

Abstract
BACKGROUND AND PURPOSE
The endocannabinoid anandamide (N-arachidonoyl ethanolamide; AEA) exerts negative inotropic and antiarrhythmic effects in ventricular myocytes.
EXPERIMENTAL APPROACH
Whole-cell patch-clamp technique and radioligand-binding methods were used to analyse the effects of anandamide in rat ventricular myocytes.
KEY RESULTS
In the presence of 1–10 μM AEA, suppression of both Na+ and L-type Ca2+ channels was observed. Inhibition of Na+ channels was voltage and Pertussis toxin (PTX) – independent. Radioligand-binding studies indicated that specific binding of [3H] batrachotoxin (BTX) to ventricular muscle membranes was also inhibited significantly by 10 μM metAEA, a non-metabolized AEA analogue, with a marked decrease in Bmax values but no change in Kd. Further studies on L-type Ca2+ channels indicated that AEA potently inhibited these channels (IC50 0.1 μM) in a voltage- and PTX-independent manner. AEA inhibited maximal amplitudes without affecting the kinetics of Ba2+ currents. MetAEA also inhibited Na+ and L-type Ca2+ currents. Radioligand studies indicated that specific binding of [3H]isradipine, was inhibited significantly by metAEA. (10 μM), changing Bmax but not Kd.
CONCLUSION AND IMPLICATIONS
Results indicate that AEA inhibited the function of voltage-dependent Na+ and L-type Ca2+ channels in rat ventricular myocytes, independent of CB1 and CB2 receptor activation.
Keywords: endocannabinoid, anandamide; Na+ channel, L-type Ca2+ channel, ventricular myocyte
Introduction
Endocannabinoids are a group of polyunsaturated fatty acid-based compounds that mimic most of the effects tetrahydrocannabinol, the active ingredient of the marijuana plant Cannabis sativa. N-arachidonoyl ethanolamide (AEA) or anandamide and 2-arachidonylglycerol are the most widely studied endogenous cannabinoids (Di Marzo et al., 2005; Hanus and Mechoulam, 2010). In recent years, extensive research focusing on the biological actions of these compounds indicated that endocannabinoids have important regulatory roles in several physiological and pathological conditions (Di Marzo et al., 2005; Pacher et al., 2006; Pertwee et al., 2010). The endocannabinoid system consists of the endocannabinoid receptors (CB1 and CB2 cannabinoid receptors; receptor nomenclature follows Alexander et al., 2013a), the enzymes regulating the synthesis (such as PLD and monoacylglycerol lipase), the degradation, such as fatty acid amide hydrolase (FAAH) and lipases. and the proteins involved in their transport across the biological membranes (Di Marzo et al., 2005; Pertwee et al., 2010). CB1 receptors are located in the brain and several peripheral tissues including the heart and the vasculature (Pertwee et al., 2010). CB2 receptors, on the other hand, are expressed primarily in the immune system, but recently their presence in the brain, myocardium and smooth muscle cells have also been demonstrated (Pertwee et al., 2010).
Recent studies suggest that endocannabinoids have important modulatory roles on the function of the cardiovascular system under various pathological conditions, such as hypertension, myocardial infarction and heart failure (see Batkai and Pacher, 2009; Montecucco and Di Marzo, 2012). AEA, the most studied endocannabinoid, exerts a complex set of actions on cardiac function. Experiments with AEA performed in isolated Langendorff rat hearts and in isolated, electrically stimulated human atrial appendages (Ford et al., 2002; Bonz et al., 2003) have revealed a negative inotropic effect, which may underlie its ability to decrease cardiac output, as observed in vivo (Wagner et al., 2001). Moreover, AEA and other cannabinoids have been reported to have antiarrhythmic effects in in vivo animal models (Ugdyzhekova et al., 2001; Krylatov et al., 2002). In a recent study, a weak protective effect of endocannabinoids during the late stages of ischaemia has been suggested to be mediated by CB1 receptors (Andrag and Curtis, 2013). Another study reported that AEA potently inhibits the function of L-type Ca2+ channels by activating CB1 receptors in rat cardiomyocytes (Li et al., 2009; ion channel nomenclature follows Alexander et al., 2013b). However, electrophysiological mechanisms underlying these cardiac actions of AEA remain largely unknown. We hypothesized that some of the negative inotropic and antiarrhythmic actions of AEA could be mediated by the modulation of voltage-gated inward Na+ and Ca2+ channels. Thus, in the present study, using whole-cell patch-clamp and radioligand-binding methods, we investigated the actions of AEA on these major inward currents underlying the shape of the action potential (AP) in acutely dissociated rat ventricular myocytes.
Methods
Isolation of ventricular myocytes from rats
All animal care and experimental procedures complied with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Animal Research Ethics Committee of the College of Medicine and Health Sciences, UAE University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 90 animals were used in the experiments described here.
The original stocks of Wistar rats were purchased from Harlan Laboratories (Oxon, England). Animals were bred at our own animal facility from the original stock. The animals were housed in polypropylene cages (43 × 22.5 × 20.5 cm; six rats per cage) in climate and access controlled rooms (22–24°C; 50% humidity). The day/night cycle was 12 h/12 h. Food and water were provided ad libitum. The food was standard maintenance diet for rats purchased from Emirates Feed Factory (Abu Dhabi, UAE). Ventricular myocytes were isolated from adult male Wistar rats (264 ± 19 g) according to previously described techniques (Howarth et al., 2002). Briefly, the animals were killed using a guillotine and the hearts were removed rapidly and mounted for retrograde perfusion according to the Langendorff method. Hearts were perfused at a constant flow of 8 mL g per heart per min and at 36–37°C with a solution containing (mM): 130 NaCl, 5.4 KCl, 1.4 MgCl2, 0.75 CaCl2, 0.4 NaH2PO4, 5 HEPES, 10 glucose, 20 taurine and 10 creatine set to pH 7.3 with NaOH. When the heart had stabilized, perfusion was continued for 4 min with Ca2+-free isolation solution containing 0.1 mM EGTA, and then for 6 min with cell isolation solution containing 0.05 mM Ca2+–, 0.75 mg·mL−1 collagenase (type 1; Worthington Biochemical Corp., Lakewood Township, NJ, USA) and 0.075 mg·mL−1 protease (type X1 V; Sigma, Taufkirchen, Germany). Ventricles were excised from the heart, minced and gently shaken in collagenase-containing isolation solution supplemented with 1% BSA. Cells were filtered from this solution at 4 min intervals and resuspended in isolation solution containing 0.75 mM Ca2+.
