

The authors investigate the consequences of miR-29a/b misregulation in the mouse brain. They use a neurotropic peptide, derived from the rabies virus glycoprotein (RVG), for targeted delivery of an miRNA inhibitor (miR-29a/b LNA anti-miR) to neuronal cells in vivo, overcoming the blood-brain barrier, and down-regulating miR-29a/b expression in the brain. They show that the mitochondrial outer membrane protein VDAC1 is a target of this miRNA and its dysregulation is a critical feature of neuronal cell death, highlighting the protective role of miR-29 in neuronal survival.
Keywords: neurodegeneration, ataxia, VDAC1, microRNA
Abstract
Several microRNAs have been implicated in neurogenesis, neuronal differentiation, neurodevelopment, and memory. Development of miRNA-based therapeutics, however, needs tools for effective miRNA modulation, tissue-specific delivery, and in vivo evidence of functional effects following the knockdown of miRNA. Expression of miR-29a is reduced in patients and animal models of several neurodegenerative disorders, including Alzheimer's disease, Huntington's disease, and spinocerebellar ataxias. The temporal expression pattern of miR-29b during development also correlates with its protective role in neuronal survival. Here, we report the cellular and behavioral effect of in vivo, brain-specific knockdown of miR-29. We delivered specific anti-miRNAs to the mouse brain using a neurotropic peptide, thus overcoming the blood-brain-barrier and restricting the effect of knockdown to the neuronal cells. Large regions of the hippocampus and cerebellum showed massive cell death, reiterating the role of miR-29 in neuronal survival. The mice showed characteristic features of ataxia, including reduced step length. However, the apoptotic targets of miR-29, such as Puma, Bim, Bak, or Bace1, failed to show expected levels of up-regulation in mice, following knockdown of miR-29. In contrast, another miR-29 target, voltage-dependent anion channel1 (VDAC1), was found to be induced several fold in the hippocampus, cerebellum, and cortex of mice following miRNA knockdown. Partial restoration of apoptosis was achieved by down-regulation of VDAC1 in miR-29 knockdown cells. Our study suggests that regulation of VDAC1 expression by miR-29 is an important determinant of neuronal cell survival in the brain. Loss of miR-29 results in dysregulation of VDAC1, neuronal cell death, and an ataxic phenotype.
INTRODUCTION
The discovery of microRNAs has added an additional layer to the complex regulatory pathways that control gene expression (Bartel 2004). MicroRNAs maintain the expression landscape of the transcriptome and the proteome, their own expression varying spatially and temporally to fine-tune the biological levels of their target molecules (McNeill and Van 2012). Several miRNAs show tissue-specific expression patterns and are either enriched or selectively depleted from the brain. They are implicated in neuronal differentiation, neurite outgrowth, synaptic plasticity, dendritic spine morphology, and memory formation (Kosik 2006).
Neurodegenerative diseases show variable neuronal vulnerabilities with functional failure or pathogenesis initiated from specific subpopulations of CNS cells. Nervous system dysfunction encompasses chronic loss of functional capability of cells and networks as well as neuronal death. A common thread which runs through these diverse mechanisms is the machinery implicated in neuronal survival and apoptosis. Apoptosis is a necessary aspect of neuronal development and the underlying mechanism by which neurodegenerative diseases are manifested. The appropriate expression of regulatory elements such as miRNA maintains the balance between survival and apoptosis (Kent and Mendell 2006; Roshan et al. 2009).
An excellent candidate miRNA which could play a role in neurodegeneration and neuronal survival is miR-29. The miR-29 family consists of miR-29a, miR-29b-1, miR-29b-2, and miR-29c, of which miR-29c has very low expression, perhaps due to a high turnover rate (Zhang et al. 2011; Kriegel et al. 2012). The mature sequences of these miRNAs are highly conserved in human, mouse, and rat. All the members of the miR-29 family have identical seed sequences, and they emerge from two gene clusters on different chromosomes (Kriegel et al. 2012). Both of the clusters contain miR-29b-1 and miR-29b-2, two genomic loci that can potentially code for miR-29b. Down-regulation of miR-29a/b has been correlated with neurodegenerative disease conditions such as Alzheimer's and Huntington's, and several types of cancer (Roshan et al. 2009). In Alzheimer's patient samples, depleted levels of the microRNA are correlated with an elevated level of β-secretase 1 (BACE1) enzyme, which contributes to formation of plaques by cleavage of the amyloid precursor protein (APP) (Hebert et al. 2008; Delay et al. 2012). In a cellular model of spinocerebellar ataxia 17 (SCA17), where we had expressed a TATA-binding protein (TBP) with a pathogenic length of CAG repeats, miRNA expression profiling had shown this miRNA to be down-regulated (Roshan et al. 2012). We have also found mir-29a to be highly expressed in primary neurons and the adult mouse brain (Jovicic et al. 2013). mir-29b expression has also been shown to increase in neuronal cells during cerebral and cortical maturation and in the sympathetic nervous system at a time when these cells are most resistant to apoptosis (Kole et al. 2011).
The miR-29a/b cluster has also been shown to affect synapse formation and plasticity by directly targeting ARPC3, a protein which has a role in actin cytoskeleton remodeling (Lippi et al. 2011). Apart from having roles in neuronal cell differentiation, miR-29a also is seen to be down-regulated in Schwann cells which are in proliferative and differentiating stages, in vitro in culture and in vivo during development, and after sciatic nerve injury (Verrier et al. 2009).
The dysregulation of the miR-29 microRNA family, thus, has implications in neuronal differentiation, proliferation, death, and communication. However, there are other lines of evidence that suggest that, in spite of its expression loss, miR-29a/b function may not be a major determinant of neurodegeneration. A study in Alzheimer's patients (including familial and sporadic cases) has shown that genetic variability in miR-29a and miR-29b and in the miRNA binding sites of the 3′ UTR of BACE1 and APP, which could potentially alter miRNA-target interaction, does not show an association with the disease condition (Bettens et al. 2009). This apparent disconnect between the in vitro effects and in vivo relevance extends to other miRNAs. For instance, knock-out of miR-34a in mice showed no abnormalities in p53 function (Concepcion et al. 2012) in spite of several in vitro studies supporting its role in the p53-mediated apoptosis (He et al. 2007). Also, correlation studies in disease patient samples point to molecular changes which not only contribute to pathogenesis but also those which could occur as a result of compensatory mechanisms. Only in vivo studies with controlled modulation of miR-29a/b expression levels can resolve these problems and reveal the biological relevance of the miRNA. The broad expression pattern of miR-29a/b spanning neuronal, hematopoetic, and lung cells (Kriegel et al. 2012) suggests that modulation should be restricted to the relevant tissue to prevent any pleiotropic effects.
