]
- Strack RL, Disney MD, Jaffrey SR 2013. A superfolding Spinach2 reveals the dynamic nature of trinucleotide repeat-containing RNA. Nat Methods 10: 1219–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack RL, Song W, Jaffrey SR 2014. Using Spinach-based sensors for fluorescence imaging of intracellular metabolites and proteins in living bacteria. Nat Protoc 9: 146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Mallikaratchy P, Yang R, Kim YM, Zhu Z, Wang H, Tan W 2008. Aptamer switch probe based on intramolecular displacement. J Am Chem Soc 130: 11268–11269 [DOI] [PubMed] [Google Scholar]
- Tuerk C, Gold L 1990. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage-T4 DNA-polymerase. Science 249: 505–510 [DOI] [PubMed] [Google Scholar]
- Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO 2002. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci 99: 5860–5865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xayaphoummine A, Bucher T, Isambert H 2005. Kinefold web server for RNA/DNA folding path and structure prediction including pseudoknots and knots. Nucleic Acids Res 33: W605–W610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, van Nimwegen E, Zavolan M, Rajewsky N, Schroeder M, Magnasco M, Darnell JE 2003. Decay rates of human mRNAs: correlation with functional characteristics and sequence attributes. Genome Res 13: 1863–1872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin P, Choi HM, Calvert CR, Pierce NA 2008. Programming biomolecular self-assembly pathways. Nature 451: 318–322 [DOI] [PubMed] [Google Scholar]
- Yurke B, Turberfield AJ, Mills AP, Simmel FC, Neumann JL 2000. A DNA-fuelled molecular machine made of DNA. Nature 406: 605–608 [DOI] [PubMed] [Google Scholar]
- Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, Dirks RM, Pierce NA 2011. NUPACK: analysis and design of nucleic acid systems. J Comput Chem 32: 170–173 [DOI] [PubMed] [Google Scholar]
- Zhang DY, Seelig G 2011a. DNA-based fixed gain amplifiers and linear classifier circuits. In DNA Computing and Molecular Programming, Vol. 6518, pp. 176–186 Springer-Verlag, Heidelberg, Berlin, Germany [Google Scholar]
- Zhang DY, Seelig G 2011b. Dynamic DNA nanotechnology using strand-displacement reactions. Nat Chem 3: 103–113 [DOI] [PubMed] [Google Scholar]
- Zhang DY, Winfree E 2009. Control of DNA strand displacement kinetics using toehold exchange. J Am Chem Soc 131: 17303–17314 [DOI] [PubMed] [Google Scholar]
- Zhang DY, Chen SX, Yin P 2012. Optimizing the specificity of nucleic acid hybridization. Nat Chem 4: 208–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Revyakin A, Grimm JB, Lavis LD, Tjian R 2014. Single-molecule tracking of the transcription cycle by sub-second RNA detection. Elife 3: e01775. [DOI] [PMC free article] [PubMed] [Google Scholar]