Measurement of Na+ and L-type Ca2+ currents
Currents were measured using whole-cell patch-clamp technique and recorded with an Axopatch 200B amplifier (Molecular Devices, Downington, PA, USA) linked to an A/D interface (Digidata 1322; Molecular Devices). Currents were filtered at 5 kHz and acquired using Clampex 8.2. Patch pipettes were fabricated from filamented GC150TF borosilicate glass (Harvard Apparatus, Holliston, MA, USA) on a horizontal puller (Sutter Instruments Co., Novato, CA, USA). Electrode resistances ranged from 2.0 to 3.0 MΩ, and seal resistances were 1–5 GΩ. After giga seal formation, the membrane was ruptured with gentle suction to obtain whole-cell voltage-clamp configuration. In recording of Na+ currents, cells were held at −80 mV, and were depolarized with 50 ms pulses from −80 to +70 mV in 10 mV increments. Extracellular solution for recordings of Na+ currents consisted of (in mM): 100 TEA Cl, 40 NaCl, 10 glucose, 1 MgCl2, 5 CsCl, 0.1 CaCl2, 1 NiCl2 and 10 HEPES (adjusted to pH 7.3 with CsOH). Intracellular solution contained (in mM) 135 CsCl, 5 NaCl, 10 EGTA, 10 HEPES and 1 Mg ATP (adjusted to pH 7.25 with CsOH). For recording of Ca2+ currents, data were elicited from a holding potential of −50 mV to membrane potentials ranging from −70 mV to +70 mV in 10 mV increments every 300 ms. The whole-cell bath solution contained (in mM): 95 NaCl, 50 TEA Cl, 2 MgCl2, 2 CaCl2, 10 HEPES and 10 glucose (adjusted to pH 7.35 with NaOH). The pipette solution contained (in mM): 140 CsCl, 10 TEA Cl, 2.0 MgCl2, 2 HEPES, 1 Mg ATP and 10 EGTA (adjusted to pH 7.25 with CsOH). Experiments were performed at room temperature (22–24°C). Changes of external solutions and application of drugs were performed using a multi-line perfusion system with a common outflow connected to recording chamber. Electrophysiological data were analysed using pClamp 10.2 (Molecular Devices, Union City, CA, USA) and Origin 7.0 (OriginLab Corp., Northampton, MA, USA) software. The amplitudes of the currents were normalized to the cell membrane capacitance to provide current densities (nA/pF). The Gi/o protein blocker Pertussis toxin (PTX; 2 μg·mL−1) was purchased from Sigma. Cells were incubated with PTX for 3 h at 37°C (control cells to this group were incubated in the same conditions with distilled water only).
Radioligand binding studies with [3H] batrachotoxin B (BTX-B)
Myocytes were prepared daily from adult rat ventricles with a yield of 8–10 × 106 myocytes per heart, of which 75–80% were viable rod-shaped striated cells. Cells were collected by gentle centrifugation (40× g) for 5 min, rinsed twice with incubation solution, stored at room temperature (21–23°C) in incubation solution and used within 30 min. Myocytes (5 × 105 per assay) in 200 μL of incubation buffer (minimal essential medium with 50 μM CaCl2) were incubated (in duplicate or triplicate in polypropylene centrifuge tubes) with 1 μM sea anemone toxin (ATX), 100 μM tetrodotoxin (TTX), and various concentrations of [3H]BTX-B for 60 min at 37°C in the cell-culture incubator. TTX was added to prevent depolarization induced by Na+ influx induced by the toxins (Sheldon et al., 1986). Various concentrations of metAEA dissolved in ethanol were added in volumes of 3–5 μL to the incubation buffer. (The same volumes of ethanol were added to the control samples.) These volumes of ethanol had no effect on the [3H]BTX-B binding). Assays were done in parallel with tubes containing 0.4 mM aconitine to define non-specific binding. At the end of the 60 min incubation period, 150 μL of the reaction mixture (cell suspension) from each sample were filtered through a Whatman GF/C 25 mm fibreglass filter under vacuum. The reaction was terminated by adding 10 mL of Krebs–Henseleit–BSA buffer (127 mM NaCl, 2.3 mM KC1, 1.30 mM KH2PO4, 1.2 mM MgSO4, 25 mM NaHCO3, 10 mM glucose, 50 μM CaCl2, 1% BSA) equilibrated with 95% O2 + 5% CO2 and incubated at 37°C for 1 mm, then filtered through a Whatman GF-C 24 mm fibreglass filter and washed four times with 5 mL of rinse buffer (25 mM Tris-Cl, pH 7.4, 130 mM NaC1, 5.5 mM KCI, 0.8 mM MgSO4, 5.5 mM glucose, 50 μM CaCl2). The filters were then dried and counted in Econofluor-2 scintillation fluid (PerkinElmer Inc., Waltham, MA, USA) and assessed for cell-bound radioactivity in a Beckman LS-6000SC liquid scintillation counter. (The counting efficiency was 48–50%.) Specific binding was calculated as the difference between total and non-specific binding. Under these conditions non-specific binding was 20–30% of total binding at 30 nM [3H]BTXB. The dissociation constant (Kd) and maximal binding (Bmax) were determined by Scatchard analysis. Specific activity of [3H]BTX-B was 41.3 Ci·mmol–1; 1 Ci = 37 GBq) and was supplied by New England Nuclear. Sea anemone toxin and TTX were obtained from Sigma-Aldrich. metAEA was dissolved weekly in ethanol at the concentration of 10 mM and stored at −20°C.
Preparation of cardiac muscle membranes
Cardiac membranes were prepared from the heart of adult male Wistar rats by some modifications of previously described methods (Dunn, 1989; Oz et al., 2000). Briefly, rat heart was minced with scissors and washed two times with ice-cold binding buffer to remove extraneous blood. The tissue (one heart in a 50 mL) was placed in plastic centrifuge tubes containing 10 mL ice-cold buffer (50 mM Tris·Cl, pH 7.2) and homogenized on ice by two 5 s bursts in a Polytron homogenizer at setting 5. The solution was decanted to a motor-driven 10 mL glass-Teflon homogenizer and homogenized on ice by 10 passes at setting 8. The homogenate was filtered through four layers of cheesecloth (to remove tissue debris) into a clear 50 mL centrifuge tube and centrifuged for 45 min at 45 000× g, 4°C. The pellet was suspended in 5 mL ice-cold binding buffer for each rabbit heart by homogenizing the pellet five times on ice with a motor-driven 10 mL glass-Teflon homogenizer at setting 5. Membrane protein concentration was determined using the Bradford method using BSA as a standard. The membranes were diluted in binding buffer to 1 mg protein·mL–1 and store in 5 mL aliquots at −70°C.
Radioligand-binding studies with [3H]isradipine
Experiments on the binding of [3H]isradipine (specific activity 58.6 Ci·mmol−1, New England Nuclear, Chadds Ford, PA, USA) were conducted similar to our earlier studies (Oz et al., 2000). Briefly, aliquots of membranes (0.1 mg) were added to different concentrations of radiolabelled ligand to give a final concentration of 0.02 mg·mL−1 membranes in a total volume of 0.8 mL. After 60 min incubation at room temperature, 0.4 mL aliquots of each sample were filtered under vacuum through Watman GF\C filters and rapidly washed with 5 mL of ice-cold assay buffer. The filters were dried and extracted in 5 mL of Hydroflourrrrr™ (National Diagnostics, St. Louis, MO, USA) scintillation fluid before counting for 3H. Triplicate 50 μL samples of the incubation mixtures were also counted directly for estimations of total binding. Non-specific binding was estimated from parallel measurements of binding in the presence of 5 μM unlabeled nifedipine. MetAEA, AEA, AM251 and AM630 were purchased from Tocris (Ellisville, MO, USA). All other materials mentioned were purchased from Sigma-Aldrich (St. Louis, MO, USA). In patch-clamp experiments, ethanol concentrations in control and in presence of AEA, metAEA and AM251 were in the range of 0.007–0.07% v/v. AM630 was dissolved in DMSO and the final concentration for DMSO used in the experiments did not exceed 0.007% (v/v). Stock solutions of AEA, AM251 and AM630 were kept at −20°C until their use. Ethanol at the concentration range used in our experiments had no effect on the specific binding of [3H]BTX-B and [3H]isradipine.