To establish the effects of neuronal miR-29 perturbations in vivo and to mimic conditions of miRNA dysregulation in neurodegenerative disorders, we down-regulated miR-29 in a brain-specific manner in mice. We used a neurotropic peptide for targeted delivery of a locked nucleic acid (LNA) antagomir (Kaur et al. 2007) specific for miR-29 to the brain. After successful down-regulation of the miRNA, we observed apoptosis in the brain cells and an ataxia-like behavioral defect in mice. We tested the levels of chosen targets of the miRNA and did not find significant up-regulation of BACE1 or the other selected pro-apoptotic genes which have been reported to be targets of miR-29. However, we observed a highly significant up-regulation in the expression level of Vdac1, a mediator of apoptosis and a novel target of miR-29 identified in an miRNA-proteomics screen that we carried out previously (Bargaje et al. 2012). In vitro, down-regulation of Vdac1 leads to partial rescue from neuronal death of cells transfected with LNA against miR-29. Here, we report the first attempt to modulate miRNA levels in a brain-specific manner to elucidate the downstream effects in vivo.
RESULTS
Rabies virus glycoprotein delivers antagomir in a brain-specific manner
The rabies virus is a neurotropic virus which overcomes the hindrances posed by the blood-brain-barrier to target itself specifically to the brain. A short peptide derived from rabies virus glycoprotein (RVG) specifically recognizes nicotinic acetylcholine receptors of the neurons to mediate viral entry into the neuronal cells and facilitates retrograde axonal and trans-synaptic transport of the encapsulated nucleic acid into the brain. RVG has been used to deliver siRNA against specific targets in a CNS-specific manner (Kumar et al. 2007). An exosomal membrane protein LAMP2B conjugated with RVG peptide has been used for targeted delivery of siRNA against Bace1 to the brain by intravenous injection (Alvarez-Erviti et al. 2011). Similarly, liposomes which protect siRNA from serum degradation have been used to deliver siRNA against PRPC protein and RVG peptide as utilized for specifically targeting this complex to the brain (Pulford et al. 2010).
We used the 41-amino acid peptide sequence of rabies virus glycoprotein with an additional nona D-arginine (henceforth referred to as RVG) stretch for better interaction with nucleic acid molecules (Kumar et al. 2007). As a delivery control, we used a similar-sized peptide derived from the rabies virus matrix protein (RVM), which is known to show no specificity for delivery to brain tissues (Table 1). We checked the neuronal specificity of the delivery of nucleic acid molecules by RVG peptide in vitro in the mouse neuroblastoma cell line, Neuro2a (N2a), and in vivo in mice. Different molar ratios of a fluorescently labeled control RNA siGLO with RVG peptide and with RVM peptide were used to assemble nucleic acid-peptide complexes and then transfected into HeLa cells and N2a cells. N2a cells showed specific uptake of the fluorescent molecule compared to the HeLa cells when RVG peptide was used, as opposed to delivery using RVM peptide, which showed no preferential localization of siGLO in either cell type (Fig. 1A–H). Although a 1:10 molar ratio of the nucleic acid and peptide was reported to be sufficient (Kumar et al. 2007), in our own experiments, a 1:15 molar ratio was required (Fig. 1).
TABLE 1.
Sequences of peptide and LNA

FIGURE 1.
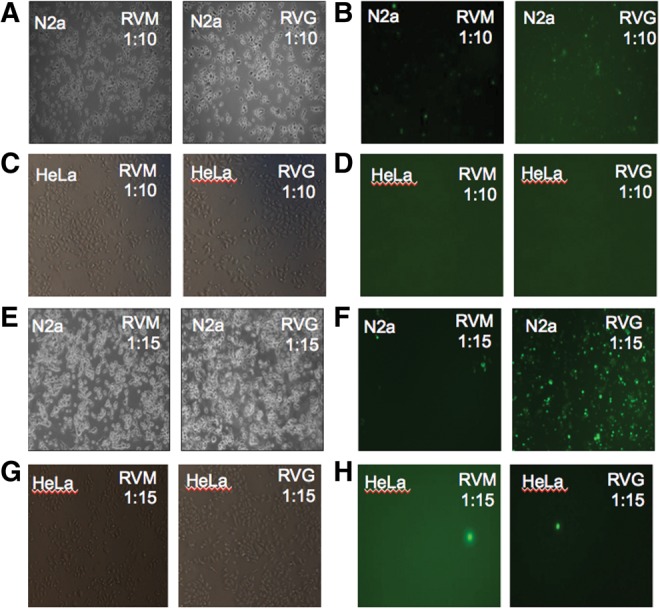
RVG peptide specifically delivers siRNA to N2a cells. N2a and HeLa cells were transfected with siGLO and RVG/RVM peptide complex at two different molar ratios. At a 1:10 molar ratio (siGLO:peptide), RVG/RVM peptide is not able to deliver siGLO efficiently into either N2a or HeLa cells (A–D). At a 1:15 molar ratio (siGLO:peptide), RVG peptide specifically delivers siGLO into N2a and not HeLa cells. RVM peptide is not able to deliver siGLO in either cell type (E–H).
In vivo, the standardized ratio was used to deliver the siGLO-peptide complex into mice by a single intravenous injection into the tail vein. When the RVM peptide was used for delivery, fluorescence was observed mostly in the liver. When complexed with RVG peptide, siGLO was localized to the brain (Fig. 2A–D). Targeting of peptide to different brain regions was also evaluated. Mouse brain was dissected into three different parts—cerebellum, cortex, and hippocampus—and single cell suspensions were prepared. Fluorescence was detected in all the three parts where mice had received siGLO-RVG complex, whereas siGLO-RVM complex injected mice showed no fluorescence (Fig. 2E–J). We counted siGLO-positive cells as a fraction of DAPI-positive cells (total cell number) to estimate percentage transfection in different brain parts. The peptide did not show any specificity for parts of the brain, with ∼30%–45% transfected cells in all parts (Fig. 2K).
