

Abstract
Purpose
To test the hypothesis that small molecule targeting of nucleophosmin (NPM1) represents a rational approach for radiosensitization.
Methods and Materials
Wild type and NPM1-deficient mouse embryo fibroblasts (MEFs) were used to determine if radiosensitization produced by the small molecule YTR107 was NPM1 dependent. The stress response to ionizing radiation was assessed by quantifying pNPM1, γH2AX, and Rad51 foci, neutral comet tail moment, and colony formation. NPM1 levels in a human-derived NSCLC tissue microarray (TMA) were determined by immunohistochemistry. YTR107-mediated radiosensitization was assessed in NSCLC cell lines and xenografts.
Results
Use of NPM1-null MEFs demonstrated that NPM1 is critical for DNA double strand break (DSB) repair, that loss of NPM1 increases radiation sensitivity, and that YTR107-mediated radiosensitization is NPM1 dependent. YTR107 was shown to inhibit NPM1 oligomerization and impair formation of pNPM1 irradiation-induced foci (IRIF) that co-localize with γH2AX foci. Analysis of the TMA demonstrated that NPM1 is over expressed in subsets of NSCLC. YTR107 inhibited DNA DSB repair and radiosensitized NSCLC lines and xenografts.
Conclusions
These data demonstrate that YTR107-mediated targeting of NPM1 impairs DNA DSB repair, an event that increases radiation sensitivity.
Introduction
Developing durable therapeutic approaches that minimize locoregional failure following radiotherapy with curative intent represents a significant challenge. Genetic composition dictates a tumor cell’s response to irradiation and genetic heterogeneity can explain why some tumors respond more favorably than others. Comprehensive sequencing of cancer cell genomes has identified a large variation in mutational spectrum [14]. A collateral consequence of the genetic heterogeneity is a divergent radiation response that increases the complexity of developing novel approaches for radiosensitization.
The current paradigm of dose escalation has not proven an effective approach. RTOG trials 9311 and 0617 have identified two weaknesses of conventional dose escalation: failure of higher doses to increase local control coupled with an increase in normal tissue toxicity [2,12,28]. Conventional dose escalation fails to discriminate between tumor and normally differentiated parenchyma. RTOG study authors suggest that the protracted treatment times required for dose escalation can promote accelerated tumor cell repopulation, which negates the tumoricidal effect of higher doses [2,12]. Thus, the challenge is to develop radiation sensitizers that are efficacious against a spectrum of disparate cancer harboring complex genetic profiles, but do not radiosensitize normal tissue.
Oncogenesis-mediated proliferation can produce significant replication stress that causes DNA damage response pathways in tumor cells to operate at near maximal capacity, limiting their ability to repair the additional DNA DSBs generated by therapeutic radiation [18]. Thus, targeting DNA repair pathways in cancer cells exposed to ionizing radiation may represent an attractive actionable approach for genetically diverse cancers.
We previously employed a forward chemical genetics screen to identify compounds that could be used to exploit DNA repair pathways in tumor cells burdened with oncogenesis-mediated replication stress [24]. We identified YTR107, a substituted 5-((N-benzyl-1H-indol-3-yl)methylene)pyrimidine-2,4,6(1H,3H,5H) trione, that amplifies replication stress, inhibits repair of DNA DSBs, and radiosensitizes a broad spectrum of tumor types harboring complex genetic profiles [24]. YTR107 is well tolerated in mice and does not produce overt toxicity [24]. Biochemical approaches led to the identification of NPM1 as a potential molecular target for YTR107 [24]. However, direct genetic evidence demonstrating that small molecule targeting of NPM1 is a radiosensitizing event remains to be established.
Herein we use a defined genetic model to demonstrate that YTR107-mediated radiosensitization requires NPM1. We found that NPM1-null mouse embryo fibroblasts (MEFs), but not wild type MEFs, are burdened with high levels of γH2AX foci, a characteristic of replication stress [6]. NPM1-null MEFs exhibit a defect in DNA DSB repair and a corresponding increase in radiation sensitivity. Because the DNA damage response pathway is continuously activated in NSCLC in response to constant replication stress [10], and with the knowledge that NPM1 expression minimizes such stress [6], we hypothesized that NSCLC tumors might require significant expression of NPM1 [6]. This hypothesis was validated by analyzing a NSCLC tumor microarray. We then showed that targeting of NPM1 is a radiosensitizing event in NSCLC cell lines and in xenograft tumor models. Thus, these data support the hypothesis that NPM1 is an actionable target for radiosensitization.