Data analysis
The results of the experiments were expressed as mean ± SEM. Statistical analysis was performed using the paired t-test (within the same cell analysis). Statistical significance among groups was determined using one-way anova followed by Benferonni post hoc analysis. Statistical analysis of the data was performed using Origin 7.0 software (OriginLab Corp., Northampton, MA, USA) and IBM spss statistics version 20. P < 0.05 was taken to show statistical significance of differences between means.
Results
The passive properties of the ventricular cells from controls were not significantly different from those of the AEA-treated cells. Resting membrane potentials (mean ± SEM) were −76.2 ± 1.3 and −78.3 ± 1.5 mV in control (n = 38) and AEA-treated (n = 53) myocytes respectively. The mean cell capacitance in the control group was 117.2 ± 14.7 pF, whereas in the AEA-treated cells was 108.3 ± 12.1 pF. The input resistance (measured close to the resting potential) was 72.4 ± 16.5 MΩ in the control cells and 79.3 ± 17.8 MΩ in AEA-treated cells. In control cells, these passive membrane properties were not altered significantly in experiments lasting up to 25–30 min. In 18 control cells measured, resting membrane potentials, cell capacitance and input resistance after 25 min of experiment were −74.8 ± 3.4 mV, 109.6 ± 14.3 pF, and 81.2 ± 14.3 MΩ respectively. These values were not significantly different from control values obtained within the first 5 min of patch-clamp experiment (n = 18; paired t-test, P > 0.05). Characteristics (threshold, maximal and reversal potentials) of current-voltage relationship remained stable during the experiments.
Effects of AEA on voltage-dependent Na+ channels
Previous studies have indicated that AEA has significant antiarrhythmic effects suggesting that this compound may affect voltage-activated inward Na+ (INa) and Ca2+ (IL,Ca) currents of ventricular myocytes. To verify these possibilities, we have conducted a series of experiments under conditions that enable reliable isolation of either INa or ICa in voltage-clamp mode. In the first set of experiments, INa was typically elicited by pulses from −80 to −20 mV for 50 ms. Figure 1A shows recordings of INa in ventricular myocytes before, during and after application of 10 μM AEA. The effect of AEA was detectable at 2–3 min and reached a steady-state level within 10–15 min (Figure 1B). The recovery was partial during the experiments lasting up to 25–30 min (Figure 1B). In our studies, AEA was dissolved in ethanol, and therefore we have tested the effect of ethanol as a vehicle. In agreement with earlier studies (Danziger et al., 1991; Bebarova et al., 2010), our results indicated that maximal amplitudes of INa were altered after 10 min vehicle application in experiments lasting up to 20–25 min. Because of the effect of the vehicle, we have tested each concentration of AEA and vehicle separately and plotted the concentration–response curve after the subtraction of vehicle effect (Supporting Information Figure S1). The effect of increasing AEA and corresponding ethanol concentrations and corrected concentration–response curve were presented in Supporting Information Figure S1. In order to exclude the possibility of the involvement of degradation products of AEA in the inhibition of Na+ current, the effect of 10 μM metAEA was tested. metAEA caused a significant inhibition of Na+ currents (36 ± 4% inhibition of controls; n = 5; paired t-test).
Figure 1.
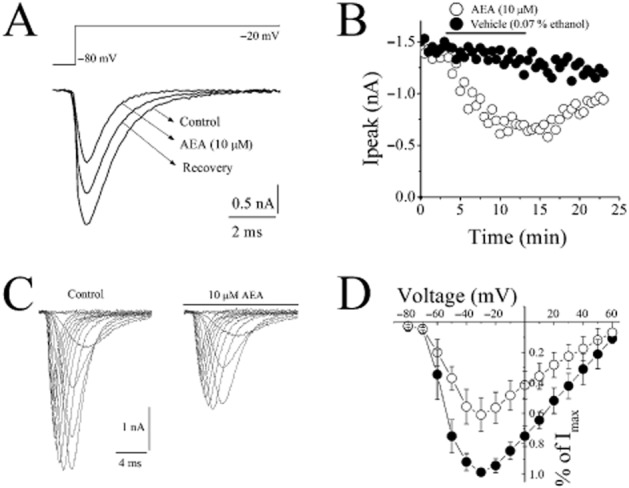
Effect of AEA on voltage-dependent Na+ currents (INa) in rat ventricular myocytes. (A) AEA inhibits INa recorded using whole-cell voltage-clamp mode of the patch-clamp technique. INa was recorded during 50 ms voltage pulses to −20 mV from a holding potential of −80 mV. Current traces were recorded before (control) and after 10 min application of 10 μM AEA. (B) Maximal currents of VGSCs presented as a function of time in the presence of vehicle (0.07% ethanol) and 10 μM AEA (n = 5–6 cells). (C) Representative recordings of INa in response to pulse protocol shown, under control conditions and after application of 10 μM AEA. (D) Normalized and averaged I–V relationships of control INa and INa in the presence of 10 μM AEA, determined by applying a series of step depolarizing pulses from −80 to +70 mV in 10 mV increments for a duration of 50 ms. Data shown are means ± SEM); n = 5–7 cells.
With 40 mM Na+ outside and Cs+ as the major intracellular cation, inward INa in response to incremental step depolarizations (Vm, 10 mV increment) applied from a holding potential Vh = −80 mV started to activate at Vm = −50 mV, and reached maximal amplitude at Vm = −30 mV. At more positive potentials the inward current decreased reversing its direction at an apparent reversal potential (Vrev) of around +60 mV. Traces of INa in the absence and presence of 10 μM AEA were presented in Figure 1C. AEA inhibited INa without causing significant changes in the I–V relationship. The current–voltage (I–V) relationship for INa was illustrated in Figure 1D. AEA inhibited INa without changing the threshold, peak and reversal potentials.
Steady-state activation (SSA) curves of INa before and after AEA application were derived by fitting the respective I–V relationships with the product of Boltzmann and Goldman–Hodgkin–Katz (GHK) equations of which the first one describes voltage dependence of SSA, and the second one the current through open channels. This allowed us to determine if AEA influences the parameters of INa SSA-the voltage of half-maximal activation (V1/2) and slope factor (k). In controls, V1/2 and k values were −45.2 mV and 7.1 mV respectively. In the presence of 10 μM AEA, V1/2 and k values were −42.3 mV, and 7.5 mV (Figure 2A). There were no statistically significant differences between controls and in the presence of AEA (anova, n = 8–10; P > 0.05).
Figure 2.