FIGURE 2.

RVG peptide delivers siRNA specifically and uniformly to the mouse brain. In vivo specificity of RVG peptide to deliver siRNA in brain was checked by tail vein injections of RVG/RVM peptide along with siGLO. RVM peptide delivered siGLO to the liver but not the brain while RVG peptide specifically delivered siGLO to the brain (A–D). siGLO/RVG complex-injected mice showed fluorescence in all the three regions, whereas siGLO/RVM complex-injected mice showed no fluorescence (E–J). Uniformity of targeting of peptide to different regions of the brain was confirmed by calculating the percentage of siGLO-positive cells in DAPI-positive cells in three fields adding up to 100 siGLO-positive cells (K).
LNA modified antagomirs
We designed two LNA modified oligonucleotides as antagomirs complementary to miR-29. We tested their efficacy by transfecting N2a cells with each of the antagomirs, comparing them with a control LNA that we had previously established to have no discernible effect (Roshan et al. 2012) on neuronal apoptosis. We achieved successful down-regulation of miR-29 in N2a cells by transfecting them with LNA (1 and 2) (see Table 1) at the standardized (1:15) ratio of the LNA-peptide complex. LNA1 down-regulates miR-29a efficiently, resulting in 90% knock-down in 24 h. LNA2 down-regulates miR-29a by ∼70% and miR-29b by ∼95%. LNA2 was more effective in bringing down the collective levels of miR-29a/b transcripts (Fig. 3A,B). miR-29c nucleotide sequence differs from miR-29a at a single position. We did not find detectable miR-29c expression in N2a cells and therefore could not assess the ability of the antagomirs to it. However, the complementarity of the LNA sequences suggests that they may be effective against miR-29c as well. Depending on the efficacy and potential targeting of the LNA, LNA1 is thus referred to as LNA29a/c and LNA2 as LNA29. The LNA:RVG peptide complex in the 1:15 molar ratio was able to down-regulate miR-29 specifically to ∼80% in N2a cells but not in HeLa cells (Fig. 3C–E). The LNA:RVM peptide complex could not down-regulate miR-29 in either cell type (Fig. 3C–E).
FIGURE 3.

RVG peptide-LNA complex can knock down miR-29a/b specifically in N2a cells. To check the knockdown efficiencies of antagomirs, LNA29a/c (LNA1 in Table 1) and LNA29 (LNA2 in Table 1) were transfected into N2a cells using lipofectamine transfection agent, and real-time PCR was performed using Taqman probes for both miR-29a and miR-29b. miR-92a was used as a normalization control. LNA29a/c was able to down-regulate the levels of miR-29 by ∼90% (A), while LNA29 down-regulates miR-29a by ∼70% and miR-29b by ∼95% (B). LNA29a/c was transfected using RVG peptide or RVM peptide in both N2a and HeLa cells at 1:10 and 1:15 molar ratios. In each case, the ratio of RVG/RVM is shown. RVG peptide was specifically able to deliver LNA29a/c in N2a cells at a 1:15 (LNA:peptide) ratio (C). All experiments were done in triplicate. Similarly, LNA29 was transfected using RVG or RVM peptide at 1:10 and 1:15 ratios in N2a and HeLa cells. RVG was able to specifically deliver LNA29 at a 1:15 ratio in N2a cells, and LNA29 was able to down-regulate levels of both miR-29a and miR-29b (D,E). RVM peptide was not able to deliver LNA at both ratios (C–E).
Down-regulation of miR-29 was achieved by RVG peptide and LNA delivery in vivo
BALB/c mice were injected with the LNA-peptide complex intravenously into their tail vein every 24 h on three consecutive days. The dosage of LNA delivered was standardized at 2 mg/g of body weight of the mice. We found a higher dose of LNA-peptide complex (4 mg/g of body weight) was toxic to the mice. Since both control LNA and LNA against miR-29 were toxic at this concentration, it is unclear whether the toxicity was because of the increased amounts of LNA or the consequently increased amounts of RVG peptide used to deliver it.
Behavioral assays for ataxia phenotype, such as hind-limb clasping, gait, and ledge tests, were performed and recorded daily. Whole brain and liver tissues were extracted from all the mice 24 h after the last injection. The brain was divided into intact right and left hemispheres. One hemisphere from each mouse was used for further cellular analysis and the other for molecular analysis. We checked the expression of miR-29a and miR-29b in different parts of the brain. From one hemisphere of the brain from each of the mice, the cerebellum, hippocampus, and cortex were dissected. RNA was isolated from each of the parts, and the expression levels of miR-29 were determined using real-time PCR (Fig. 4A).
FIGURE 4.

Schema for in vivo experiments. RVG peptide-LNA29 complex leads to down-regulation of miR-29a/b levels in mice brain. Tail vein injections were used to deliver control LNA, LNA29a/c, or LNA29 against miR-29 in mouse brain over a 4-d time course followed by daily phenotypic assays (A). Endogenous levels of miR-29 were checked in mouse brain parts after control LNA treatment. Real-time PCR was performed for miR-29a, miR-29b, and miR-92a (normalization control) in cerebellum, cortex, and hippocampus using specific Taqman probes. miR-29a levels were found to be significantly higher in all brain parts compared to levels of miR-29b (n = 9) (B). Control LNA or LNA29 were injected intravenously using RVG peptide, and real-time PCR was done for miR-29a (C), miR-29b (D), and normalized to miR-92a.
We found that the endogenous levels of miR-29a are much higher than that of miR-29b. Expression of miR-29a is higher than that of miR-29b across the different regions of the brain (Fig. 4B). This is in agreement with our observations from in situ hybridization (Roshan et al. 2012) and reports from other groups in HeLa cells (Hwang et al. 2007) and in renal medulla of rats. miR-29a transcripts have also been shown to be more stable than miR-29b transcripts (Zhang et al. 2011). In vivo, LNA29 effectively down-regulated miR-29a and miR-29b in different parts of the brain (Fig. 4C,D).
miR-29a down-regulation causes cell death in the brain
We performed terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assays on the fixed histological sections to visualize cellular DNA fragmentation. DNA damage, which occurs as a result of cell death pathways, was observed throughout the brain sections of the miR-29a knockdown mice. The massive cell death observed in the brain sections of miR-29 knockdown mice confirms the physiological relevance of miR-29a in vivo and confirms that the perturbations of the levels of miR-29a can have downstream effects on neuronal cell death and/or survival (Fig. 5).
