

Abstract
BACKGROUND AND PURPOSE
Dysfunction and injury of endothelial cells in the pulmonary artery play critical roles in the hypertension induced by chronic hypoxia. One consequence of hypoxia is increased activity of 15-hydroxyprostaglandin dehydrogenase (PGDH). Here, we have explored, in detail, the effects of hypoxia on the proliferation of pulmonary artery endothelial cells.
EXPERIMENTAL APPROACH
We used bromodeoxyuridine incorporation, cell-cycle analysis, immunohistochemistry and Western blot analysis to study the effects of hypoxia, induced 15-PGDH) activity and its product, 15-keto-6Z, 8Z, 11Z, 13E-eicosatetraenoic acid (15-KETE), on endothelial cell proliferation. Scratch-wound and tube formation assays were also used to study migration of endothelial cells.
KEY RESULTS
15-KETE increased DNA synthesis and enhanced the transition from the G0/G1 phase to the S phase in hypoxia. Inhibition of 15-PGDH or siRNA for 15-PGDH reversed these effects. 15-KETE also activated the ERK1/2 signalling pathway. 15-KETE-induced cell migration and tube formation were reversed by blocking ERK1/2, but not the p38 MAPK pathway.
CONCLUSIONS AND IMPLICATIONS
Hypoxia-induced endothelial proliferation and migration, an important underlying mechanism contributing to hypoxic pulmonary vascular remodelling, appears to be mediated by 15-PGDH and 15-KETE, via the ERK1/2 signalling pathway.
Keywords: pulmonary artery endothelial cells, pulmonary vascular remodelling, 15-hydroxyprostaglandin dehydrogenase, 15-keto-6Z, 8Z, 11Z, 13E-eicosatetraenoic acid, ERK1/2
Introduction
The vascular endothelium constitutes the major barrier to the movement of molecules across the blood vessel wall (Partovian et al., 2000). Dysfunction and injury of pulmonary artery endothelial cells (PAECs) along with increased permeability and the expression of inflammation may trigger the pathogenesis of pulmonary hypertension (PH) (Archer and Rich, 2000). Endothelium plays a critical role in mediating many of the changes observed in the process of hypoxia-induced hypertension and chronic hypoxia exposure increases the intimal thickness in the pulmonary circulation by causing hypertrophy and hyperplasia of the PAECs (Stenmark et al., 2006). Although the contribution of hypoxia to the proliferation of endothelium has been studied over the past decades, the cellular and molecular mechanisms for the effects of hypoxia on PAECs have not yet been fully described.
The NAD+-dependent enzyme 15-hydroxyprostaglandin dehydrogenase (15-PGDH) catalyses the oxidation of 15(S)-hydroxyl group of PGs and lipoxins to form 15-keto metabolites (Tai et al., 2006; nomenclature follows Alexander et al., 2013). 15-PGDH has been purified and cloned in different mammals, such as humans, rats, mice and bovines, and the enzyme is widely distributed across mammalian tissues, including the renal proximal tubules, placenta and uterus (Ensor and Tai, 1995; Matsuo et al., 1996; Miyaki et al., 2009). Several studies have identified a tumour suppressor activity of 15-PGDH in colon, lung, bladder, gastrointestinal and breast cancer (Yan et al., 2004; Backlund et al., 2005; Ding et al., 2005; Myung et al., 2006). The critical physiological role of this enzyme is to inactivate the PGs by conversion to the corresponding 15-keto-PGs. These metabolites exert greatly reduced biological activities in angiogenesis, invasion and metastasis of cancer (Tai et al., 2002; Tseng-Rogenski et al., 2012). However, the biological actions of 15-PGDH on other 15-hydroxyeicosanoids may have wider effects than previously recognized.
Earlier, we showed that hypoxia activated 15-lipoxygenase (15-LO) in PAECs, and increased production of 15-hydroxyeicosatetraenoic acid (15-HETE), along with pulmonary vascular remodelling (PVR) (Zhu et al., 2003; Ma et al., 2011). Proliferative and angiogenic effects of 15-HETE on PAECs could be important underlying mechanisms for the pathological features of PVR (Ma et al., 2011). The 15-hydoxyl group of 15-HETE is easily oxidised by NADPH-independent cytochrome P450 (CYPs) and NADPH-dependent 15-PGDH (Weiss et al., 1987; Chang et al., 1996; Yeh et al., 2007; Chawengsub et al., 2009). Furthermore, macrophage-derived 15-PGDH can convert 15(S)-HETE to 15-keto-6Z, 8Z, 11Z, 13E-eicosatetraenoic acid (15-KETE) (Bergholte et al., 1987). We were interested in the possibility that the angiogenic effects of 15-HETE were, in fact, mediated by this metabolite. Therefore, we examined the effects of hypoxia on expression of 15-PGDH and on 15-KETE generation, on disordered proliferation of PAECs and, finally, on the development of PVR.
We evaluated the expression and localization of 15-PGDH in hypoxic lungs of rats and in PAECs by immunostaining and Western blot analysis. We also determined the role of 15-KETE in regulation of cell-cycle progression, along with effects on cell migration. We found that 15-KETE induced ERK1/2 phosphorylation both in normoxic and hypoxic conditions. Our results showed that 15-KETE could mediate the excessive proliferation and migration of PAECs, through the ERK1/2 pathway.
Methods
Animals
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee of Harbin Medical University.
Adult male Wistar rats with a mean weight of 200 g were supplied by the Experimental Animal Center of Harbin Medical University. Rats were randomized into 9 days of normoxic or hypoxic environments with fractional inspired oxygen (FiO2) of 0.21 and 0.12, respectively, as previously described (Zhu et al., 2003; Ma et al., 2011).
Immunohistochemistry
At the end of the exposure period, we anaesthetized each rat with pentobarbital (120 mg·kg−1, i.p.), removed the lungs and immersed them in 4% paraformaldehyde for overnight fixation. Then the tissues were dehydrated, cleared and embedded in paraffin wax. The paraffin blocks were cut into 5 μm thick sections. Some sections were stained with haematoxylin and eosin (H&E). For immunohistochemistry, sections were deparaffinized and rehydrated in graduated alcohol. Then they were placed in sodium citrate buffer (0.1 mol L−1, pH 6.0) and heated for 2 min for antigen retrieval, and then the sections were incubated with anti-15-PGDH (1:100) and anti hypoxia-inducible factor (HIF)-1α (1:100) antibodies. After overnight incubation, the sections were exposed to the secondary antibodies (1:200) for the IgGs. Sections were visualized with 3,3-diaminobenzidine and counterstained using haematoxylin. Brown and yellow colours indicated positive stains.