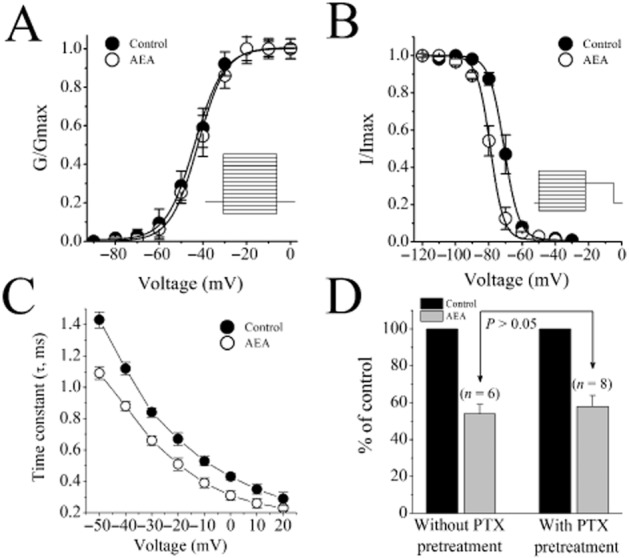
Effect of AEA on SSA and SSI of INa and of PTX pretreatment on AEA inhibition of INa in rat ventricular myocytes. (A) SSA and (B) SSI curves of INa in the absence and presence of 1 μM AEA. Fit of experimental data points with Boltzmann equation (see text for parameters). (C) Voltage dependence of INa inactivation time constant (τi) under control conditions and in the presence of 1 μM AEA Data shown are means ± SEM; n = 5 cells. (D) Effect of PTX pretreatment on AEA inhibition of the maximal INa amplitudes. Data shown are means ± SEM; n = 5–7 cells.
In order to determine if AEA influences the properties of voltage-gated sodium channels (VGSCs) inactivation, we compared steady-state inactivation (SSI) dependencies of INa in the absence and presence of AEA. SSI curves were acquired using a standard voltage protocol consisting of prolonged (400 ms) conditioning pre-pulse to various Vm in the range of −100 to +70 mV, which was immediately followed by the constant INa-activating test pulse to Vm = −20 mV. SSI dependency was plotted as normalized amplitude of INa at Vm = −20 mV against the value of conditioning Vm (normalization was performed to the amplitude of INa at conditioning Vm = −100 mV). The fit of the obtained data points using the Boltzmann equation (Figure 2B) has indicated that under control conditions, SSI of INa is V1/2 = −70.2 mV and k = 5.8 mV, and in the presence of 10 μM of AEA, V1/2 = −81.4 mV and k = 5.1 mV. Thus, AEA induced a significant hyperpolarizing shift in the voltage-dependence of SSI of cardiac VGSCs (−11.4 mV; paired t-test, P < 0.05). Comparison of INa currents in the absence and presence of AEA revealed noticeable acceleration of the current's inactivation kinetics by AEA. Quantification of the time constant of INa inactivation (τi, Figure 2C) by fitting the currents’ decay phase with exponential functions showed that AEA (10 μM) significantly reduced τi in the range of Vm between −50 and +20 mV (Figure 2C).
The rat heart expresses CB1 and CB2 receptors (Bouchard et al., 2003). Therefore, it was likely that the effect of AEA is mediated by the activation of these receptors. For this purpose, we chose two antagonists (AM251 and AM630) for their relative selectivity for CB1 rather than CB2 receptors (Lan et al., 1999; Ross et al., 1999). In the presence of 0.3 μM AM251, a CB1 receptor antagonist with Ki of 7.5 nM (Lan et al., 1999) and 0.3 μM AM630, a CB2 receptor antagonist with Ki of 32.1 nM (Ross et al., 1999), AEA (10 μM) inhibition of INa remained unaltered (n = 6–8, data not shown). As the CB1 and CB2 receptors are coupled to PTX-sensitive Gi/o type G-proteins (see Pertwee et al., 2010), we tested whether the inhibitory effect of AEA on INa could be modulated by PTX pretreatment. Our results show that the extent of the inhibitory effect of AEA on the maximal amplitudes of INa was not affected by PTX pretreatment (Figure 2D). There were no statistically significant difference in the inhibition by AEA in control and PTX-treated groups (n = 6–8; anova; P > 0.05). In positive control experiments, PTX, as reported earlier (Zhang et al., 2005), effectively attenuated the inhibitory actions of BRL-37344, a β3 adrenoceptor agonist, on L-type voltage-gated Ca2+ currents recorded in ventricular myocytes (Supporting Information Figure S2). These results indicate that G-proteins are functionally coupled to their target receptors.
Effect of metAEA on [3H]BTX-B binding
The results of electrophysiological experiments suggest that AEA can directly interact with VGSCs. For this reason, we conducted radioligand-binding experiments to test the possibility of direct interaction between AEA and VGSCs in cardiomyocytes. Binding of [3H] BTX in cardiomyocytes (Sheldon et al., 1986; Kang and Leaf, 1996) and neuronal structures (Postma and Catterall, 1984) has been well characterized in earlier investigations. In this study, because of long incubation times (up to 60 min), we have tested the effect of methanandamide (metAEA), a non-hydrolysable AEA analogue (Abadji et al., 1994), to avoid likely confounding effects of degradation products and oxygenated metabolites on the specific binding of [3H]BTX-B in rat ventricular cardiomyocytes.
Equilibrium curves for the binding of [3H]BTX-B, in the presence and absence of metAEA are presented in Figure 3A (n = 8–11). At a concentration of 10 μM, metAEA caused a significant inhibition of the specific binding of [3H]BTX-B. In controls and in the presence of 10 μM metAEA, maximum binding activities (Bmax) were 76 ± 6 and 41 ± 4 fM, and Kd values were 32 ± 4 and 35 ± 3 nM respectively. There was a statistically significant difference between control and metAEA-treated groups with respect to Bmax values (n = 8–11; anova; P < 0.05). Effect of metAEA on saturation binding was further analysed by Scatchard analysis (Figure 3B), which yielded a Bmax of 92 fmol and a Kd of 21 nM [3H]BTX-B for controls and a Bmax of 43 fmol and a Kd of 24 nM for cells treated with metAEA. To further determine the effects of metAEA on the binding of [3H]BTX-B to cardiac Na+ channels, myocytes were incubated with [3H]BTX-B in the presence of increasing metAEA concentrations. The results indicated that the metAEA inhibited the specific binding of [3H]BTX-B in a dose-dependent manner with an IC50 of 8.6 μM and slope of 1.1 (Figure 3A). We have also investigated the effect of the CB receptor antagonists, AM251 (1 μM) and AM630 (1 μM) on the specific binding of 10 nM [3H]BTX-B. Results presented in Figure 3D indicate that the effect of metAEA (10 μM) on the specific binding of [3H]BTX-B was not significantly altered in the presence of AM251 or AM630 (compared with AEA alone, n = 7–11; anova; P > 0.05). Incubation with AM251 or AM630 alone did not significantly alter the specific [3H]BTX-B binding in cardiac muscle membranes (anova; n = 7–11; P > 0.05).
Figure 3.