FIGURE 5.
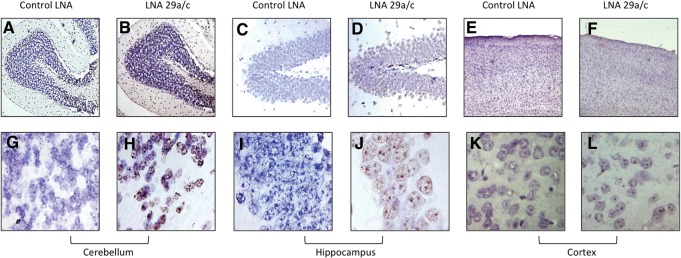
miR-29 knock-down leads to neuronal apoptosis. A TUNEL assay was done on brain sections from Control LNA- and LNA29a/c-injected mice on sections of cerebellum (A,B; G,H), hippocampus (C,D; I,J), and cortex (E,F; K,L). Using an rTdT reaction, slides were developed by DAB solution, and nuclei were counter-stained with hematoxylin. A–F images are at 4× magnification, while G–L are at 100× magnification.
Differential targeting of apoptotic genes by miR-29a
To integrate our observation of cell death with the molecular changes downstream to miR-29a, we investigated the effect of its down-regulation on its well-known direct targets such as Bace1, Puma, Bak, and Bim in vivo. The mRNA levels of these genes were quantified by real-time PCR in the cerebellum, hippocampus, and cortex.
BACE1 (β-site APP-cleaving enzyme) has been shown to have a role in the pathophysiology of Alzheimer's disease. It is highly expressed in the brain tissues of AD patients and is known to contribute to plaque formation by cleaving amyloid precursor protein (APP) and releasing amyloid-β peptide (Cole and Vassar 2007). We have previously validated the increased expression of PUMA, BAK, and BACE1 in a cellular model of SCA17, which expresses a pathogenic form of the TATA-binding protein with extended CAG repeats. BAK, along with BAX and BOK, directly facilitates apoptosis by mediating cytochrome c release from the mitochondrial membrane, which are, in turn, sequestered and antagonized by BCL-2 proper, BCL-2 XL, MCL-11, and TCL-1. Upstream initiators of apoptosis, called the BH3-only proteins, such as PUMA and BIM, are efficient binders of these anti-apoptotic proteins, thereby releasing BAX, BAK, and BOK (Youle and Strasser 2008). N-Bak, a splice variant of Bak, restricted to neurons and not expressed in other neural cells like glia or other cells of the body, also acts as an upstream apoptotic initiator (Uo et al. 2005). Several of these BH3-only proteins have been shown to be targets of miR-29a/b (Kole et al. 2011). However, in our in vivo study, Bim, Bak, Puma, and Bace1 show no significant up-regulation at the mRNA level (Fig. 6A–D).
FIGURE 6.

Expression levels of (A) Bim, (B) Bak, (C) Puma, and (D) Bace1 in miR-29a/b knockdown mice. To check the effect of miR-29 down-regulation on its targets, real-time PCR was performed using total RNA isolated from control LNA- and LNA29a-injected mice. Real-time PCR was done using SYBR Green, and for relative quantification, the reference gene β-2-microglobulin (b2m) was used.
miR-29 down-regulation leads to up-regulation of Vdac1 (voltage-dependent anion-selective-channel protein 1)
We have recently reported the identification of a novel target of miR-29a/b, voltage-dependent anion-selective channel protein 1 (Vdac1) (Supplemental Fig. 1; Bargaje et al. 2012). VDAC1 is an outer mitochondrial membrane protein porin implicated in apoptosis. It is believed that VDAC1 interacts with Bcl-2 family members (Arbel and Shoshan-Barmatz 2010) and is the major channel through which cytochrome c release into the cytoplasm is mediated in apoptotic cells (Shimizu et al. 1999). We have previously carried out a proteomics screen in HEK293 cells in which miR-29 was knocked down, to identify up-regulated targets of the microRNA. VDAC1 was identified in this screen and was shown to contain a target site in its 3′ UTR, although this target site is not predicted by miRNA target prediction programs—PicTar, Targetscan, or miRanda. We performed luciferase assays to experimentally show direct targeting of the Vdac1 3′ UTR by miR-29a in vitro (Bargaje et al. 2012). We have earlier shown that VDAC1 is up-regulated in a cellular model of SCA17 in which N2a cells expressed aberrant TATA-binding protein with a pathogenic expansion of a polyglutamine repeat stretch (TBP-59Q) (Ghosh et al. 2007). Since then, VDAC1 has also been shown to be up-regulated in the hippocampus of Alzheimer's transgenic mice models and in postmortem brain tissue from Alzheimer's patients (Yoo et al. 2001; Cuadrado-Tejedor et al. 2011).
We checked the mRNA levels of VDAC1 in different parts of the brain of the miR-29 knockdown mice and found Vdac1 to be strongly up-regulated in the cerebellum, cortex, and hippocampus (Fig. 7). miRNAs generally result in 40%–80% down-regulation of targets. The large magnitude of induction suggests that, besides direct miRNA-target interaction, other redundant downstream mechanisms also play a role in the observed induction of Vdac1 mRNA.
FIGURE 7.

Vdac1 is up-regulated in miR-29a knockdown mice. To check the effect of miR-29a knock-down on Vdac1, total RNA was isolated from cerebellum, cortex, and hippocampus of LNA29a/c- or control LNA-injected mice. Real-time PCR was done using SYBR Green, and for relative quantification, the reference gene β-2-microglobulin (b2m) was used. There was significant up-regulation observed in Vdac1 expression in miR-29a knockdown mice compared to control LNA-injected mice in all three brain regions. The change in median Ct value corresponds to a fold change of 99.16 (P-value = 0.09; n = 5) in cerebellum, a fold change of 57.54 (P-value = 0.03; n = 5) in the cortex, and a fold change of 31.8 (P-value = 0.023; n = 5) in the hippocampus of the LNA 29a/c-injected mice compared to control LNA-injected mice.