Materials and Methods
Cell lines and antibodies
Wild type and NPM1-null MEFs have been described previously [16]. Calu1, HCC827, A549, H460, and H226 NSCLC cells were obtained from ATCC. PC9/BRC-1 and PC9/BRC-4 NSCLC lines are well described [5] and were a gracious gift from Dr. William Pao. Lewis Lung Carcinoma (LLC) cells were a gracious gift from Dr. Dennis Hallahan. The following antibodies were used: pS139 H2AX (Millipore), pT199 NPM1 (Abcam), NPM1 (Life Technologies), and Rad51 H-92 (Santa Cruz Biotech).
Colony formation assays were performed according to [9]. Known numbers of cells were inoculated into T25 flasks containing 5 ml of growth medium, incubated overnight at 37°C, and then irradiated at room temperature using a 137Cs Mark 1 irradiator (2 Gy/min).
Immunofluorescence
Immunofluorescence staining was performed as described [24]. Images were acquired using an Olympus FV1000 or a LSM 510 laser confocal microscope. Ionizing radiation-induced foci (IRIF) were quantified using ImageJ software PZ_FOCIEZ.
Irradiation
Cells were irradiated with a Mark 1 137Cs irradiator (2.0 Gy per min). A Pantek 300 kVp/10 mA X-ray machine (2.3 Gy per min) was used to irradiate xenograft tumors.
Neutral Comet Assay
The assay was performed using the Comet Assay kit following the manufacturer’s instructions (Trevigen). Comet tail moment (defined in [21]) is reported as the mean of the log2 transformed moment, as suggested by Wiklund and Agurell [27].
Tissue microarray
A NSCLC tissue microarray (TMA) containing 100 cores compiled from 10 normal lung tissue samples and 40 cases of NSCLC was obtained from US Biomax (Catalog # LC10011). The TMA was formalin-fixed and paraffin-embedded. Clinical data (Grade, Stage, and Type) was provided by the manufacturer. The array was immunostained for NPM1 and lightly counterstained with Mayer’s hematoxylin. Immunohistochemical staining for NPM1 was semiquantitatively scored from 1 (no cytoplasmic staining) to 3 (intense cytoplasmic staining).
Tumor growth measurement
These studies were approved by the XXX Institutional Animal Care and Use Committee and performed under guidelines outlined in The Guide for the Care and Use of Laboratory Animals. Hindlimbs of immunocompetent 6–8 wk old C57BL/6J mice (Charles Rivers) were subcutaneously implanted with 106 LLC tumor cells and the hindlimbs of athymic Fox1 nu/nu mice (Harlan Sprague Dawley) were subcutaneously implanted with 5×105 A549 tumor cells. When tumors were palpable mice were randomized to the following protocols: daily i.p. injections of (a) DMSO (25 μl) or (b) YTR107 in DMSO (25 μl). Mice harboring LLC tumors were administered 10 mg/kg YTR107. Mice with A549 tumors received 20 mg/kg. Thirty min after injection tumors were administered (c) 0 Gy or (d) 2.4 Gy of x-rays. Mice were shielded such that only the tumors were irradiated. Digital calipers were used to obtain the length and width of each tumor. Tumor volume was calculated 3 times per week, according to the formula: ).
YTR107
Synthesis and characterization of YTR107 was performed as previously described [24].
Statistical Methods
Statistical analysis was carried out using either GraphPad Prism5 or Excel 2010 software. P values less than 0.05 were considered significant.
Results
We used a genetic model consisting of p53−/− NPM1+/+ (denoted as wt-MEFs) and p53−/− NPM1−/− (denoted as NPM1-null) MEFs [16] to test the hypothesis that targeting of NPM1 by the small molecule YTR107 can be a radiosensitizing event. In this model it is necessary to use a p53 null phenotype because NPM1 deletion can interfere with cell viability due to the induction of the tumor suppressor p53 [7].
Persistence of γH2AX foci in the absence of NPM1
Approximately 8 +/− 2% (SD) of non-irradiated wt-MEFs contained more than 10 γH2AX foci per cell and after counting 250 cells we did not observe a single cell with more than 20 foci (Fig 1). In contrast, 14% +/− 3% (SD) of unirradiated NPM1-null MEFs exhibited between 10 and 20 γH2AX foci per cell and an additional 14 +/−3% (SD) contained more than 21 foci per cell. Thus, proliferating NPM1-null MEFs have significantly more γH2AX foci than wild type (P < 10−3, 2 tailed Fisher’s Exact Test).
Fig 1. Loss of NPM1 results in more constitutive and IR-induced γH2AX foci.