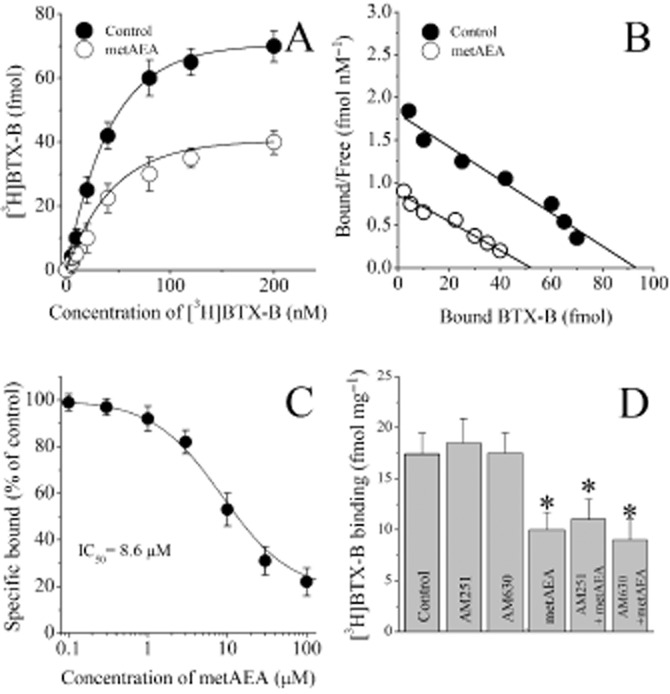
Effects of metAEA and cannabinoid receptor antagonists AM251 and AM630 on the specific binding of [3H]BTX-B to rat ventricular myocytes. (A) Specific binding as a function of the concentration of [3H]BTX-B in the absence and presence of metAEA (10 μM). (B) Scatchard analysis of the effects of metAEA on saturation binding of [3H]BTX-B to ventricular myocytes. The slope and intercept of the linear regression curve indicate the Kd and Bmax values respectively. Data points were determined from a set of binding experiments presented in the panel A. (C) Effect of increasing metAEA concentration on the specific binding of [3H]BTX-B to ventricular myocytes. Binding was measured as described in the methods. Data are the means of 5–6 experiments. (D) Effects of cannabinoid receptor antagonists AM251 (1 μM) and AM630 (1 μM) and their co-application with metAEA (10 μM) on the specific binding of [3H]BTX-B to cardiomyocytes. Data shown are means ± SEM; n = 7–11 cells. *P < 0.05; significant effect of metAEA.
Effect of AEA on L-type Ca2+ currents
We have also investigated the effect of AEA (1 μM) on the L-type Ca2+ currents (IL,Ca). Figure 4A shows a typical record of IL,Ca elicited by applying a single 300 ms voltage pulse to +10 mV from a holding potential of −50 mV in rat ventricular myocyte before and after 10 min superfusion with 1 μM AEA. The time course of the effect of AEA on the density of IL,Ca is presented in Figure 4B. Current density of IL,Ca was also altered after 10 min vehicle application in experiments lasting up to 20–25 min. Effects of increasing AEA and corresponding ethanol concentrations on IL,Ca and corrected concentration–response curve were presented in Supporting Information Figure S3. The effect of 1 μM metAEA was also tested. metAEA caused a significant inhibition of Ca+2 currents (46 ± 4 inhibition of controls; n = 5; paired t-test).
Figure 4.
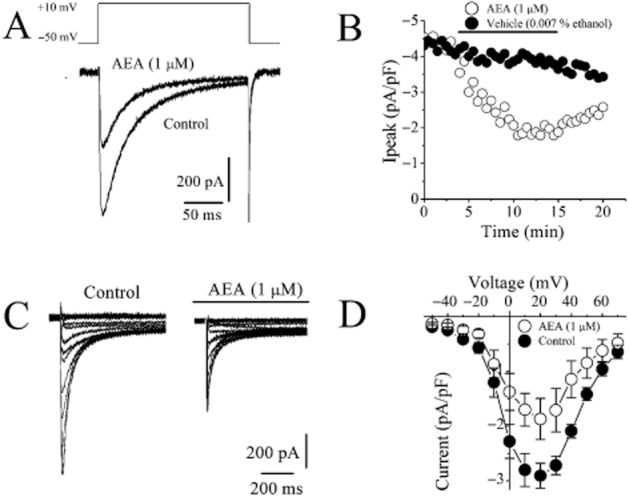
Effect of AEA on Ca2+ currents mediated by L-type Ca2+ channels in rat ventricular myocytes. (A) AEA inhibits L-type Ca2+ currents recorded using whole-cell voltage-clamp mode of patch-clamp technique. Current traces recorded before (control) and after 10 min application of 1 μM AEA. ICa were recorded during 300 ms voltage pulses to +10 mV from a holding potential of −50 mV. (B) Averages of the maximal currents of VGCCs presented as a function of time in the presence of vehicle and 1 μM AEA; n = 5 cells. Application time for the agents is shown as a horizontal bar. (C) Representative recordings of ICa in response to the depicted pulse protocol under control conditions and after application of 1μM AEA. (D) Normalized and averaged I–V relationships of control ICa and ICa in the presence of 10 μM AEA determined by applying a series of step depolarizing pulses from −70 to +70 mV in 10 mV increments for a duration of 300 ms. Data shown are means ± SEM; n = 5–7 cells.
The effects of AEA were also investigated on the biophysical properties of IL,Ca in rat ventricular myocytes. IL,Ca was recorded in the presence of intracellular Cs+ and extracellular TEA+ to suppress K+ currents while retaining 95 mM Na+ in the extracellular solution. Elimination of contaminating Na+ current during recording of IL,Ca was achieved by applying voltage step-pulses from relatively depolarized Vh of −50 mV, which produced steady-state INa inactivation (Voitychuk et al., 2012). As evident from original recordings and I–V relationships (Figure 4C and D), IL,Ca had much slower kinetics in response to step depolarization and activated at more positive potentials than INa: it started to appear at Vm = −30 mV, reached maximum at around Vm = +10 mV, and decreased at higher voltages approaching zero at about Vm = +60 mV (Figure 4D).
AEA also produced a depolarizing shift of IL,Ca SSA by 12.6 mV (i.e. from control value V1/2 = −9.4 ± 0.3 mV to V1/2 = +3.2 ± 0.2 mV in the presence of AEA) and hyperpolarizing shift of IL,Ca SSI by 4.3 mV (i.e. from control value V1/2 = −18.9 ± 0.1 mV to V1/2 = −23.2 ± 0.1 mV in the presence of AEA) with little influence on the slopes of respective dependencies (k = 7. 2 ± 0.4 mV and k = −5.3 ± 0.3 mV for the control activation and inactivation, respectively, vs. k = 6.9 ± 0.3 mV and k = −5.1 ± 0.2 mV for the AEA-modified activation and inactivation, respectively), which altogether resulted in the notable reduction of IL,Ca ‘window current’ responsible for the stationary Ca2+ entry in the range of membrane potentials from −40 to +10 mV (Figure 5A and B). Thus, the mechanism of AEA action on cardiac L-type voltage-gated calcium channel (VGCC) most likely involves changes in channel gating that reduce ‘window current’ as well as part blockade of the ion-conducting pathway that decreases current amplitude. In time-matching controls measured after 10 min of ethanol application, activation and inactivation parameters were not significantly different compared with time-matched controls (without ethanol application after 10 to 15 min of patching time). V1/2 values for activation and inactivation were −10.3 ± 0.4 mV and −20.6 ± 0.7 for controls, and −9.7 ± 0.6 mV and −19.6 ± 0.8 in the presence of 0.007% (1.5 mM) ethanol, respectively (n = 7–9; anova, P > 0.05).
Figure 5.