Partial rescue of cell death was observed by reducing the elevated levels of Vdac1
Next, we wanted to test if we could rescue the cell death phenotype caused by miR-29a/b by reducing the elevated levels of Vdac1 in miR-29a/b knockdown cells. Neuro-2a cells were transfected with control or Vdac1 siRNA and harvested after 36 h. Compared to control siRNA, ∼90% down-regulation in the levels of Vdac1 was obtained in Vdac1 siRNA-treated cells (Supplemental Fig. 2). Under similar conditions, miR-29a was also down-regulated using LNA29a/c. Around 40% cell death was obtained in LNA29a/c knockdown cells, while down-regulation of Vdac1 under similar conditions rescued cell death by ∼50% (Fig. 8A,B). Vdac1 siRNA alone had no significant effect on cell viability. Thus, partial restoration of apoptosis, caused by miR-29a/b knockdown, was achieved by reducing the levels of Vdac1. This implies that Vdac1 is a critical player in miR-29a/b-induced neuronal cell death. Besides, the massive neurodegeneration observed in brain sections suggests that knockdown of miR-29a might act through several targets including, but not limited to, VDAC1.
FIGURE 8.
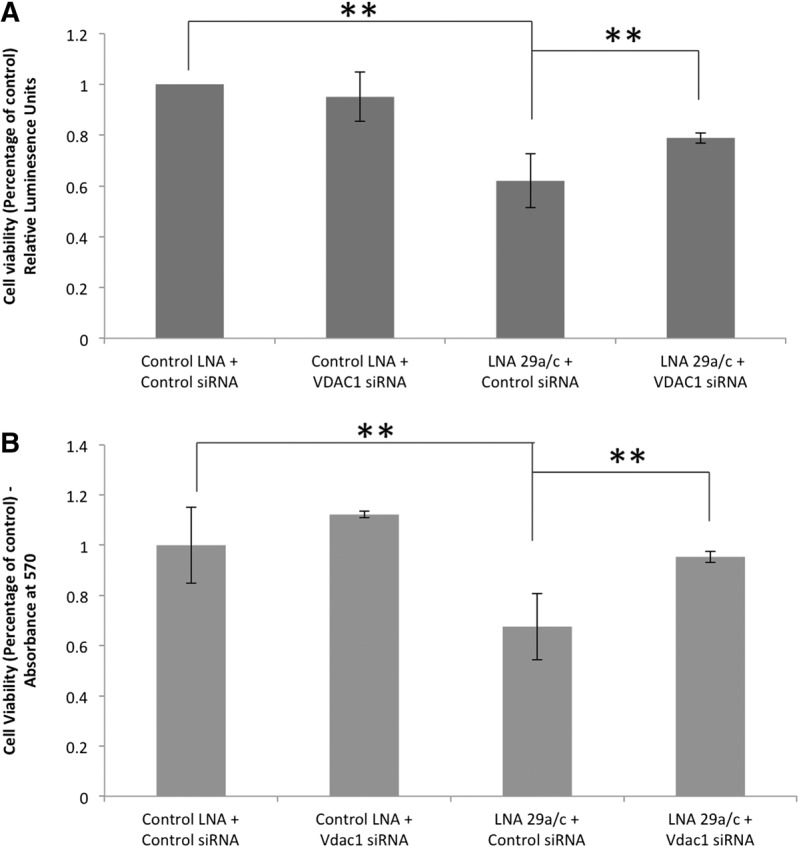
Partial rescue of cell death by reducing the elevated levels of Vdac1. To check the effect of Vdac1 siRNA on cell death, a luminescence-based cell viability assay (A) and MTT assay (B) were performed. Neuro-2a cells were transfected with 40 nM Control LNA or LNA29a/c and 50 nm of Control siRNA/Vdac1 siRNA for 36 h. Upon treatment of LNA29a/c and control siRNA, a 40% reduction in cell viability was observed, whereas by down-regulating the elevated levels of Vdac1 transcripts, close to 48% rescue of cell death was observed (A). A similar trend was obtained by an MTT assay, where down-regulating elevated Vdac1 levels after LNA 29a/c treatment lead to a rescue of up to 64% of cell death (B), whereas Vdac1 down-regulation alone had no significant effect on cell viability. (**) P-value < 0.01; n = 3.
Ataxia phenotype was observed in mice injected with LNA29
We have previously reported that miR-29 is down-regulated in neuronal cells expressing pathogenic polygultamine-TBP protein in a cellular model of SCA17. Therefore, we assayed the mice for an ataxia phenotype using standard tests for motor control, gait assays, spinal deformity, and hind-limb clasping (Guyenet et al. 2010). Mice injected with LNA29a/c showed no abnormalities in the tests performed. In contrast, mice injected with LNA29 showed gait abnormalities typical of ataxia, which include difficulty in walking on plain surfaces, a ledge, and a straight rod. We recorded the footsteps of the mice and observed a significant reduction in step-length in the miR-29a/b knockdown mice (Fig. 9B,C). LNA29a/c, which specifically affects miR-29a, did not show the ataxia phenotype (Fig. 9A,C). This is, perhaps, due to redundancy in the functions of miR-29a and miR-29b. Alternatively, miR-29b may be specifically expressed in regions of the brain that are important for motor coordination.
FIGURE 9.