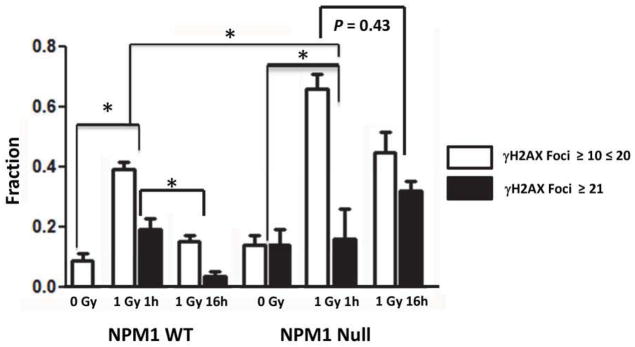
NPM1-wt and NPM1-null MEFs were administered 0 or 1 Gy. Irradiated cells were incubated at 37°C for 1 or 6 hrs. Histogram showing the fraction of cells with 11–20 (white bars) or >20 (black bars) γH2AX foci N= 200–250 nuclei per point. *P < 10−3 2 tailed Fisher’s Exact Test.
One hour after administering 1 Gy to NPM1 wt-MEFs, 58% +/− 11% (SD) harbored more than 10 γH2AX foci per cell, but this number diminished significantly after 16 hours of repair (P < 10−3; Fig 1, representative foci are shown in Fig S1A). On the other hand, 81+/− 16% (SD) of NPM1-null MEFs contained 10 or more γH2AX foci per cell 1h hour after administering 1 Gy and there was not a statistically significant decrease in foci number over the 16 hour repair period (P = 0.43). The failure to disassemble γH2AX foci following IR in NPM1-null MEFS implies a defect in DNA repair.
NPM1 is phosphorylated at T199 [13] in order to regulate centrosome duplication [20] and DNA repair [13]. To investigate the role of NPM1 T199 phosphorylation in γH2AX foci formation and disassembly, we transiently transfected NPM1-null MEFs with plasmids expressing Myc-tagged wild type NPM1 or a T199 non-phosphorylatable NPM1 mutant (Myc-tagged T199A, Fig S1B), administered 1 Gy and assayed γH2AX foci 1 hr later. Expression of wild type NPM1 yielded significantly fewer cells harboring 10 or more γH2AX foci per cell compared to cells expressing T199A NPM1 (25 +/− 6% vs 57 +/− 10%, P = 0.014, N=76, 2 tailed Fisher’s Exact Test, representative images shown in Fig S1C). These results are consistent with the results of Koike et al [13] and demonstrate that the increase in γH2AX foci observed in irradiated NPM1-null cells is a consequence of a loss of pT199 NPM1.
Inactivation of NPM1 enhances Rad51 foci formation in response to irradiation
Paull et al [22] have shown that there is a strong correlation between formation of γH2AX foci and formation of Rad51 foci in proliferating S and G2 cells. Therefore, MEFs were irradiated with 4 Gy and Rad51 foci formation assessed 0–6 hrs later. Representative images of Rad51 foci are shown in Fig S2. The data presented in Fig 2 demonstrates that whereas very few non-irradiated wt MEFs cells contained Rad51 foci, roughly 10% of NPM1-null MEFs contained 5 or more foci (P < 0.001 Student’s t test). In addition, whereas only 15–25% of wt MEFs accumulated more than 5 Rad51 foci/cell 2–6 hrs following 4 Gy, ~40% of NPM1-null cells contained more than 5 Rad51 foci following irradiation (P< 0.001 two tailed ANOVA). The results represent the average of 3 independent experiments.
Fig 2. Rad51 foci formation.
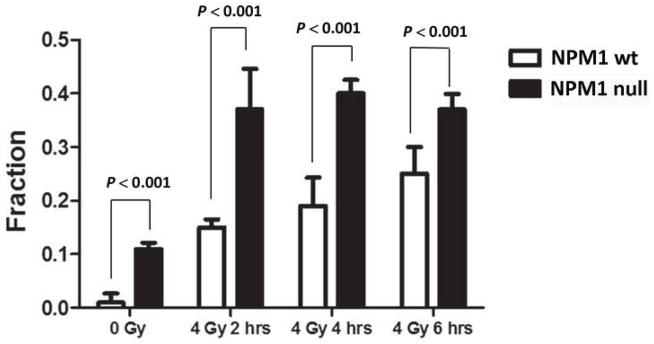
NPM1-wt and NPM1-null MEFs were administered 0 or 4 Gy. Irradiated cells were incubated at 37°C for 1–6 hrs. Histogram showing the fraction of nuclei with greater than 5 Rad51 foci as a function of time after 4Gy irradiation. N=100 nuclei per point. The results represent the average (error bars: +/− SD) from three independent experiments.