Cell culture
Primary cultures of PAECs were prepared as previously described (Zhu et al., 2003; Ma et al., 2011). Calf lungs, from a local slaughterhouse, used in the study were approved by the Ethical Committee of Laboratory Animals at Harbin Medical University. Pulmonary arteries were carefully dissected from the lungs and washed with HEPES buffer (0.01 mol L−1, pH 7.3) to remove blood. They were then slit open along their lengths and gently scratched along the intimal surface with a surgical blade. The cells were immediately transferred to centrifuge tube with a collagenase solution (1 mg·mL−1 in DMEM) incubated at 25°C for 8–9 min. Then the cells were vortexed and centrifuged at 200× g for 10 min to obtain cell pellets. The purity and identity of PAECs were confirmed by positive immunofluorescence staining using antibodies to CD31 (Santa Cruz Biotechnology Inc.). Cells in hypoxic culture were incubated in a Tri-Gas Incubator (Heal Force, Shanghai, China) with a gas mixture containing 92% N2 – 5% CO2 – 3% O2 for 24 h.
3-(4,5-Dimethylthiazol- 255 2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability was determined by the MTT assay, PAECs were cultured in a 96-well culture plate and then subjected to growth arrest for 24 h before treatment with different compounds, according to the experimental groups. At the end of incubation, the cells were incubated for 4 h in a medium containing 0.5% MTT. The reaction was terminated after adding 150 μL DMSO to the cells for 10 min. Absorbance at 490 nm was recorded by an elisa plate reader.
siRNA design and transfection
To block the expression of 15-PGDH and HIF-1α protein, PAECs were transfected with the corresponding siRNAs, which were designed and synthesized by GenePharma using X-tremeGene siRNA Transfection Reagent (Roche Applied Science, Mannheim, Germany). Non-targeted control siRNA (siNC) was used as the negative control. The sense sequence of siRNA against 15-PGDH and siNC sequence was listed below: accession no. ds-siRNA sequence corresponding nucleotides, 15-PGDH: (NM_001034419.2) 5′-GCUUUGAAUGGUGCUAUUATT-3′, siNC: 5′-UUCUCCGAACGUGUCACGUTT-3′. The sense sequence of siRNA against HIF-1α and siNC sequence was listed below: accession no. ds-siRNA sequence corresponding nucleotides, HIF-1α: (NM_174339.3) 5′-GCUCCUGAGGAAGAACUAATT-3′, siNC: 5′-UUCUCCGAACGUGUCACGUTT-3′. Cells were cultured till 30–50% confluence, 1.5 μg siRNA and 7.5 μL X-tremeGene siRNA Transfection Reagent were diluted in the serum-free Opti-MEM-1 medium, and after 5 min, we mixed them together. Next, we incubated the mixture (siRNA/transfection reagent) for 20 min then added it directly to the cells. After 4–6 h exposure to siRNA, the transfection reagents were removed and cell culture continued in DMEM containing 5% FBS for another 24 h under normoxic (21% O2/5% CO2/balance N2) or hypoxic (3% O2/5% CO2/balance N2) growth conditions as required.
BrdU incorporation
PAECs were plated in 96-well plates at the density of 1 × 104 cells per well, and treatment with different agents in 5% FBS DMEM. We measured BrdU incorporation using a BrdU proliferation assay kit according to the manufacturer's protocol. Briefly, the cells were labelled with 10 ng·mL−1 of BrdU. Then, cells were fixed, air-dried and incubated 1 h with mouse anti-BrdU monoclonal antibody (diluted 1:200). The cells were washed three times and then incubated with peroxidase goat anti-mouse IgG (1:2000) at room temperature. Thereafter, 100 μL substrate was added to each well and incubated for 30 min in darkness. The absorbance at dual-wave lengths of 450–540 nm was then measured.
Cell-cycle analysis
DNA fluorescence and flow cytometry were measured using BD FACSCalibur Flow Cytometer (Bedford, MA, USA). Briefly, after the cells were exposed to the indicated compounds or hypoxia for 24 h, they were harvested by trypsinization and fixed using 70% cold ethanol. Before detection, the cells were resuspended in 200 μL PBS, and 200 μL RNase A was added, then incubated at room temperature for 10 min. Next, 200 μL of propidium iodide was added and mixed in dark. Finally, the cells were filtered and detected by flow cytometer.
Western blotting
The PAECs were treated with 15-KETE (1 μM), siRNA for 15-PGDH or 15-KETE plus siRNA for 15-PGDH, in DMEM with 5% FBS and exposed to hypoxia for 24 h. Cells were washed with PBS, and a whole-cell lysate was prepared using lysis buffer containing (in mmol L−1) Tris 50, pH 7.4, NaCl 150, Triton X-100 1%, EDTA 1, and PMSF 2. The proteins of interest were separated by SDS-PAGE and transferred to nitrocellulose membranes and then blocked with 5% non-fat dry milk powder. The membranes were incubated initially with polyclonal antibodies directed against PCNA (rat, 1:500), cyclin A (rabbit, 1:400), cyclin D (rat, 1:1000), ERK (rabbit, 1:1000) and p-ERK (rabbit, 1:200) overnight at 4°C, followed by reaction with HRP-conjugated secondary antibodies and enhanced chemiluminescence reagents, with β-actin as an internal control.
Immunocytochemistry
PAECs were cultured on coverslips, which were covered in 24-well culture plates with polylysine. After treatment, cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100 for 10 min, blocked with 3% normal bovine serum at 37°C for 30 min, and incubated with anti-15-PGDH primary antibody (rabbit, 1:50) at 4°C overnight. After washing three times with PBS, the cells were incubated with FITC-conjugated secondary antibody (1:100) for 2 h and DAPI for 10 min. in the dark. The images were recorded by digital photomicrography (Olympus, Tokyo, Japan).
Endothelial cell migration and tube formation assay
The migratory activity of PAECs was assessed using a scratch-wound assay. Confluent cultures of PAECs in six-well plates were ‘wounded’ by scratching the monolayer with a pipette tip (1 mm width). After that, cells were treated with vehicle or compounds, as indicated. After 24 h of incubation, photos were taken from the same areas as those recorded at zero time. For the tube formation assay, 96-well culture plates were coated with 30 μL growth factor-reduced Matrigel (BD Biosciences) and solidified for 30 min at 37°C. PAECs were trypsinized and resuspended at 5 × 104 mL−1, followed by adding 200 μL of this cell suspension into each well. Tube formation was observed under an inverted microscope (Nikon, Tokyo, Japan). Tube length was measured using the Image-Pro Plus 6.0 (Media Cybernetics, Rockville, MD, USA).
Data analysis
Data are presented as means ± SEM. Statistical analysis was performed with Student's t-test or one-way anova followed by Tukey's test where appropriate. P < 0.05 was considered statistically significant.