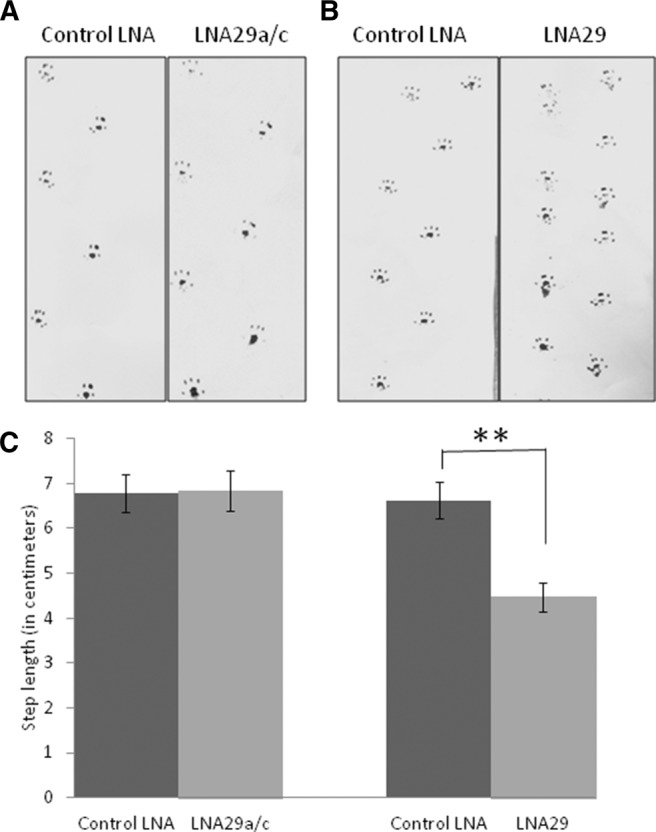
Behavioral effect on miR-29a/b knockdown mice. Footprint analysis was done with Control LNA-, LNA29a/c-, or LNA29-injected mice. Ink was applied on hind paws of the mice, and they were allowed to walk on a strip of paper (A,B). The distance between adjacent footprints was measured (C). (**) P value < 0.001; n = 3.
DISCUSSION
Several targets of the microRNA of the miR-29 family have been identified and validated in vitro. miR-29a/b has been reported to target members of the Bcl-2 protein family, which includes the BH3-only family of pro-apoptotic proteins, and it also translationally inhibits anti-apoptotic proteins such as MCL-1 and TCL-1 (Pekarsky et al. 2006; Mott et al. 2007). These contradictory roles in apoptosis are not yet well understood in the neuronal system. miR-29 has also been shown to target Cdc42 and p85α, thus acting as a positive regulator of p53 (Park et al. 2009). DNA methyltransferases 3A (Dnmt3a) and 3B (Dmnt3b) are also reported targets in a study in which miR-29 was shown to reduce the growth of lung cancer cells in vitro and in mice (Fabbri et al. 2007). Another class of targets of miR-29 includes key proteins involved in extracellular matrix formation (Kriegel et al. 2012). This is empirically correlated with the aberrant expression profile of miR-29 in different tissues in fibrotic conditions (Kriegel et al. 2012). It is not clear how many of the large number of miR-29 targets reported in diverse cell types are relevant in vivo. Knock-down of the endogenous miRNA in a tissue-specific manner allows us to test the validity of the targets in vivo. The target identified in this study—Vdac1—seems to be a major player in miR-29a/b-mediated neuronal apoptosis. Vdac1 is known to be one of the key contributors in apoptosis, but the exact mechanism is still unclear (Rostovtseva et al. 2005). We observed partial restoration of cell death while reducing the elevated levels of Vdac1. This could be because the elevation of Vdac1 is several fold in the LNA29a/c condition, and siRNA against Vdac1 is not able to restore the level to the normal state. The residual Vdac1 can contribute to cell death. Secondly, it is possible that an activator of Vdac1 also could be a target of miR-29a, which leads to such a massive elevation. Also certain unexplored apoptotic genes can be targeted by miR-29a. All these possibilities remain to be explored in the future to characterize the cell death caused by miR-29.
Our results indicate that several of the apoptosis-related targets of miR-29a are not elevated in expression following miR-29a knockdown, at least, at the RNA level. However, in view of the massive cell death in the brain following miR-29a/b knock-down, it is likely that the apoptosis-related known targets of miR-29a/b are affected at the protein level. The amount of tissue does not allow us to test the protein levels of these targets at this time. There are several possible reasons for the apparent lack of effect on known targets of miR-29a. Firstly, miR-29a may block the translation of these targets, leaving transcript levels unchanged. Secondly, the relative affinity and accessibility of the targets may determine which of the several targets reported in vitro are of relevance in vivo. We are currently not aware of any selectivity that the peptide may show for different neurons. However, this possibility has not been explicitly ruled out and may explain the escape of targeting of mRNAs. In neuropathological conditions, changes not only reflect intrinsic molecular or functional disturbances in the cell but have implications in other regions of the brain with which the affected cells communicate and function in a concerted manner. In-built redundancies or degeneracy can also buffer changes within the system.
It is pertinent to note that the single miR-29a target site present in Vdac1 is a low-affinity site that is not predicted by target prediction algorithms. Antisense-mediated partial knock-down of an miRNA is likely to selectively impact low-affinity targets, since high-affinity targets may sequester the residual miRNA. This is a possible explanation for the apparent lack of effect on other targets of miR-29a. This is an important aspect of the development of anti-miRNA therapeutics, since any partial knockdown is likely to result in selective targeting of low-affinity sites.
Since microRNAs are vital modulators of various physiological processes, perturbations in their levels or any other form of disruption of their function have important implications in human diseases. There are several paradigms of gene dysregulation by miRNA in neurodegenerative conditions (for review, see Roshan et al. 2009). For instance, a pathogenic expansion of CAG repeats in the coding region of the Ataxin1 gene causes spinocerebellar ataxia 1, and the levels of the transcripts of this gene are directly controlled by the microRNAs (miR-101, miR-19, miR-130) which have target sites in its 3′ UTR (Lee et al. 2008). Aberrant miRNA expression can also upset general homeostasis or maintenance of cellular or network function in the nervous system by targeting vital pathways involved in synaptic transmission, excitatory or inhibitory responses, oxidative stress, inflammation, cell death, cell survival, or other processes. Expression of miR-9, which regulates transcription factor REST and coREST, has been shown to be significantly down-regulated in tissue samples from Huntington's patients (Packer et al. 2008). miR-133b has been found to be reduced in the midbrain of post-mortem tissue samples from Parkinson's patients. This miRNA is engaged in a regulatory feedback loop with PITX3 (Kim et al. 2007), a transcription factor which has a role in dopamine neuron development. Our study provides a methodology for knocking down the expression of miRNAs in the brain using anti-miRs delivered through the trans-vascular route. In the future, it will be of interest to test the applicability of this method to introduce miRNA mimics in the brain and its ability to correct neurodegeneration in transgenic mouse models.
MATERIALS AND METHODS
Animals
Experimental protocols were approved by the Institutional Animals Ethics Committee. BALB/c mice (5–6 wk old, weighing 20–22 g) were obtained from the Central Drug Research Institute, Lucknow, India, and acclimatized for 1 wk under laboratory conditions (25 ± 2°C, 55% humidity) before starting the experiments.