NPM1 is required for repair of DNA DSBs
To clarify why large numbers of γH2AX and Rad51 foci were observed in the absence of NPM1, neutral comet assays, which measure DNA DSBs, were performed on wt MEFs and NPM1-null MEFs. Formation of DNA DSBs can be quantified by irradiating at 4°C and then immediately subjecting cells to a neutral comet assay. Under conditions that do not allow repair, we did not observe a statistically significant difference in mean tail moment (P > 0.05, Fig 3). However, when repair was allowed to proceed at 37°C there was a statistically significant difference between wild type and NPM1-null MEFs following 45 (P=0.042) and 90 minutes (P<0.0001) of repair. Repair kinetics followed a log linear relationship in wild type NPM1 cells. In contrast, the difference between log2 mean tail moment was not statistically significant between 45 and 90 min in NPM1 null cells (P=0.258, Fig 3). Repair of DNA DSBs was essentially stalled in NPM1 null MEFs. These results support the hypothesis that NPM1 promotes resolution of Rad51 foci and repair of DSBs.
Fig 3. NPM1-null MEFs harbor a defect in DNA DSB repair.

NPM1-wt and NPM1-null MEFs were irradiated (4Gy) at 4°C and then immediately analyzed using a neutral comet assay or incubated at 37°C for 45 or 90 min prior to assaying. N = 50–75 nuclei per point.
Loss of NPM1 confers radiosensitivity but YTR107-mediated radiosensitization requires NPM1
We measured cell survival of wt- and NPM1-null MEFs by colony formation assay (Fig 4A). Radiation sensitivity was significantly increased by loss of NPM1 (P < 0.05 two way ANOVA). The survival values were best fitted by the equation [26] (chi square = 0.978) as compared to S = e−αD−βD2 (chi square = 0.930). The data were well fit to a single Do of 2.4 +/− 0.4 (SD). In wt-MEFs the extrapolation number, n, a qualitative measure of repair capacity was 5.52 +/− 0.27 (SD). In NPM1-null MEFs, n= 3.04 +/− 0.09 (SD). This is a 55% decrease in n (P< 0.01, F test, Table S1).
Fig 4. Loss of NPM1 confers radiosensitivity in MEFs and YTR107-mediated radiosensitization requires NPM1.
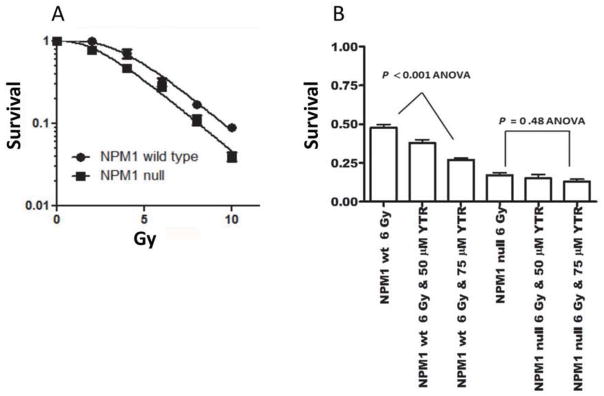
A) Cell survival of NPM1- wt- and NPM1-MEFs. Error bars: +/− SD. B) NPM1 wt- and null MEFs were exposed to the indicated concentrations of YTR107 for 30 mins, administered 6 Gy, incubated at 37°C for 90 min then washed and assayed for colony formation. Error bars: +/− SD.
Having established that NPM1-null MEFs are deficient in DNA repair and are radiosensitive, we then used this model to test the hypothesis that YTR107-mediated radiosensitization is a consequence of targeting NPM1. Wt- and NPM1-null MEFs were exposed to various concentrations of YTR107 and irradiated. YTR107 exposure resulted in a significant dose-dependent radiosensitization of wt-MEFs (P < 0.001, ANOVA), but importantly, did not radiosensitize NPM1-null cells (P=0.48 ANOVA, Fig 4B). There is no statistically significant difference in cell survival between wt-MEFs exposed to 100 μM YTR107 + 6 Gy and NPM1-null MEFs exposed to 6 Gy (P > 0.05, Student’s t test). We conclude that YTR107-mediated radiation sensitivity is dependent on its ability to target NPM1.