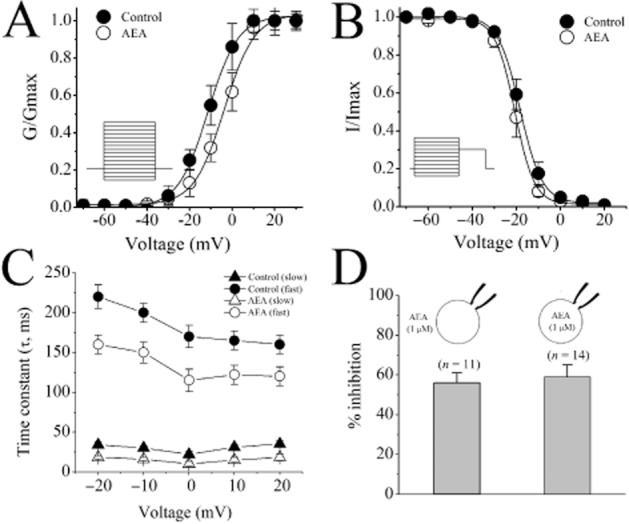
Effect of AEA on SSA and SSI of ICa and of sidedness of AEA application on ICa in rat ventricular myocytes. (A) SSA and (B) SSI curves of ICa in the absence and presence of 1 μM AEA. Data shown are means ± SEM; n= 5 cells. Fit of experimental data points with Boltzmann equation. (C) Voltage-dependent fast and slow inactivation time constants (τi) of IL,Ca under control conditions and in the presence of 1 μM AEA. Data shown are means ± SEM; n = 5–6 cells. (D) Comparison of the intracellular and extracellular application of AEA on the maximal inhibition of ICa. Data shown are means ± SEM; n = 5–8 cells.
In line with earlier reports (Soldatov et al., 1998), kinetic analysis of IL,Ca currents were fit to double-exponential function with fast (τf) and slow (τs) inactivation time constants. Comparison of IL,Ca currents in the absence and presence of AEA revealed noticeable acceleration of the current's inactivation kinetics by AEA. Quantification of the time constants of IL,Ca inactivation showed that AEA (1 μM) significantly reduced τi in the range of Vm −30 mV and +10 mV (Figure 5C).
In earlier electrophysiological studies, sidedness of AEA actions on various ion channels has been reported (Oz, 2006). For this reason, we have tested the effect of intracellular application of AEA by including AEA (1 μM) inside the patch electrode (Figure 5D). The extent of AEA inhibition (compared after 15 min of AEA exposure) was not significantly different between intracellular and extracellular AEA applications (anova; n = 11–14; P > 0.05). We have also conducted experiments with URB597, a potent inhibitor of FAAH, which is the enzyme that hydrolyses AEA (incubation of 1 μM URB597 for 45 min at 37°C vs. controls incubated with 0.007% ethanol alone for 45 min at 37°C). Our results indicate that the effect of 1 μM AEA on L-type Ca2+ currents remained unaltered in URB597-incubated cells. The extent of AEA inhibition was in the presence and absence of URB597 treatment was 35 ± 4% and 32 ± 3%, respectively (n = 5–7; anova, P > 0.05).
In the next series of experiments, we tested if the modulation of Ca2+ binding site can mediate the effect of AEA on the inactivation kinetics of L-type VGCCs. For this purpose, we have replaced extracellular Ca2+ with Ba2+ and tested the effect of AEA on Ba2+ currents (IBa) through L-type VGCCs. In line with earlier studies (Soldatov et al., 1998), inactivation of IBa fit to mono-exponential decay function (Figure 6A) with significant voltage-dependency. In the presence of AEA, inactivation time constant (Figure 6A and B), and the maximal amplitudes of IBa were significantly inhibited compared with control values (paired t-test; n = 7; P < 0.05).
Figure 6.
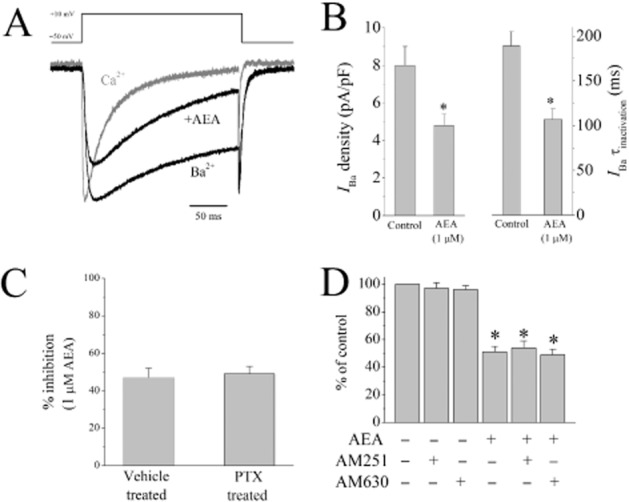
Effect of AEA on Ba2+ currents mediated by L-type Ca2+ channels, and effects of PTX pretreatment and cannabinoid receptor antagonists on AEA inhibition of L-type Ca2+ channels. (A) Traces of normalized Ca2+ and Ba2+ currents through L-type VGCCS. Normalized Ba2+ current in the presence 1 μM AEA was presented in the figure. (B) Effect of 1 μM AEA on the maximal amplitudes and the inactivation kinetics of Ba2+ currents. Data shown are means ± SEM; n = 7–8 cells. (C) Percentage of inhibition of L-type Ca2+ currents after vehicle (distilled water) and PTX (2 μg·μL−1, 3 h). Data shown are means ± SEM; n = 6–8 cells. (D) Effects of CB1 antagonist AM251 (0.3 μM) and CB2 antagonist AM630 (0.3 μM) on AEA (1 μM) inhibition of L-type Ca2+ currents. Data shown are means ± SEM; n = 6–9 cells. *P < 0.05, significantly different from control.
We have also investigated whether the inhibitory effects of AEA on L-type VGCCs are mediated by the activation of cannabinoid receptors by checking their sensitivity to inhibition by PTX, as CB1 and CB2 receptors are coupled to PTX-sensitive Gi/o type G-proteins Our results show that the inhibitory effect of AEA on the maximal amplitudes of IL,Ca was not affected by PTX pretreatment (Figure 6C). We have conducted further experiments in which 1 mM GTP was included in the pipette solution. In the presence of GTP, AEA (10 min bath application) continued to inhibit L-type Ca2+ currents to 62 ± 4% of controls (n = 6). Furthermore, in the presence of 0.3 μM AM251, a CB1 receptor antagonist with Ki of 7.5 nM (Lan et al., 1999) and 0.3 μM AM630, a CB2 receptor antagonist with Ki of 32.1 nM (Ross et al., 1999), AEA (1 μM) inhibition of IL,Ca remained unaltered (Figure 6D). There were no statistically significant difference in the % inhibition by AEA among AEA, AEA + AM251, and AEA + AM630-treated groups (n = 6–9; anova; P > 0.05). Application of AM251 or AM630 alone did not have a significant effect on the amplitudes of IL,Ca (Figure 6D) (n = 8; paired t-test; P > 0.05).