Peptide-LNA complex
For in vitro delivery experiments, 1 d before transduction, 0.3 × 106 cells were plated in a six-well plate. On the next day, 40 nM of LNA/siGLO were incubated with a 1:10 and 1:15 molar ratio of LNA:peptide for 30 min at room temperature in serum-free media. The complex was added drop-wise on cells and incubated at 37°C for 24 h. For in vivo delivery, female BALB/c mice, aged 6–8 wk, were used. Fifty micrograms of LNA/siGLO and 850 µg peptide were used per injection at a 1:15 molar ratio of LNA:peptide. Two hundred to three hundred microliters of LNA and peptide complex in 5% glucose were incubated at RT for 30 min and used for tail vein injections.
Real-time PCR for miRNA-29a/b
For real-time PCR, total RNA was isolated from N2a/HeLa cells using TRIzol reagent (Invitrogen), following the manufacturer's protocol. Mouse brain was microdissected into three different parts—cerebellum, cortex, and hippocampus. For real-time PCR, total RNA was isolated by crushing brain parts in TRIzol reagent using liquid nitrogen, following the manufacturer's protocol. For cDNA synthesis and real-time PCR, TaqMan assays (Applied Biosystems; Cat.No. 000412[miR-29a], 000413[miR-29b], 000430[miR-92]) specific for mature mmu-miR-29a, mmu-miR-29b, and mmu-miR-92 was used. mmu-miR-92 was used as an endogenous control. A relative quantification method was used for data analysis (Livak and Schmittgen 2001).
Real-time PCR for targets
After total RNA isolation using TRIzol, 2 µg of RNA were used for DNase I treatment (Fermentas, #EN0521). cDNA was synthesized using random hexamer primers (Invitrogen) and M-MuLV reverse transcriptase (NEB, BO253S) at 42°C for 1 h. Real-time PCR was done using SYBR Green (Roche). Gapdh and β-2-microglobulin (B2m) were used as endogenous controls. For analysis, a relative quantification method was used. Sequence of primers is given in Table 2 below.
TABLE 2.
Sequence of primers

TUNEL assay
Terminal deoxynucleotidyltransferase dUTP nick end labeling was done on mouse brain sections using the DeadEnd Colorimetric TUNEL System (Promega) using the manufacturer's protocol. The fixed, paraffin-embedded sections, after de-paraffinization and hydration, were incubated with Proteinase K (1 mg/mL) for 15 min at 37°C. After PBS washes, the slides were treated with kit-provided equilibration buffer for 7 min, followed by treatment with a reaction mixture containing recombinant Terminal Deoxynucleotidyl Transferase and biotinylated nucleotides for 1 h at 37°C. The slides were then treated with saline-sodium citrate buffer and with 6% hydrogen peroxide in PBS. Next, the slides were incubated with Streptavidin-HRP conjugate for 30 min at 37°C and then in a DAB substrate solution for 5 min. Cells were counter-stained in hematoxylin solution for 5 min.
Down-regulation of Vdac1 and cell viability assay
Neuro-2a cells were transfected with 50 nM control siRNA (siGENOME smart, Dharmacon)/Vdac1 siRNA (Ambion, AM16704) and 40 nM LNA using lipofectamine transfection reagent. Thirty-six hours post-transfection, a CellTiter-GLO luminescent cell viability assay (Promega) was performed as per the manufacturer's protocol. For the MTT assay, 20 µL of MTT solution (5 mg/mL) were added per 100 µL of media. Cells were incubated for 1 h at 37°C, and 100 µL of DMSO were added in every well to solubilize the colored purple formazan. Absorbance at 570 nm and, for reference, absorbance at 650 nm were measured.
Behavioral assays
The gait of mice was observed and recorded. The mice were allowed to walk on a rod and cage for 2 min each. For observing hind-limb clasping, mice were held by their tail for 15 sec. To record their footprints, the hind paws were dipped in ink and the mice were made to walk on paper in a straight line.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Munia Ganguli and Souvik Maiti for critical comments on the manuscript. We also acknowledge Rangeetha J. Naik, Rakesh Dey, and Bijay Pattnaik for their help with experimental methods. This work was funded by the Council of Scientific and Industrial Research (BSC0123). R.R. acknowledges fellowships from the Council of Scientific and Industrial Research, India. M.A.S. acknowledges fellowship from the University Grants Commission, India.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.044008.113.