Materials
15-KETE (CAY-10397) and 15-PGDH antibody were obtained from Cayman Chemical Company (Ann Arbor, MI, USA). CycleTEST™ PLUS DNA Reagent Kit was obtained from BD Biosciences (Bedford, MA, USA). Bromodeoxyuridine (BrdU) proliferation assay kit was purchased from Millipore Corporation (Billerica, MA, USA). Antibodies against cyclin A, rabbit anti-ERK and phosphor-specific antibodies against ERK were from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Proliferating cell nuclear antigen (PCNA) and cyclin D3 were purchased from Cell Signaling Technology (Danvers, MA, USA). All other reagents were from common commercial sources.
Results
Hypoxia increases the expression of 15-PGDH in the pulmonary vasculature
H&E stain and HIF-1α staining were applied to pulmonary vessels to examine the responses to hypoxia in our model. Figure 1A shows the thickening of the vascular walls in medium-size vessels obtained from rats exposed to hypoxia for 9 days. Moreover, we observed that the expression of HIF-1α in lung tissue was up-regulated by hypoxia, compared with normoxic rats, showing the expected responses to hypoxia in our model (Figure 1B, C). We then localized 15-PGDH in the lung using immunohistochemistry on tissue sections. The expression of 15-PGDH was up-regulated in hypoxia, which was mainly localized in vascular intima (Figure 1D). This was confirmed by immunoblot analysis, showing that hypoxia increased 15-PGDH expression the lung (Figure 1E).
Figure 1.

Morphometric analysis of pulmonary arteries exposed in vivo to normoxia and hypoxia. (A) H&E staining of sections from rats after 9 days of normoxia (Nor; 20% O2) or hypoxia ( Hyp; 12% O2). (B and C) Expression of HIF-1α evaluated by immunohistochemistry and Western blot. (D) Immunoreactive 15-PGDH was mainly expressed in pulmonary vascular intima, with quantitative analyses of positive staining per vascular area (adventitia + media + intima + lumen). (E) The effects of hypoxia on 15-PGDH expression in lung tissue. Data shown are means ± SEM; (n = 6). *P < 0.05, significantly different from normoxia. Scale bars are 20 μm.
Expression and subcellular distribution of 15-PGDH under hypoxic condition in cultured PAECs
To ascertain whether hypoxia is capable of regulating the 15-PGDH function in vitro, we examined the expression of 15-PGDH protein after silencing HIF-1α in cultured PAECs. First, to assess the efficiency and specificity of the siRNA, the intracellular expression of HIF-1α was measured. Expression of HIF-1α treated with non-targeted control siRNA (NC) was not different from that in untreated control cells (Blank) or cells treated with the transfection reagent alone (Mock) (Figure 2A). Increased expression of 15-PGDH was detected in PAECs exposed to hypoxia for 24 h. Furthermore, after treating PAECs with siRNA for HIF-1α, hypoxia-induced expression of 15-PGDH was inhibited (Figure 2B). Increased expression of HIF-1α after hypoxia was not affected by CAY-10397, a potent and selective inhibitor of 15-PGDH (Figure 2C). As shown in Figure 2D, 15-PGDH expression after hypoxia was down-regulated by an inhibitor of 15-LO cinnamyl-3,4-dihydroxy-a'-cyanocinnamate (5 μmol·L−1), and by an inhibitor of COX-2, NS-398 (10 μmol·L−1). Using immunofluorescent staining with 15-PGDH antibody, we found that 15-PGDH was distributed in both cytosol and nucleus in the normoxic group whereas, after hypoxia, 15-PGDH accumulated in the nucleus. This translocation was blocked by CAY-10397, suggesting that an additional regulatory mechanism of 15-PGDH stimulation by hypoxia may occur via cytoplasmic-nuclear shuttling (Figure 2E).
Figure 2.
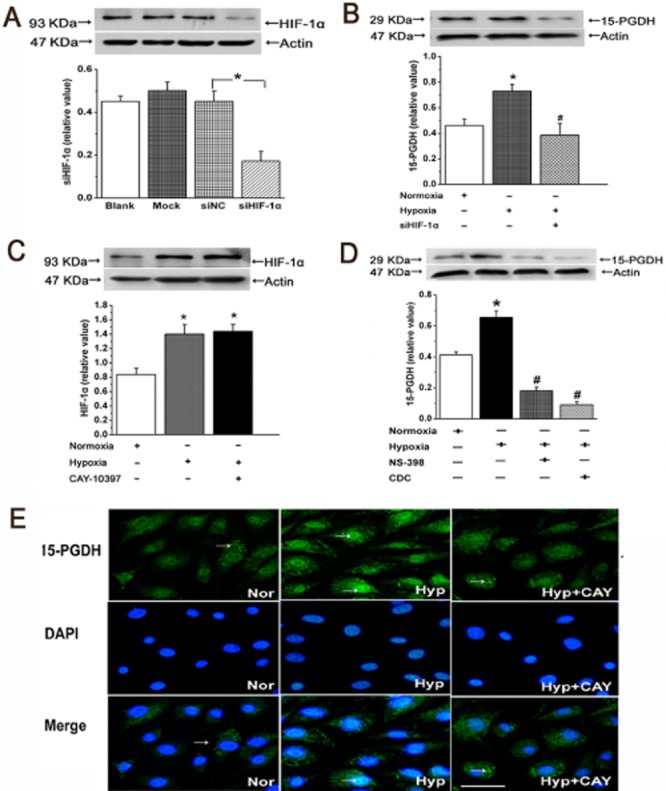
Hypoxia promotes the expression and translocation of 15-PGDH in PAECs. (A) The efficiency and specificity of siRNA directed against HIF-1α (siHIF-1α; n = 4). (B) Expression of 15-PGDH inpAECs was decreased after treatment with siRNA against HIF-1α (n = 4). (C) Expression of HIF-1α in hypoxia was not affected by administration of CAY-10397 (n = 4). (D) NS-398 and cinnamyl-3,4-dihydroxy-a'-cyanocinnamate (CDC) decreased the expression of 15-PGDH in hypoxia (n = 4). Data shown are means ± SEM; *P < 0.05, significantly different from normoxia, #P < 0.05, significantly different from hypoxia. (E) Cells were fixed and stained with anti-15-PGDH (green) and DAPI to stain nucleus (blue). Nuclear translocation of 15-PGDH in PAECs was induced by hypoxia (n = 6). Scale bars are 25 μm.