Effect of metAEA on [3H]isradipine binding
In cardiac tissue, AEA is hydrolysed to arachidonic acid (AA), and AA is known to inhibit the function of cardiac L-type Ca2+ channels and cause negative inotropic actions (Li et al., 2009; for a review of earlier studies, Oz, 2006). Therefore, it was possible that AA, rather than AEA, interacts with L-type Ca2+ channels. For this reason, we tested the effect of metAEA, the metabolically stable chiral analogue of AEA, which is resistant to hydrolytic inactivation by FAAH (Abadji et al., 1994) on the specific binding of [3H]isradipine. Equilibrium curves for the binding of [3H]isradipine, in the presence and absence of the metAEA are presented in Figure 7A (n = 8–11). At a concentration of 10 μM, metAEA caused a significant inhibition of the specific binding of [3H]isradipine. In controls and in the presence of 10 μM AEA, maximum binding activities (Bmax) were 126 ± 19 and 58 ± 24 fM·mg−1 protein, and Kd values were 75 ± 12 pM and 86 ± 14 pM respectively. There was a statistically significant difference between control and metAEA-treated groups with respect to Bmax values (P < 0.05, anova, n = 8–11). Effect of metAEA on saturation binding was further analysed by Scatchard analysis (Figure 7B), which yielded a Bmax of 178 ± 19 fmol·mg−1 protein and a Kd of 78 ± 8 pM [3H]isradipine for controls and a Bmax of 68 ± 9 fmol·mg−1 protein and a Kd of 76 ± 8 pM for membranes treated with metAEA. The effect of increasing metAEA concentrations was also investigated on the specific [3H]isradipine binding from cardiac muscle membranes (Figure 7C). In the concentration range used (0.1–30 μM), metAEA caused a significant inhibition of the specific binding of [3H]isradipine. The values for IC50 and slope factor for metAEA were 3.6 μM and 1.4 respectively.
Figure 7.
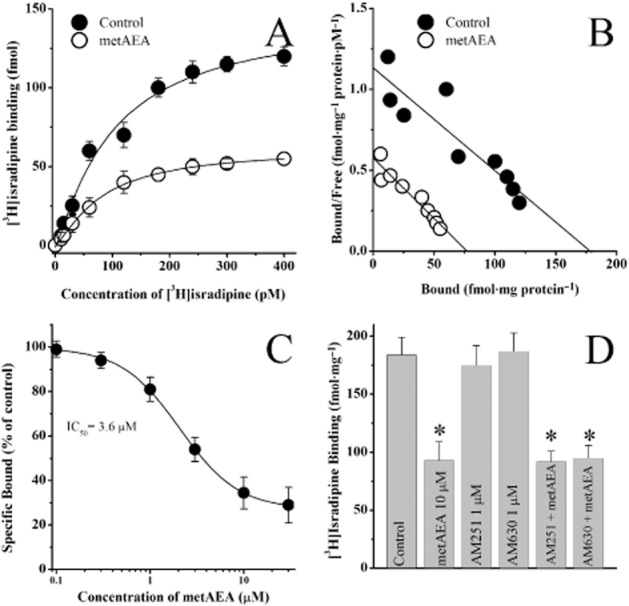
Effects of metAEA and cannabinoid receptor antagonists AM251 and AM630 on the specific binding of [3H]isradipine to rat ventricular muscle membranes. (A) Specific binding as a function of the concentration of [3H]isradipine in the absence and presence of metAEA (10 μM). (B) Scatchard analysis of the effects of metAEA on saturation binding of [3H]isradipine binding to cardiac muscle membranes. The slope and intercept of the linear regression curve indicate the Kd and Bmax values respectively. Data points were determined from a set of binding experiment presented in the panel A. (C) Effects of increasing the concentration of metAEA on the specific binding of [3H]isradipine to cardiac muscle membranes. Data shown are means ± SEM; n = 5–6 experiments. (D) Effects of cannabinoid receptor antagonists AM251 (1 μM) and AM630 (1 μM) and their co-application with metAEA (10 μM) on the specific binding of [3H]isradipine to T-tubule membranes. Data shown are means ± SEM; n = 5–6 experiments.
Effects of CB receptor antagonists on the specific [3H]isradipine binding from cardiac muscle membranes, were also investigated. Compared with controls, inhibitory effect of metAEA (10 μM) on the specific binding of [3H]isradipine was not altered by incubation (30 min) with AM251 or AM630 (1 μM) (Figure 7D) (anova; n = 8–10; P > 0.05). Incubation with AM251 or AM630 alone did not significantly alter the specific [3H]isradipine binding from cardiac muscle membranes (anova; n = 8–10; P > 0.05)
Discussion
The results of this study indicate for the first time that previously reported actions of AEA on cardiac muscle contractility (negative inotropic effect) and the AP configuration involves direct inhibition of voltage-dependent Na+ and Ca2+ channels in ventricular myocytes. The actions of the endocannabinoids on the cardiovascular system affect a wide range of organ systems and involve complex set of cellular and molecular mechanisms (Randall et al., 2004; Malinowska et al., 2012). In addition to cannabinoid receptor-mediated actions, several other factors such as the activation of autonomic reflexes, the presence of endothelial cells and fatty acid-based metabolic products have been reported to contribute to the complexity of endocannabinoid actions on the heart. On the other hand, acutely dissociated ventricular myocytes have several advantages over in vivo and traditional in vitro conditions, as this preparation excludes the influences of reflex pathways, autonomic nerve endings, neurotransmitter uptake system, gap-junction connections, and coronary perfusion status.
In our experiments, AEA caused a significant reduction in the maximal amplitudes of both Na+ and Ca2+ currents. Inhibitory effect of AEA, on VGSC, which is the major inward current during the upstroke (phase 0) of the AP is in agreement with our recently published current-clamp studies (Al Kury et al., 2014) indicating that AEA decreases the amplitude and initial rate of rise of the AP in ventricular myocytes. The results suggest that the effect of AEA on VGSC is not mediated by CB1 or CB2 cannabinoid receptors. Furthermore, radioligand-binding experiments indicated that AEA interacted directly with VGSC in ventricular myocytes. In cardiac muscles, VGSCs are almost exclusively represented by their TTX-resistant Nav1.5 isoform (SCN5A gene; Catterall et al., 2005a). Therefore, the changes in the biophysical properties of INa by AEA, namely induction of the hyperpolarizing shift in the voltage-dependence of its SSI can be attributed to their effects on Nav1.5 channel gating.
A hyperpolarizing shift of the SSI indicates that a higher proportion of VGSCs would be inactivated at resting membrane potential, and therefore substantially fewer channels would be available for activation, resulting in decreased amplitude and rate of rise during the upstroke of the AP.
Our radioligand studies indicated that [3H]BTX-B binding was inhibited by AEA in an allosteric manner. The binding site for [3H]BTX-B has also been shown to interact allosterically with local anaesthetics and class I antiarrhythmics (Sheldon et al., 1994). Therefore, it is likely that AEA synthesized during cell stress can bind to Nav1.5 channels and modulate the actions of local anaesthetics and class I antiarrhythmics with Nav1.5 channels. Although this, to our knowledge, is the first demonstration of the direct inhibitory action of AEA on a muscle type voltage-dependent Na+ channel, in several earlier investigations, AEA, at similar or higher concentrations, has been shown to directly inhibit the function of voltage-gated Na+ channels in neuronal structures (Nicholson et al., 2003; Kim et al., 2005; Duan et al., 2008; Theile and Cummins, 2011). In agreement with our findings, both AEA (Theile and Cummins, 2011) and its metabolic product arachidonic acid (Bendahhou et al., 1997) have been shown to increase inactivation of Na+ channels. Inhibition of VGSCs would slow the conduction of depolarization in the heart. However, it is unlikely that the slowed conduction because of Na+ channel inhibition alone would result in the widening of the QT interval observed in rat hearts (Farkas and Curtis, 2003).