Next, we refined our knowledge of NPM1-YTR107 interactions by showing that YTR107 binds to amino acid residues 1–122 in recombinant NPM1 (Fig S3B). These residues form a globular domain necessary for NPM1 oligomerization, which is required for most, but not all of NPM1’s biological functions [3]. Specifically, monomeric human NPM1 folds into pentameric rings [15] and this oligomerization can be visualized using 4–16% NativePage Bis Tris gels (Fig S3C, lane 1, arrow). Incubation of YTR107 with human recombinant NPM1 disrupts pentamer formation (Fig S3C, lane 2), similar to another known inhibitor of NPM1 oligomerization NSC348884, which causes dimer formation ([23] and Fig S2C, lane 3, arrow). ImageJ software was used quantify the staining intensity of NPM1. Exposure to YTR107 reduced pentamer formation by 85%. Thus, one could hypothesize that YTR107 binding to NPM1 would impair function.
pT199 NPM1 (pNPM1) is recruited to irradiation induced DNA DSB repair foci (Fig. S4C, (red foci denoted by white arrow) in a RNF8/RNF168 ubiquitin ligase-dependent manner [13]. Fig. S5 illustrates pNPM1 co-localization with γH2AX foci (yellow foci denoted by white arrow). Exposure to YTR107 inhibits pNPM1 foci formation (Figs. S4, S5, and S6). The consequence of inhibiting pNPM1 foci formation appears to be a failure to repair DNA DSBs, as denoted by the elevated levels of γH2AX foci shown in Fig. S5. These data support the hypothesis that YTR107 impairs NPM1 function with regard to DNA damage repair.
NPM1 is overexpressed in subsets of NSCLC
The data presented above, combined with that in ref [13], support the hypothesis that NPM1 is involved in Rad51-mediated repair of DNA DSBs. Others have shown that NPM1 is an important component of the replication stress response pathway [6,17], a pathway predominately dependent upon Rad51-mediated repair [19]. As the replication stress response pathway is constantly activated in NSCLC [10], one might expect that NSCLC would require significant expression of NPM1 [6]. To test this hypothesis a TMA composed of 100 cores from 40 samples (80 cores) of human NSCLC of various AJCC tumor stages and 10 samples (20 cores) of matched normal lung parenchyma was immunohistochemically stained for NPM1. Representative cores are shown in Fig. 5A. Seventy eight percent of the normal lung parenchyma cores exhibited very low levels of NPM1 immunoreactivity (staining intensity of 1.0, Fig. 5B). In contrast, 64% (n = 11) of cores obtained from Stage IIIa and IIIb tumors exhibited a staining intensity of 3. The relationship observed between stage III tumors and NPM1 immunoreactivity in normal tissue, stage I, and stage II is statistically significant (P < 0.0008 Kruskal-Wallis). The Oncomine database was used to independently confirm the relationship between NPM1 and subsets of human NSCLC. A representative study is shown in Fig. S7 and illustrates the overexpression of NPM1 mRNA in subsets of human NSCLC. Taken together, the TMA and Oncomine database analysis suggest that NPM1 is overexpressed in subsets of NSCLC.
Fig 5. NPM1 is overexpressed in a panel of NSCLC cancer.
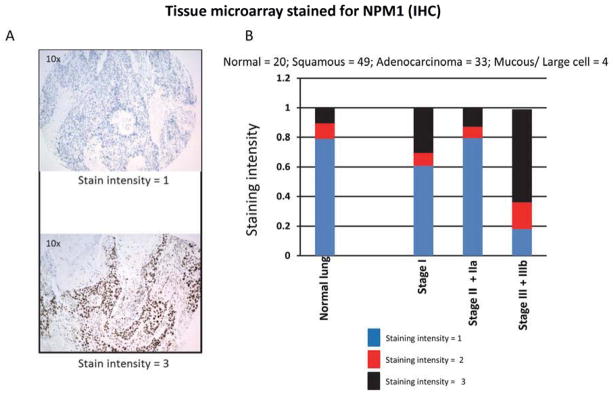
A) Representative staining of individual cores with anti-NPM1 antibody. B) Semiquantification of staining intensity as a function of pathological stage.
YTR107 inhibits the repair of DNA DSBs and radiosensitizes NSCLC in vitro and in vivo
Having shown that NPM1 is overexpressed in NSCLC and because the replication stress response pathway is hyperactivated in NSCLC, we asked if YTR107 targeting of NPM1 would impair the response of NSCLC cells to DNA damage generated by ionizing radiation. A neutral comet assay was used to assess formation and repair of DNA DSBs in H460 lung carcinoma cells. We compared the log(2)-transformed mean tail moment [27] from cells exposed to 0 or 25 μM YTR107 for 90 min/37°C prior to administering 4 Gy at 4°C. Assaying cells immediately after irradiation at 4°C revealed that more DSBs breaks were generated in H460 cells irradiated in the presence of YTR107 compared to cells irradiated in DMSO (P = 0.0065, Fig 6A). To investigate the effect of YTR107 on repair, cells were irradiated in the presence of YTR107, incubated at 37°C for 45 min, and then subjected to a comet assay. The assay indicated that YTR107 inhibited DNA DSB repair (P = 0.0028, Fig 6A).
Fig 6. YTR107 inhibits DNA DSBs repair and radiosensitizes NSCLC cells and LLC tumors.