Cell proliferation and tube formation are associated with the 15-PGDH/15-KETE pathway
To further assess the mechanisms of endothelial dysfunction in hypoxia, cell viability was analysed by MTT assay in vitro. As shown in Figure 3A, PAEC cultures were incubated with either 15(S)-hydroperoxy-5,8,11,13-eicosatetraenoic acid (15-HPETE), which is the precursor of 15-HETE, 5,15-(6E,8Z,11Z,13E)-dihydroxyeicosatetraenoic acid (5,15-diHETE), which is the oxidation product of 15-HETE generated by 5-LO and 15-KETE, which is the oxidation product of 15-HETE, catalysed by 15-PGDH. Only 15-KETE (1 μmol·L−1) increased cell viability. Similarly, in the tube formation assay, only 15-KETE increased the tube length (Figure 3B). Next, we used specific siRNA to silence 15-PGDH gene expression in PAECs, confirming with Western blot analysis that 15-PGDH was adequately knocked down (Figure 3C). As shown in Figure 3D, exogenous 15-KETE stimulated the expression of PCNA under normoxic conditions and this effect was reversed by transient transfection with 15-PGDH siRNA. These results show that 15-PGDH/15-KETE may be involved in mediating proliferation of PAECs in vitro.
Figure 3.
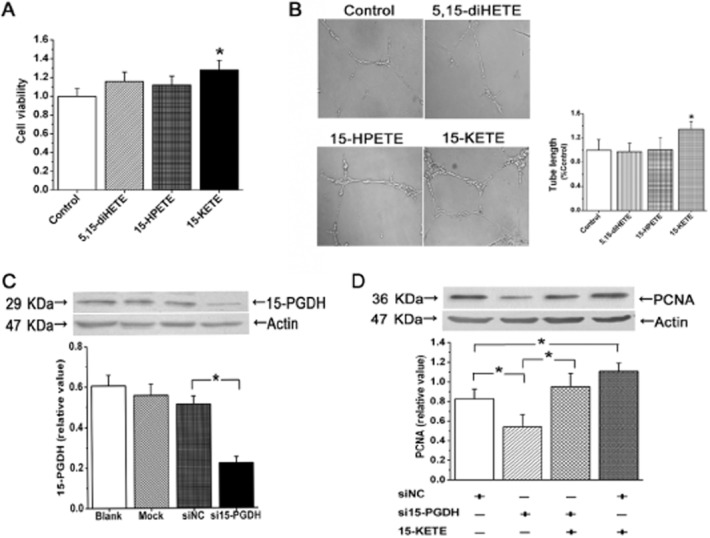
PAEC proliferation in PVR is due, at least in part, to 15-KETE, the product of 15-PGDH. (A and B) Only 15-KETE, not 5,15-diHETE or 15-HPETE, increased cell viability and tube formation (n = 6). (C) 15-PGDH protein expression was down-regulated after transfecting the 15-PGDH siRNA sequence into PAECs (n = 4). (D) Cell proliferation was decreased after 15-PGDH silencing in normoxia (n = 4). Data shown are means ± SEM; *P < 0.05, significantly different from control.
Stimulation of the proliferation and migration of PAECs by 15-KETE in hypoxia
First, to confirm the functional and biochemical activation of PAECs by hypoxic conditons, we transfected cells with HIF-1α siRNA and measured PCNA expression. As shown in Figure 4A, hypoxia enhanced PCNA expression, compared with that in normoxia, and this increased expression was suppressed after treating the cells with HIF-1α siRNA. Viability of PAECs, determined by MTT assay, was increased by 15-KETE after hypoxia. Cell viability after incubation with CAY-10397, to decrease the formation of endogenous 15-KETE, was slightly less than that of the hypoxic group and this decline was reversed by adding exogenous 15-KETE to the cells (Figure 4B). Proliferation of PAECs, as measured by DNA synthesis using BrdU incorporation and PCNA expression, was increased by incubation with exogenous 15-KETE (Figure 4C). Moreover, expression of PCNA was decreased in cells incubated with CAY-10397 or with siRNA for 15-PGDH, in hypoxia. These decreases were partly reversed by exogenous 15-KETE (Figure 4D, E). As shown in Figure 4F, in the scratch-wound assay, 15-KETE increased the migratory capability of PAECs, which was inhibited by CAY-10397. Together, these data suggest that 15-PGDH and/or 15-KETE was involved in the proliferation and migration of PAECs, induced by hypoxia.
Figure 4.

15-KETE increased PAECs proliferation and migration in hypoxia. (A) Under hypoxic conditions, expression of PCNA was increased, and was reversed by siHIF-1α (n = 3). (B) CAY-10397 inhibited the protective effect of 15-KETE (1 μmol·L−1) on cell viability in hypoxic condition (n = 6). (C) BrdU incorporation assay was detected after blocking 15-PGDH with CAY-10397 (n = 4). (D and E) Results showed the effects of hypoxia on expression of PCNA protein expression were blocked by CAY-10397 or 15-PGDH silencing (n = 4). (F) Confluent PAECs monolayers were wounded either in the absence or presence of 15 μmol·L−1 CAY-10397 (n = 3). Scale bars are 50 μm. Data are presented as means ± SEM. *P < 0.05; **P < 0.01, significantly different as indicated.
15-KETE regulates expression of cell-cycle–related proteins and promotes cell-cycle progression
We tested the effects of 15-KETE on the cell cycle in PAECs (Fig 5). CAY-10397 induced cell-cycle arrest with more than 80% of cells in the G0/G1 phase, and hypoxia with 15-KETE exposure increased cell proportions in the S and G2/M phase (Figure 5A). The D-type cyclins play an important role in the stimulation of cyclin-dependent kinases and thus regulate the transition of cell cycle from G1 to S phase, and cyclin A is known to regulate the S to G2/M phase transitions (Liu et al., 2009; Chibazakura et al., 2011). Hypoxia increased the expression of cyclin A and cyclin D (Figure 5B, D) and these effects were attenuated after treatment with the siRNA for 15-PGDH (Figure 5C, E).
Figure 5.

Effects of hypoxia and the 15-PGDH/15-KETE pathway on the cell cycle in PAECs. (A) Cells accumulated in S and G2/M after hypoxia. This was reversed by 15-PGDH inhibition. (B) Hypoxia increased cyclin A expression and this was reversed by CAY-10397 or (C) by siRNA for PGDH. (D and E) Expression of cyclin D in PAECs was similarly affected by hypoxia or 15-KETE. Values are denoted as means ± SEM from at least three separate experiments. *P < 0.05; **P < 0.01; significantly different as indicated.
Both endogenous and exogenous 15-KETE increases ERK1/2 phosphorylation in PAECs
To determine whether ERK signalling was altered along with PAEC proliferation and migration, we measured levels of phospho-ERK1/2 and total ERK1/2 in PAECs. Exogenous 15-KETE increased ERK1/2 phosphorylation under normoxic conditions (Figure 6A) and hypoxia alone increased phospho-ERK1/2 but this increase was inhibited by adding CAY-10397. The inhibitory effect was partly reversed in the presence of exogenous 15-KETE (Figure 6B).