REFERENCES
- Arbel N, Shoshan-Barmatz V 2010. Voltage-dependent anion channel 1-based peptides interact with Bcl-2 to prevent antiapoptotic activity. J Biol Chem 285: 6053–6062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, Wood MJ 2011. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 29: 341–345 [DOI] [PubMed] [Google Scholar]
- Bargaje R, Gupta S, Sarkeshik A, Park R, Xu T, Sarkar M, Halimani M, Roy SS, Yates J, Pillai B 2012. Identification of novel targets for miR-29a using miRNA proteomics. PLoS One 7: e43243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116: 281–297 [DOI] [PubMed] [Google Scholar]
- Bettens K, Brouwers N, Engelborghs S, Van Miegroet H, De Deyn PP, Theuns J, Sleegers K, Van Broeckhoven C 2009. APP and BACE1 miRNA genetic variability has no major role in risk for Alzheimer disease. Hum Mutat 30: 1207–1213 [DOI] [PubMed] [Google Scholar]
- Cole SL, Vassar R 2007. The Alzheimer's disease β-secretase enzyme, BACE1. Mol Neurodegener 2: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Concepcion CP, Han YC, Mu P, Bonetti C, Yao E, D'Andrea A, Vidigal JA, Maughan WP, Ogrodowski P, Ventura A 2012. Intact p53-dependent responses in miR-34-deficient mice. PLoS Genet 8: e1002797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuadrado-Tejedor M, Vilarino M, Cabodevilla F, Del Rio J, Frechilla D, Perez-Mediavilla A 2011. Enhanced expression of the voltage-dependent anion channel 1 (VDAC1) in Alzheimer's disease transgenic mice: an insight into the pathogenic effects of amyloid-β. J Alzheimers Dis 23: 195–206 [DOI] [PubMed] [Google Scholar]
- Delay C, Mandemakers W, Hebert SS 2012. MicroRNAs in Alzheimer's disease. Neurobiol Dis 46: 285–290 [DOI] [PubMed] [Google Scholar]
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. 2007. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci 104: 15805–15810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh T, Pandey N, Maitra A, Brahmachari SK, Pillai B 2007. A role for voltage-dependent anion channel Vdac1 in polyglutamine-mediated neuronal cell death. PLoS One 2: e1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyenet SJ, Furrer SA, Damian VM, Baughan TD, La Spada AR, Garden GA 2010. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J Vis Exp 10.3791/1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, et al. 2007. A microRNA component of the p53 tumour suppressor network. Nature 447: 1130–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert SS, Horre K, Nicolai L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B 2008. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/β-secretase expression. Proc Natl Acad Sci 105: 6415–6420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HW, Wentzel EA, Mendell JT 2007. A hexanucleotide element directs microRNA nuclear import. Science 315: 97–100 [DOI] [PubMed] [Google Scholar]
- Jovicic A, Roshan R, Moisoi N, Pradervand S, Moser R, Pillai B, Luthi-Carter R 2013. Comprehensive expression analyses of neural cell-type-specific miRNAs identify new determinants of the specification and maintenance of neuronal phenotypes. J Neurosci 33: 5127–5137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur H, Babu BR, Maiti S 2007. Perspectives on chemistry and therapeutic applications of Locked Nucleic Acid (LNA). Chem Rev 107: 4672–4697 [DOI] [PubMed] [Google Scholar]
- Kent OA, Mendell JT 2006. A small piece in the cancer puzzle: microRNAs as tumor suppressors and oncogenes. Oncogene 25: 6188–6196 [DOI] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A 2007. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 317: 1220–1224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole AJ, Swahari V, Hammond SM, Deshmukh M 2011. miR-29b is activated during neuronal maturation and targets BH3-only genes to restrict apoptosis. Genes Dev 25: 125–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS 2006. The neuronal microRNA system. Nat Rev Neurosci 7: 911–920 [DOI] [PubMed] [Google Scholar]
- Kriegel AJ, Liu Y, Fang Y, Ding X, Liang M 2012. The miR-29 family: genomics, cell biology, and relevance to renal and cardiovascular injury. Physiol Genomics 44: 237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Wu H, McBride JL, Jung KE, Kim MH, Davidson BL, Lee SK, Shankar P, Manjunath N 2007. Transvascular delivery of small interfering RNA to the central nervous system. Nature 448: 39–43 [DOI] [PubMed] [Google Scholar]
- Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY 2008. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci 11: 1137–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippi G, Steinert JR, Marczylo EL, D'Oro S, Fiore R, Forsythe ID, Schratt G, Zoli M, Nicotera P, Young KW 2011. Targeting of the Arpc3 actin nucleation factor by miR-29a/b regulates dendritic spine morphology. J Cell Biol 194: 889–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔCT) method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- McNeill E, Van Vactor D 2012. MicroRNAs shape the neuronal landscape. Neuron 75: 363–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mott JL, Kobayashi S, Bronk SF, Gores GJ 2007. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 26: 6133–6140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Packer AN, Xing Y, Harper SQ, Jones L, Davidson BL 2008. The bifunctional microRNA miR-9/miR-9* regulates REST and CoREST and is downregulated in Huntington's disease. J Neurosci 28: 14341–14346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Lee JH, Ha M, Nam JW, Kim VN 2009. miR-29 miRNAs activate p53 by targeting p85 α and CDC42. Nat Struct Mol Biol 16: 23–29 [DOI] [PubMed] [Google Scholar]
- Pekarsky Y, Santanam U, Cimmino A, Palamarchuk A, Efanov A, Maximov V, Volinia S, Alder H, Liu CG, Rassenti L, et al. 2006. Tcl1 expression in chronic lymphocytic leukemia is regulated by miR-29 and miR-181. Cancer Res 66: 11590–11593 [DOI] [PubMed] [Google Scholar]
- Pulford B, Reim N, Bell A, Veatch J, Forster G, Bender H, Meyerett C, Hafeman S, Michel B, Johnson T, et al. 2010. Liposome-siRNA-peptide complexes cross the blood-brain barrier and significantly decrease PrP on neuronal cells and PrP in infected cell cultures. PLoS One 5: e11085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roshan R, Ghosh T, Scaria V, Pillai B 2009. MicroRNAs: novel therapeutic targets in neurodegenerative diseases. Drug Discov Today 14: 1123–1129 [DOI] [PubMed] [Google Scholar]
- Roshan R, Ghosh T, Gadgil M, Pillai B 2012. Regulation of BACE1 by miR-29a/b in a cellular model of Spinocerebellar Ataxia 17. RNA Biol 9: 891–899 [DOI] [PubMed] [Google Scholar]
- Rostovtseva TK, Tan W, Colombini M 2005. On the role of VDAC in apoptosis: fact and fiction. J Bioenerg Biomembr 37: 129–142 [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y 1999. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399: 483–487 [DOI] [PubMed] [Google Scholar]
- Uo T, Kinoshita Y, Morrison RS 2005. Neurons exclusively express N-Bak, a BH3 domain-only Bak isoform that promotes neuronal apoptosis. J Biol Chem 280: 9065–9073 [DOI] [PubMed] [Google Scholar]
- Verrier JD, Lau P, Hudson L, Murashov AK, Renne R, Notterpek L 2009. Peripheral myelin protein 22 is regulated post-transcriptionally by miRNA-29a. Glia 57: 1265–1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo BC, Fountoulakis M, Cairns N, Lubec G 2001. Changes of voltage-dependent anion-selective channel proteins VDAC1 and VDAC2 brain levels in patients with Alzheimer's disease and Down syndrome. Electrophoresis 22: 172–179 [DOI] [PubMed] [Google Scholar]
- Youle RJ, Strasser A 2008. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol 9: 47–59 [DOI] [PubMed] [Google Scholar]
- Zhang Z, Zou J, Wang GK, Zhang JT, Huang S, Qin YW, Jing Q 2011. Uracils at nucleotide position 9–11 are required for the rapid turnover of miR-29 family. Nucleic Acids Res 39: 4387–4395 [DOI] [PMC free article] [PubMed] [Google Scholar]