A) DSB formation and repair. Formation of DSBs was measured at 4°C to prevent repair: H460 cells were exposed to DMSO (black bar) or 25 μM YTR107 (white bar) for 90 min at 37°C, administered 4 Gy at 4°C, and immediately assayed for formation of DNA DSBs using a neutral comet assay (N = 110). Repair of DSBs: Cells were also were exposed to DMSO (bar with horizontel lines) or 25 μM YTR107 (bar with vertical lines) for 30 min at 37°C, administered 4 Gy, incubated at 37°C for 45min and then subjected to a neutral comet assay (N=100). B) YTR107-mediated radiosensitization. H460 cells were exposed to 25 μM YTR107 prior to, during, and for 90 min after irradiation. Colony formation was used to assess survival (+/− SD). C) YTR107-mediated radiosensitization following multiple 2 Gy fractions. For five consecutive days H460 and Calu1 cells were exposed to 0 or 15 μM YTR107 for 30 min prior to, during a 2 Gy fraction, and for 90 after irradiation. Colony formation was used to assess survival (+/− SD). D) Growth of syngeneic Lewis Lung Carcinoma (LLC) tumors is delayed following exposure to YTR107 and factionated x-irradiation. For five consecutive days mice bearing LLC tumors were injected with DMSO or 10 mg/kg YTR107 and administered 2.4 Gy. Fold change in tumor volume +/− SD is shown. Symbols are larger than SD error bars. N= 8 mice per point.
Based on the strong correlation between DSBs and radiation sensitivity [1,8], one would predict that YTR107 would function as a radiation sensitizer. We tested this hypothesis on H460, A549, and H226 NSCLC cell lines (Fig 6B, Fig S8A & B, Table S1). Cells were exposed to 25 μM YTR107 for 30 min prior to, during and for 90 min after irradiation. Sensitivity was significantly increased by exposure to YTR107 (P < 0.001 two way ANOVA). Survival data were fitted to the equation (Table S1). A YTR107-mediated sensitizer enhancement ratio calculated at 10% survival (SER10%) was determined for each cell line. Exposure to YTR107 produced a SER10% of approximately 1.5 for each cell line, with a corresponding decrease in the survival fraction at 2 Gy (SF2, Table S1). Fig S8C illustrates a waterfall plot for NSCLC cell lines: Calu1, HCC827, PC9/BRC-1, and PC9/BRC-4 NSCLC cells irradiated in the absence or presence of YTR107. PC-9/BRC-1 and PC-9/BRC-4 cell lines were derived from the parental PC-9 cell line that harbors a deletion of exon19 in the EGFR gene. PC-9/BRC-1 and PC-9/BRC-4 cells exhibit a 1000-fold resistance to the EGFR inhibitor BIBW-2992, due to the presence of an additional T790M mutation [5]. The YTR107 exposure produced significant radiosensitization in these cell lines (P < 0.01, Fig 8C).
Next, we assessed YTR107-mediated radiosensitization following five 2 Gy fractions (total 10 Gy). For five consecutive days exponetially growing H460 and Calu1 cells were exposed to 0 or 15 μM YTR107 for 30 min prior to, during administration of 2 Gy, and for 90 min after irradiation. Ninety minutes after the last 2 Gy dose, cells were trypsinized, counted and plated for colony formation. Sham-irradiated cells served as a correction for plating efficiency. As illustrated in Fig 6C, exposure to YTR107 during the five 2 Gy fractions produced a statistically significant decrease in survival in both cell lines.
Lewis Lung Carcinoma (LLC) tumors growing subcutaneously in the legs of syngeneic C57BL mice represent a rigorous model for testing efficacy. When LCC tumors reached approximately 225 mm3 in size the tumor-bearing mice were injected intraperitoneally with DMSO or 10 mg/kg of YTR107 and 30 minutes later the tumors were administered 0 or 2.4 Gy. This protocol was administered for five consecutive days (Fig 6D). Administration of YTR107 significantly enhanced radiation-induced tumor growth delay (P < 0.05, two way ANOVA). Data points were fitted to Y = ax2 + bx + c [25] [4] and the resulting fits used to estimate growth delay (Table S2). For DMSO treated mice tumor volume increased 5-fold in four days. The irradiation protocol doubled the time required to reach a 5-fold increase. Administration of YTR107 plus irradiation resulted in a 2.75 fold increase in time required to increase volume 5-fold (P< 0.01, F test). A similar analysis was obtained when A549 xenografts were administered 7 daily fractions of 2.4 Gy in the absence or presence of 20 mg/kg YTR107 and the time required to increase tumor volume by 10 fold was estimated (P < 0.01, F test, Table S2 and Fig S8D).