Figure 6.
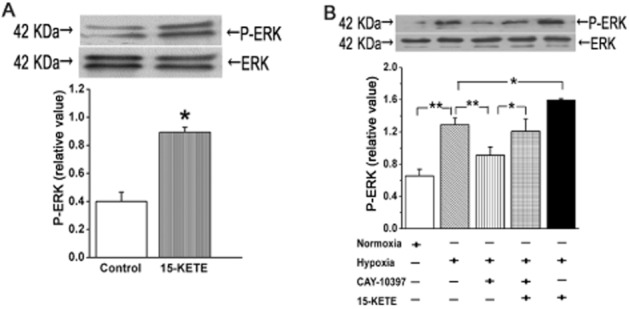
ERK1/2 activity was modulated by 15-KETE in PAECs. (A) Under normoxic conditions, 15-KETE increased phosphorylation of ER1/2. (B) In PAECs exposed to hypoxia for 60 min, both endogenous and exogenous 15-KETE (1 μmol·L−1 ) increased phospho-ERK1/2 expression. Data shown are means ± SEM from at least three separate experiments. *P < 0.05; **P < 0.01; significantly different as indicated.
15-KETE increases cell proliferation and migration in an ERK1/2-dependent manner, but not involving the p38 MAPK pathway
To elucidate the signalling pathways participating in the 15-KETE-mediated cell survival, we incubated PAECs with the p38 MAPK inhibitor SB203580 or the ERK1/2 inhibitor PD-98059. Only PD-98059 (5 μmol·L−1) blocked the protective effect of 15-KETE on cell viability (Figure 7A, B). We confirmed the involvement of the ERK1/2 pathway by testing another inhibitor, U0126, and found that both inhibitors decreased DNA synthesis (Figure 7C, D). However the decrease of PCNA expression following ERK1/2 inhibition was not reversed by 15-KETE (Figure 7E, F). Subsequently, the inhibition of ERK1/2 alone attenuated the migration induced by hypoxia. Furthermore, the effect of exogenous 15-KETE was diminished after blocking the ERK1/2 pathway with PD-98059 (Figure 7G). These data suggest that the effects of 15-KETE on PAECs proliferation and migration are mediated by the ERK1/2 pathway.
Figure 7.
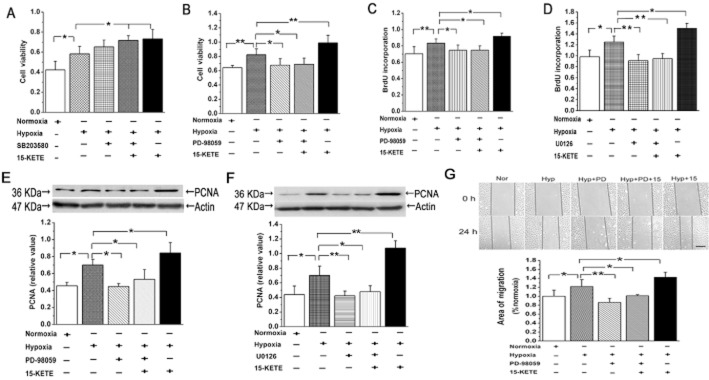
15-KETE increased proliferation and migration of PAECs in a ERK1/2-dependent manner. (A) 15-KETE increased viability of PAECs after growth-arrest for 24 h and this was not blocked by SB-203580 (n = 6). (B) Pretreatment with PD-98059 blocked the effects of 15-KETE on cell viability. (C and D) The increased BrdU incorporation induced by hypoxia and exogenous 15-KETE was suppressed after treating the cells with PD-98059 and U0126. (E and F) The increased PCNA expression after 15-KETE in hypoxic conditions, was blocked by the ERK1/2 inhibitors.(G) 15-KETE induced PAECs migration is mediated by the ERK1/2 pathway. Scale bars are 50 μm. Data shown are means ± SEM from at least three separate experiments. *P < 0.05; **P < 0.01; significantly different as indicated.
Role of ERK signalling in 15-KETE-induced cell-cycle progression
We then assessed the involvement of the ERK1/2 pathway in the effects of exogenous 15-KETE on the cell cycle. Treatment of PAECs with PD-98059 increased the fraction of cells in the first peak (G0/G1) and correspondingly decreased the fraction of cells in the S and G2/M phases. In the presence of PD-98059, exogenous 15-KETE had no effect on the cell-cycle profile (Figure 8A). As shown in Figure 8B and C, cyclin A and cyclin D expression were substantially increased after hypoxia but, in the PD-98059-treated cells, these cyclins were not increased by hypoxia plus 15-KETE.
Figure 8.
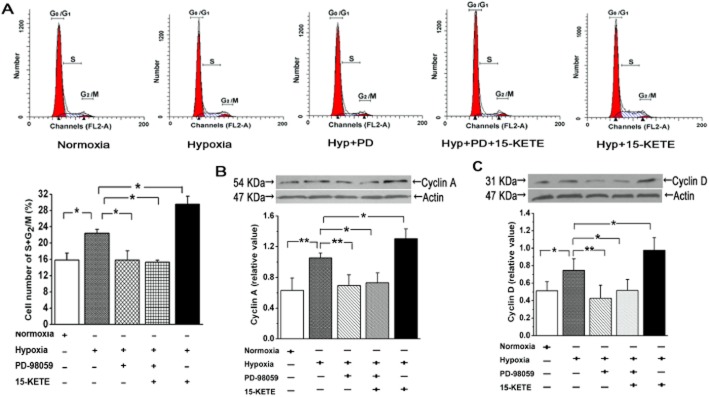
Effects of 15-KETE on the cell cycle in PAECs were mediated through the ERK pathway. (A) Flow cytometry for cell-cycle analysis indicated that 15-KETE stimulated PAEC progression into G2/M + S phase and this effect was blocked by PD-98059.. (B) PD-98059 also blocked the increase in cyclin A or (C) cyclin D, induced by 15-KETE.. Data shown are means ± SEM from at least three separate experiments. *P < 0.05; **P < 0.01; significantly different as indicated.
Discussion
In the present study, we have presented evidence for the up-regulation of 15-PGDH expression under hypoxic condition in pulmonary arteries, in which 15-PGDH is mainly localized in PAECs and translocated into the nucleus by hypoxia. Moreover, we demonstrated that 15-KETE, a product of 15-PDGH action on 15-HETE, stimulated proliferation and migration of PAECs, regulated expression of cell cycle–related proteins and modulated the cell cycle. Furthermore, the ERK signalling pathway was involved in the proliferation and migration as well as the cell cycle in PAECs, induced by 15-KETE. This study identified, for the first time, that hypoxia affects 15-PGDH expression in the pulmonary vasculature and that 15-KETE stimulates proliferation and migration of PAECs, via ERK1/2 signalling.