Discussion
The goal of this investigation was to test the hypothesis that YTR107 targeting of NPM1 represents a radiosensitizing event. Using a defined genetic model consisting of NPM1-null and wild type MEFs we show that an NPM1 deficiency impairs a cell’s ability to respond to DNA DSBs, with an outcome of enhanced radiosensitization.
NPM1’s role in the cellular response to DNA damage is poorly characterized. pNPM1 is recruited to sites of DNA DSBs and is thought to assist in homologous recombination through a poorly understood mechanism [6]. Quantification of DNA DSB ligation kinetics via the comet assay revealed that DSBs are rapidly repaired in wt MEFs but repair appears to stall in cells lacking NPM1. We suggest that the failure to repair DSBs in a timely fashion in NPM1-null cells impacts γH2AX and Rad51 foci resolution, resulting in accumulation of large numbers of foci. Rad51 foci represent the assembly of Rad51 into protein filaments that coat single strand DNA and initiate a series of steps required for homologous recombination [11]. Because γH2AX and Rad51 foci assemble in the absence of NPM1 but DNA ligation is impaired, it is tempting to speculate that NPM1 acts at a late step that links ligation with resolution of DSBs. The exact nature of pNPM1s role in repair requires further investigation.
We show that YTR107 binds to NPM1 and impairs structure and function. The NPM1-null and wt MEF model allowed us to demonstrate that YTR107-mediated radiosensitization requires NPM1. Neutral comet assays demonstrated that YTR107 increased the number of DNA DSBs generated by irradiation and impaired their repair, thus providing a molecular mechanism for the observed radiosensitization. The observation that more breaks were formed in the presence of YTR107 (Fig 6A, no repair 4°C) can be explained by the knowledge that YTR107 produces a replication stress in proliferating cells [24].
The NSCLC TMA analysis indicated that tumor subsets overexpressed NPM1, perhaps in response to replication stress [10]. Importantly, YTR107 inhibited repair of DSBs, radiosensitized NSCLC cells in vitro, and promoted radiation-induced growth delay of LLC and A549 NSCLC xenografts administered clinically relevant doses. Taken all together these data support the hypothesis that NPM1 is targetable and that it represents a potentially rational target for the radiosensitization of NSCLC.
Supplementary Material
Summary.
There is an urgent need for effective radiation sensitizers. We show that NPM1 is required for DNA DSB repair, NPM1 is targetable, and that targeting is a radiosensitizing event. We leveraged this knowledge by demonstrating that NPM1 targeting can radiosensitize cell-based and xenograft models of NSCLC due to defective repair of DNA DSBs.
Acknowledgments
This research was supported in part by RO1CA140409, R01CA140774, T32CA093240, CA152662 and CA102353, P50CA095103, Vanderbilt-Ingram Cancer CTR Grant P30 CA68485 and the Vanderbilt SPORE in lung cancer P50CA090949. Additional support for experiments performed using the VUMC Cell Imaging Shared Resource was provided in part by CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126.
Footnotes
Conflict of interest statement; “Conflict of Interest – KRS, PNR, PAC, and MLF are co-inventors on the patent covering YTR107”.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bedford JS, Mitchell JB, Griggs HG, et al. Radiation-induced cellular reproductive death and chromosome aberrations. Radiat Res. 1978;76:573–586. [PubMed] [Google Scholar]
- 2.Bradley J, Graham MV, Winter K, et al. Toxicity and outcome results of rtog 9311 A phase i–ii dose-escalation study using three-dimensional conformal radiotherapy in patients with inoperable non-small-cell lung carcinoma. Int J Radiat Oncol Biol Phys. 2005;61:318–328. doi: 10.1016/j.ijrobp.2004.06.260. [DOI] [PubMed] [Google Scholar]
- 3.Chan PK, Chan FY. Nucleophosmin/b23 (npm) oligomer is a major and stable entity in hela cells. Biochim Biophys Acta. 1995;1262:37–42. doi: 10.1016/0167-4781(95)00044-h. [DOI] [PubMed] [Google Scholar]
- 4.Cheng JD, Valianou M, Canutescu AA, et al. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol Cancer Ther. 2005;4:351–360. doi: 10.1158/1535-7163.MCT-04-0269. [DOI] [PubMed] [Google Scholar]