Hypoxia induced the expression of 15-LO-1 and 15-LO-2, and 15-HETE formation, which plays an important role in PVR (Ma et al., 2010; Liu et al., 2012). Direct evidence that the 15-PGDH/15-KETE pathway affected PVR and angiogenesis was lacking until we wereable to show a new regulatory function for 15-PGDH/15-KETE in the pulmonary vessels. Here, hypoxia increased the expression of 15-PGDH in the lung and, in cultured PAECs, hypoxia no longer induced 15-PGDH expression after treating cells with siHIF-1α, implying that the increased 15-PGDH expression induced by hypoxia was HIF-1α-dependent.
Interestingly, inhibition of 15-PGDH by CAY-10397 had no effects on HIF-1α expression, suggesting that HIF-1α may be a novel upstream target for the regulation of the 15-PGDH pathway and that there was no feedback loop between 15-PGDH and HIF-1α. However, this conclusion requires further investigation. Evidence from our study indicated that the 15-PGDH/15-KETE pathway contributed to the increased growth of PAECs following hypoxia. However, this effect is certainly not entirely dependent on 15-KETE activity. As shown in Figure 2D, blockade of cyclooxygenase or 15-LO also blocked the expression of 15-PGDH, suggesting that PGs and 15-HETE also can activate the 15-PGDH/15-KETE pathway to induce stimulation of PAECs migration and proliferation.
The half-life of 15-KETE in murine macrophages was 11 min (Wei et al., 2009), which is relatively short. However, we started our experiments after 24 h, because hypoxia for 24 h will up-regulate the expression of 15-PGDH. Furthermore, we consider that cell hypoxia could increase endogenous 15-KETE. Although 15-PGDH exists in the pulmonary arterial wall, its main product 15-KETE has not been detected in plasma. These results may be affected by limitations of the experimental method, for instance, the small sample quantity. Moreover, the action of 15-KETE is probably limited to intracellular or autocrine functions. From this viewpoint, and relative to the concentration of 15-KETE in plasma, the concentrations of 15-KETE (1 μmol·L−1) we used in the experiment was very high. Although progress has been made regarding the role of the 15-PGDH/15-KETE pathway in PVR, especially in cultured cells in vitro, less progress ahs been made in vivo. The potential relevance of 15-KETE used in vitro to its effects on PVR in vivo should be addressed in future studies.
Diminished oxygen concentration in blood induces a vascular remodelling process, which is characterized by thickening of all three layers of the blood vessel wall resulting in narrowing and obstruction of pulmonary arteries (Jeffery and Wanstall, 2001; Cahill et al., 2012). The underlying structural changes are caused by increased migration and proliferation of smooth muscle cells and fibroblasts, as well as abnormal hypertrophy of the endothelial cells and plexiform lesions (Humbert et al., 2004). Our earlier studies have indicated that 15-HETE increased PAECs migration and angiogenesis and promoted the proliferation of pulmonary artery smooth muscle cells, eventually contributing to PVR (Ma et al., 2011). However, whether 15-HETE or its final metabolite is a terminal mediator in hypoxia-induced proliferation has not been investigated. Our data demonstrate that among 15-HPETE, 15-KETE and 5,15-diHETE, only 15-KETE with a stable keto group mediated the hypoxia-induced proliferation of PAECs, suggesting that 15-PGDH action may affect the function and action of lipoxins, in terms of the disordered proliferation of PAECs in hypoxic PVR. Meanwhile, 15-KETE increased cell DNA synthesis in proliferation and promoted cell migration in angiogenesis, supporting the hypothesis that the 15-PGDH/15-KETE pathway was a major contributor to hypoxic PVR.
The results are supported by our cell-cycle studies, in which 15-KETE promoted cell transition from the first gap phase (G1 phase) to the DNA replication phase (S phase) and mitotic phase (M phase). The cell-cycle progression from the G0/G1 phase to S phase is through direct or indirect stimulation of cyclins including the accumulation of cyclin A and cyclin D (Lopez-Beltran et al., 2004; Chibazakura et al., 2011). In our study, hypoxia increased the expression of cyclin A and cyclin D, which was diminished by the 15-PGDH inhibitor or by 15-PGDH siRNA. Therefore, the effects of hypoxia are thought to result at least in part from 15-PGDH-dependent actions. The results suggest that the actions of 15-KETE on the cell-cycle progression are important in hypoxia-induced proliferation of PAECs and in the development of PVR, a novel observation worthy of further investigation.
The ERKs are members of the MAPK family that also includes the p38s and the JNKs. Activation of p42/p44 ERK is an important intracellular mediator in the kinase cascades leading to cell proliferation in response to growth factors, agonists and hormones (Rao et al., 2007; Giachini et al., 2010). As our previous reports have indicated, 15-HETE stimulates phosphorylation of ERK1/2 inhibiting apoptosis in the pulmonary arterial smooth muscle cells (Lu et al., 2006; Jiang et al., 2010). The ERK1/2 signalling stimulated by hypoxia could involve, at least in some part, the 15-PGDH/15-KETE pathway. The present study provided direct evidence that 15-KETE affected ERK1/2 activation, which was related to PAEC proliferation, assayed by BrdU incorporation, PCNA expression and cell-cycle progression. However, in our study, p38 MAPK was clearly not necessary for PAECs proliferation because the p38 MAPK inhibitor did not attenuate the effects of 15-KETE-induced proliferation.
Prostacyclin has protective effects on pulmonary vessels, including vasodilation and inhibition of cell proliferation and epoprostenol has become recognized as a therapeutic agent for PH (Schroeder et al., 2000; Obata et al., 2008). Due to complications related to the central venous catheter used for the administration of epoprostenol, other prostacyclin agonists such as treprostinil and inhaled iloprost have been developed (Mirza and Foley, 2012). In clinical studies, treprostinil showed improvements in exercise capacity, functional class, haemodynamics and survival. Moreover, iloprost inhalation resulted in significant reductions in pulmonary arterial pressure from 110.6 ± 21.8 to 105.5 ± 22.3 mmHg, and increases in pulmonary blood flow from 6.7 ± 3.3 to 9.4 ± 5.8 L·min–1 (Caojin et al., 2012). Correlated with our finding, the reduction of the vasodilators in the lungs may be relevant to increased 15-PGDH expression in pulmonary vascular intima. Because prostacyclin has a very short half-life (Badesch et al., 2004), research is now focused on developing a new type of prostacyclin agonist or analogue. Perhaps, our findings open another window for future experiments to focus on the reduction of prostacyclin oxidation, through inhibition of 15-PGDH activity.