- 5.Chmielecki J, Foo J, Oxnard GR, et al. Optimization of dosing for egfr-mutant non-small cell lung cancer with evolutionary cancer modeling. Science translational medicine. 2011;3:90ra59. doi: 10.1126/scitranslmed.3002356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colombo E, Bonetti P, Lazzerini Denchi E, et al. Nucleophosmin is required for DNA integrity and p19arf protein stability. Mol Cell Biol. 2005;25:8874–8886. doi: 10.1128/MCB.25.20.8874-8886.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colombo E, Marine JC, Danovi D, et al. Nucleophosmin regulates the stability and transcriptional activity of p53. Nature cell biology. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 8.Dewey WC, Miller HH, Leeper DB. Chromosomal aberrations and mortality of x-irradiated mammalian cells: Emphasis on repair. Proc Natl Acad Sci U S A. 1971;68:667–671. doi: 10.1073/pnas.68.3.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Franken NA, Rodermond HM, Stap J, et al. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- 10.Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907–913. doi: 10.1038/nature03485. [DOI] [PubMed] [Google Scholar]
- 11.Holthausen JT, Wyman C, Kanaar R. Regulation of DNA strand exchange in homologous recombination. DNA repair. 2010;9:1264–1272. doi: 10.1016/j.dnarep.2010.09.014. [DOI] [PubMed] [Google Scholar]
- 12.Jeffrey D, Bradley RP, Komaki Ritsuko, Masters Gregory A, Forster Kenneth, Schild Steven E, Bogart Jeffrey, Garces Yolanda I, Narayan Samir, Kavadi Vivek, Nedzi Lucien Alexander, Michalski Jeff M, Johnson Douglas, MacRae Robert Malcolm, Curran Walter John, Choy Hak. A randomized phase iii comparison of standard-dose (60 gy) versus high-dose (74 gy) conformal chemoradiotherapy with or without cetuximab for stage iii non-small cell lung cancer: Results on radiation dose in rtog 0617. J Clin Oncology. 2013;31:abstract 7501. [Google Scholar]
- 13.Koike A, Nishikawa H, Wu W, et al. Recruitment of phosphorylated npm1 to sites of DNA damage through rnf8-dependent ubiquitin conjugates. Cancer Res. 2010 doi: 10.1158/0008-5472.CAN-10-0382. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee HH, Kim HS, Kang JY, et al. Crystal structure of human nucleophosmin-core reveals plasticity of the pentamer-pentamer interface. Proteins. 2007;69:672–678. doi: 10.1002/prot.21504. [DOI] [PubMed] [Google Scholar]
- 16.Li Z, Boone D, Hann SR. Nucleophosmin interacts directly with c-myc and controls c-myc-induced hyperproliferation and transformation. Proc Natl Acad Sci U S A. 2008;105:18794–18799. doi: 10.1073/pnas.0806879105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lundin C, Erixon K, Arnaudeau C, et al. Different roles for nonhomologous end joining and homologous recombination following replication arrest in mammalian cells. Mol Cell Biol. 2002;22:5869–5878. doi: 10.1128/MCB.22.16.5869-5878.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell. 2009;136:823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mladenov E, Magin S, Soni A, et al. DNA double-strand break repair as determinant of cellular radiosensitivity to killing and target in radiation therapy. Frontiers in oncology. 2013;3:113. doi: 10.3389/fonc.2013.00113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Okuda M, Horn HF, Tarapore P, et al. Nucleophosmin/b23 is a target of cdk2/cycline in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 21.Olive PL, Banath JP. The comet assay: A method to measure DNA damage in individual cells. Nat Protoc. 2006;1:23–29. doi: 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 22.Paull TT, Rogakou EP, Yamazaki V, et al. A critical role for histone h2ax in recruitment of repair factors to nuclear foci after DNA damage. Current biology : CB. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 23.Qi W, Shakalya K, Stejskal A, et al. 348884, a nucleophosmin inhibitor disrupts oligomer formation and induces apoptosis in human cancer cells. Oncogene. 2008;27:4210–4220. doi: 10.1038/onc.2008.54. [DOI] [PubMed] [Google Scholar]
- 24.XXXX.
- 25.Taetle R, Rosen F, Abramson I, et al. Use of nude mouse xenografts as preclinical drug screens: In vivo activity of established chemotherapeutic agents against melanoma and ovarian carcinoma xenografts. Cancer treatment reports. 1987;71:297–304. [PubMed] [Google Scholar]
- 26.Tubiana M, Dutreix J, Wambersie A. Introduction to radiobiology. London; New York: Taylor & Francis; 1990. [Google Scholar]
- 27.Wiklund SJ, Agurell E. Aspects of design and statistical analysis in the comet assay. Mutagenesis. 2003;18:167–175. doi: 10.1093/mutage/18.2.167. [DOI] [PubMed] [Google Scholar]
- 28.Zosia C. Chapter closed on higher-dose radiation for late lung cancer. Vol. 2013. Medscape Today; 2013. http://www.medscape.com/viewarticle/804282. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.