In summary, our study demonstrated that hypoxia activated 15-PGDH in pulmonary arterial vascular walls and that the enzyme was primarily localized in vascular intima. Our results also showed that 15-KETE could contribute to the excessive proliferation and migration of PAECs, through regulating the cell-cycle progression. Furthermore, our experiments provide evidence of a critical role of ERK1/2 signalling in the proliferation of hypoxic endothelial cells induced by 15-KETE, which contributed to neointima formation. These observations may generate novel potential targets for the treatment of hypoxic PVR aimed at the inhibition of abnormal proliferation of vascular cells.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 31071007 and 81270113). Graduate Innovation Foundation of Heilongjiang Province (No. YJSCX2012-246HLJ). Science and Technique Foundation of Harbin (Nos. 2008AA3AS097 and 2006RFXXS029).
Glossary
- 15-HETE
15-hydroxyeicosatetraenoic acid
- 15-HPETE
15(S)-hydroperoxy-5,8,11,13-eicosatetraenoic acid
- 15-keto-6Z
8Z, 11Z, 13E-eicosatetraenoic acid
- 15-LO
15-lipoxygenase
- 15-PGDH
15-hydroxyprostaglandin dehydrogenase
- 5,15-diHETE
5,15-(6E,8Z,11Z,13E)-dihydroxyeicosatetraenoic acid;FiO2, fractional inspired oxygen
- HIF
hypoxia-inducible factor
- PAECs
pulmonary artery endothelial cells
- PCNA
proliferating cell nuclear antigen
- PH
pulmonary hypertension
- PVR
pulmonary vascular remodelling
Conflict of interest
None.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research ‘work in progress’. Circulation. 2000;102:2781–2791. doi: 10.1161/01.cir.102.22.2781. [DOI] [PubMed] [Google Scholar]
- Backlund MG, Mann JR, Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005;69:28–32. doi: 10.1159/000086629. [DOI] [PubMed] [Google Scholar]
- Badesch DB, McLaughlin VV, Delcroix M, Vizza CD, Olschewski H, Sitbon O, et al. Prostanoid therapy for pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:56S–61S. doi: 10.1016/j.jacc.2004.02.036. [DOI] [PubMed] [Google Scholar]
- Bergholte JM, Soberman RJ, Hayes R, Murphy RC, Okita RT. Oxidation of 15-hydroxyeicosatetraenoic acid and other hydroxy fatty acids by lung prostaglandin dehydrogenase. Arch Biochem Biophys. 1987;257:444–450. doi: 10.1016/0003-9861(87)90589-3. [DOI] [PubMed] [Google Scholar]
- Cahill E, Costello CM, Rowan SC, Harkin S, Howell K, Leonard MO, et al. Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation. 2012;125:920–930. doi: 10.1161/CIRCULATIONAHA.111.038125. [DOI] [PubMed] [Google Scholar]
- Caojin Z, Yigao H, Tao H, Wenhui H, Chunli X, Xinsheng H. Comparison of acute hemodynamic effects of aerosolized iloprost and inhaled nitric oxide in adult congenital heart disease with severe pulmonary arterial hypertension. Intern Med. 2012;51:2857–2862. doi: 10.2169/internalmedicine.51.7927. [DOI] [PubMed] [Google Scholar]
- Chang MS, Boeglin WE, Guengerich FP, Brash AR. Cytochrome P450-dependent transformations of 15R- and 15S-hydroperoxyeicosatetraenoic acids: stereoselective formation of epoxy alcohol products. Biochemistry. 1996;35:464–471. doi: 10.1021/bi952081v. [DOI] [PubMed] [Google Scholar]
- Chawengsub Y, Gauthier KM, Nithipatikom K, Hammock BD, Falck JR, Narsimhaswamy D, et al. Identification of 13-hydroxy-14,15-epoxyeicosatrienoic acid as an acid-stable endothelium-derived hyperpolarizing factor in rabbit arteries. J Biol Chem. 2009;284:31280–31290. doi: 10.1074/jbc.M109.025627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibazakura T, Kamachi K, Ohara M, Tane S, Yoshikawa H, Roberts JM. Cyclin A promotes S-phase entry via interaction with the replication licensing factor Mcm7. Mol Cell Biol. 2011;31:248–255. doi: 10.1128/MCB.00630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Y, Tong M, Liu S, Moscow JA, Tai HH. NAD+-linked 15-hydroxyprostaglandin dehydrogenase (15-PGDH) behaves as a tumor suppressor in lung cancer. Carcinogenesis. 2005;26:65–72. doi: 10.1093/carcin/bgh277. [DOI] [PubMed] [Google Scholar]
- Ensor CM, Tai HH. 15-Hydroxyprostaglandin dehydrogenase. J Lipid Mediat Cell Signal. 1995;12:313–319. doi: 10.1016/0929-7855(95)00040-w. [DOI] [PubMed] [Google Scholar]
- Giachini FR, Sullivan JC, Lima VV, Carneiro FS, Fortes ZB, Pollock DM, et al. Extracellular signal-regulated kinase 1/2 activation, via downregulation of mitogen-activated protein kinase phosphatase 1, mediates sex differences in desoxycorticosterone acetate-salt hypertension vascular reactivity. Hypertension. 2010;55:172–179. doi: 10.1161/HYPERTENSIONAHA.109.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43:13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Jeffery TK, Wanstall JC. Pulmonary vascular remodeling: a target for therapeutic intervention in pulmonary hypertension. Pharmacol Ther. 2001;92:1–20. doi: 10.1016/s0163-7258(01)00157-7. [DOI] [PubMed] [Google Scholar]
- Jiang J, Wang S, Wang Z, Ma J, Liu S, Li W, et al. The role of ERK1/2 in 15-HETE-inhibited apoptosis in pulmonary arterial smooth muscle cells. J Recept Signal Transduct Res. 2010;31:45–52. doi: 10.3109/10799893.2010.512013. [DOI] [PubMed] [Google Scholar]
- Liu P, Slater DM, Lenburg M, Nevis K, Cook JG, Vaziri C. Replication licensing promotes cyclin D1 expression and G1 progression in untransformed human cells. Cell Cycle. 2009;8:125–136. doi: 10.4161/cc.8.1.7528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ma C, Zhang Q, Yu L, Ma J, Zhang L, et al. The key role of transforming growth factor-beta receptor I and 15-lipoxygenase in hypoxia-induced proliferation of pulmonary artery smooth muscle cells. Int J Biochem Cell Biol. 2012;44:1184–1202. doi: 10.1016/j.biocel.2012.04.009. [DOI] [PubMed] [Google Scholar]
- Lopez-Beltran A, Luque RJ, Alvarez-Kindelan J, Quintero A, Merlo F, Requena MJ, et al. Prognostic factors in survival of patients with stage Ta and T1 bladder urothelial tumors: the role of G1-S modulators (p53, p21Waf1, p27Kip1, cyclin D1, and cyclin D3), proliferation index, and clinicopathologic parameters. Am J Clin Pathol. 2004;122:444–452. doi: 10.1309/LTFU-3UUM-BY09-5HUM. [DOI] [PubMed] [Google Scholar]
- Lu C, Liu Y, Tang X, Ye H, Zhu D. Role of 15-hydroxyeicosatetraenoic acid in phosphorylation of ERK1/2 and caldesmon in pulmonary arterial smooth muscle cells. Can J Physiol Pharmacol. 2006;84:1061–1069. doi: 10.1139/y06-057. [DOI] [PubMed] [Google Scholar]
- Ma C, Li Y, Ma J, Liu Y, Li Q, Niu S, et al. Key role of 15-lipoxygenase/15-hydroxyeicosatetraenoic acid in pulmonary vascular remodeling and vascular angiogenesis associated with hypoxic pulmonary hypertension. Hypertension. 2011;58:679–688. doi: 10.1161/HYPERTENSIONAHA.111.171561. [DOI] [PubMed] [Google Scholar]
- Ma J, Liang S, Wang Z, Zhang L, Jiang J, Zheng J, et al. ROCK pathway participates in the processes that 15-hydroxyeicosatetraenoic acid (15-HETE) mediated the pulmonary vascular remodeling induced by hypoxia in rat. J Cell Physiol. 2010;222:82–94. doi: 10.1002/jcp.21923. [DOI] [PubMed] [Google Scholar]
- Matsuo M, Ensor CM, Tai HH. Cloning and expression of the cDNA for mouse NAD(+)-dependent 15-hydroxyprostaglandin dehydrogenase. Biochim Biophys Acta. 1996;1309:21–24. doi: 10.1016/s0167-4781(96)00123-6. [DOI] [PubMed] [Google Scholar]
- Mirza S, Foley RJ. Clinical utility of treprostinil and its overall place in the treatment of pulmonary arterial hypertension. Clin Med Insights Circ Respir Pulm Med. 2012;6:41–50. doi: 10.4137/CCRPM.S8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyaki A, Yang P, Tai HH, Subbaramaiah K, Dannenberg AJ. Bile acids inhibit NAD+-dependent 15-hydroxyprostaglandin dehydrogenase transcription in colonocytes. Am J Physiol Gastrointest Liver Physiol. 2009;297:G559–G566. doi: 10.1152/ajpgi.00133.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myung SJ, Rerko RM, Yan M, Platzer P, Guda K, Dotson A, et al. 15-Hydroxyprostaglandin dehydrogenase is an in vivo suppressor of colon tumorigenesis. Proc Natl Acad Sci U S A. 2006;103:12098–12102. doi: 10.1073/pnas.0603235103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata H, Sakai Y, Ohnishi S, Takeshita S, Mori H, Kodama M, et al. Single injection of a sustained-release prostacyclin analog improves pulmonary hypertension in rats. Am J Respir Crit Care Med. 2008;177:195–201. doi: 10.1164/rccm.200703-349OC. [DOI] [PubMed] [Google Scholar]
- Partovian C, Adnot S, Raffestin B, Louzier V, Levame M, Mavier IM, et al. Adenovirus-mediated lung vascular endothelial growth factor overexpression protects against hypoxic pulmonary hypertension in rats. Am J Respir Cell Mol Biol. 2000;23:762–771. doi: 10.1165/ajrcmb.23.6.4106. [DOI] [PubMed] [Google Scholar]
- Rao R, Redha R, Macias-Perez I, Su Y, Hao C, Zent R, et al. Prostaglandin E2-EP4 receptor promotes endothelial cell migration via ERK activation and angiogenesis in vivo. J Biol Chem. 2007;282:16959–16968. doi: 10.1074/jbc.M701214200. [DOI] [PubMed] [Google Scholar]
- Schroeder RA, Wood GL, Plotkin JS, Kuo PC. Intraoperative use of inhaled PGI(2) for acute pulmonary hypertension and right ventricular failure. Anesth Analg. 2000;91:291–295. doi: 10.1097/00000539-200008000-00008. [DOI] [PubMed] [Google Scholar]
- Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res. 2006;99:675–691. doi: 10.1161/01.RES.0000243584.45145.3f. [DOI] [PubMed] [Google Scholar]
- Tai HH, Ensor CM, Tong M, Zhou H, Yan F. Prostaglandin catabolizing enzymes. Prostaglandins Other Lipid Mediat. 2002;68–69:483–493. doi: 10.1016/s0090-6980(02)00050-3. [DOI] [PubMed] [Google Scholar]
- Tai HH, Cho H, Tong M, Ding Y. NAD+-linked 15-hydroxyprostaglandin dehydrogenase: structure and biological functions. Curr Pharm Des. 2006;12:955–962. doi: 10.2174/138161206776055958. [DOI] [PubMed] [Google Scholar]
- Tseng-Rogenski S, Gee J, Ignatoski KW, Kunju LP, Bucheit A, Kintner HJ, et al. Loss of 15-hydroxyprostaglandin dehydrogenase expression contributes to bladder cancer progression. Am J Pathol. 2012;176:1462–1468. doi: 10.2353/ajpath.2010.090875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C, Zhu P, Shah SJ, Blair IA. Blair15-oxo-eicosatetraenoic acid, a metabolite of macrophage 15-hydroxyprostaglandin dehydrogenase that inhibits endothelial cell proliferation. Mol Pharmacol. 2009;76:516–525. doi: 10.1124/mol.109.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss RH, Arnold JL, Estabrook RW. Transformation of an arachidonic acid hydroperoxide into epoxyhydroxy and trihydroxy fatty acids by liver microsomal cytochrome P-450. Arch Biochem Biophys. 1987;252:334–338. doi: 10.1016/0003-9861(87)90039-7. [DOI] [PubMed] [Google Scholar]
- Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, et al. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci U S A. 2004;101:17468–17473. doi: 10.1073/pnas.0406142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh HC, Tsai AL, Wang LH. Reaction mechanisms of 15-hydroperoxyeicosatetraenoic acid catalyzed by human prostacyclin and thromboxane synthases. Arch Biochem Biophys. 2007;461:159–168. doi: 10.1016/j.abb.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Medhora M, Campbell WB, Spitzbarth N, Baker JE, Jacobs ER. Chronic hypoxia activates lung 15-lipoxygenase, which catalyzes production of 15-HETE and enhances constriction in neonatal rabbit pulmonary arteries. Circ Res. 2003;92:992–1000. doi: 10.1161/01.RES.0000070881.65194.8F. [DOI] [PubMed] [Google Scholar]