In addition to INa, AEA caused a significant inhibition of cardiac IL,Ca, which is mediated by Cav1.2 (CACNA1C gene) isoform of L-type VGCCs (Catterall et al., 2005b). This current contributes to the plateau of the cardiac AP (phase 2); therefore, its suppression causes both the decrease of the amplitude of the plateau and the shortening of the AP duration. In earlier studies, AEA has been shown to decrease the amplitude of the plateau and cause shortening of the AP duration (Li et al., 2009; Al Kury et al., 2014). Our results show that AEA affects activation and inactivation gating of cardiac L-type VGCCs, producing a significant reduction in IL,Ca ‘window current’ in the range of Vm between −40 and +10 mV, and inducing partial blockade of the ion-conducting pathway leading to decreased amplitude of ICa.
Diminished stationary Ca2+ entry as a consequence of smaller ‘window current’ may in part explain AEA-evoked enhancement of cardiomyocytes viability via prevention of Ca2+ overload and reduction of necrosis, whereas inhibition of IL,Ca may largely determine the decrease of the AP plateau amplitude and AP shortening observed in the presence of AEA. In cardiac muscle, extracellular Ca2+ required to trigger Ca2+ release from SR enters through L-type voltage-dependent Ca2+ channels opened during the AP. Collectively, these results suggest that during excitation-contraction coupling, the inhibition of L-type Ca2+ channels and the decrease in Ca2+-induced Ca2+ release from sarcoplasmic reticulum causes negative inotropic effect of AEA reported in earlier studies. In line with this hypothesis, although caffeine-induced contractures and myofilament sensitivity to Ca2+ remained unchanged, electrically induced Ca2+ transients were significantly depressed by AEA (Al Kury et al., 2014), further suggesting that Ca2+-induced Ca2+ release was impaired in the presence of AEA. Overall, AEA-mediated suppression of voltage-activated IL,Ca would provide a mechanism for the negative inotropic effects observed in earlier studies.
The mechanism of the inhibitory effect of AEA did not seem to involve Ca2+-induced inactivation process, because Ba2+ currents through L-type VGCCs were effectively inhibited by AEA. In addition, AEA was equally effective upon intracellular or extracellular applications, suggesting that there is no sidedness for AEA actions on L-type VGCCs. Considering the highly lipophilic nature of AEA, it is not surprising that AEA can effectively access its binding site from both extra and intracellular sites. The results of radioligand-binding and electrophysiological studies indicated that AEA directly interacts with and inhibits the function of L-type Ca2+ channels. Although, to our knowledge, this is the first demonstration of the direct effects of AEA on the L-type VGCC in cardiac muscle, similar results demonstrating the effects AEA on skeletal muscle L-type VGCCs have also been described in biochemical studies (Oz et al., 2000; 2004,). In our earlier studies in rabbit skeletal muscle, we have demonstrated that AEA inhibits the specific binding of [3H]isradipine to skeletal T-tubule membranes and directly inhibits the functions of skeletal muscle L-type Ca2+ channels (Oz et al., 2000; 2004,) in a manner that is independent of known cannabinoid receptors. In fact, earlier studies searching for endogenous modulators of L-type Ca2+ channels have also identified AEA as a ligand for L-type Ca2+ channels (Johnson et al., 1993). Later investigations indicated that effects of AEA are not limited to L-type VGCCs in muscles, and other types of Ca2+ currents in neuronal structures are also inhibited directly by endocannabinoids such as AEA (see Lozovaya et al., 2009).
Involvement of cannabinoid receptors in the negative inotropic actions of cannabinoids has been investigated in several earlier studies (see Batkai and Pacher, 2009). However, the results of these investigations have not been conclusive (see Randall et al., 2004; Mendizabal and Adler-Graschinsky, 2007; Malinowska et al., 2012). Both cannabinoid receptor-dependent and -independent mechanisms have been suggested (Malinowska et al., 2012). Experiments with AEA and the synthetic cannabinoid HU-210 performed in isolated Langendorff rat hearts and in isolated, electrically stimulated human atrial appendages (Ford et al., 2002; Bonz et al., 2003) have revealed a negative inotropic effect of cannabinoids that may underlie the ability of AEA and HU-210 to decrease cardiac output as observed in studies performed in vivo (Wagner et al., 2001). In a recent study, a synthetic cannabinoid A-955840 inhibited the function of L-type Ca2+ channels in rabbit heart in a manner not sensitive to CB1 or CB2 receptor antagonists (Su et al., 2011). On the other hand, in another recent study, AEA was reported to inhibit L-type Ca2+ channels by the activation of CB1 receptors (Li et al., 2009). In this study, AEA in the concentration range of 10 nM to 1 μM potently inhibited the function of Ca2+ channels, and the effect of AEA was reversed by CB1 receptor antagonists. In our experiments, AEA was not effective at concentration lower than 1 μM. In addition, in our study, the inhibitory effect of AEA was not reversed by the antagonists of CB1 or CB2 receptors. Differences between two studies could be due to different strains of rats used (Sprague-Dawley in their study vs. Wistar rats in the present study). Our findings suggest that neither CB1 or CB2 receptors are involved in AEA inhibition of L-type Ca2+ channels in rat cardiomyocytes. Although PTX-sensitive signal transduction is well documented for cannabinoid agonists, cannabinoid coupling to PTX-insensitive Gq has been reported in several studies (Ishii and Chun, 2002; Straiker et al., 2002; McIntosh et al., 2007). Therefore, the effect of AEA through PTX-insensitive pathwaysf cannot be excluded.
AEA belongs to a group of fatty acid-based molecules called long-chain N-acylethanolamines (NAEs) which are produced abundantly in response to tissue necrosis and cellular stress (Hansen et al., 2000; Berger et al., 2004). In fact, accumulation of NAEs was first observed in experimental myocardial infarction induced by ligation of coronary arteries in canine heart (Epps et al., 1979; 1982,; see Schmid and Berdyshev, 2002). NAE content increased up to 500 nmol·g−1 (approximately 500 μM) in infarcted areas of canine heart during ischaemia (Epps et al., 1979). Although AEA constitutes a minor (1–3%) part of total NAE levels (Schmid and Berdyshev, 2002), the results of this study may have important implications regarding the contractile responses of ventricular myocytes to ischaemia and cellular stress (Hansen et al., 2000; Schmid and Berdyshev, 2002; Berger et al., 2004). We have previously reported that major NAE species, including AEA, produced during ischaemia have significant effects on the amplitudes and kinetics of APs and accompanying ionic currents that could account for the negative ionotropic actions of these compounds on ventricular myocytes (Voitychuk et al., 2012). For example, functions of voltage-gated Ca2+ (Oz et al., 2000); (Oz et al., 2004; 2005,; Alptekin et al., 2010; Voitychuk et al., 2012) and Na+ (Gulaya et al., 1993; Voitychuk et al., 2012) channels are modulated by NAEs. In the concentration range used in our study, AEA inhibited the function of various cardiac ion channels in cannabinoid receptor-independent manner. For example, AEA blocks T- type Ca2+ channels (Cav3.1 and Cav 3.2) (Chemin et al., 2007) and cardiac Kv1.5 (Barana et al., 2010) and Kv4.3 (Amoros et al., 2010) channels in a receptor-independent manner. These effects may contribute to the overall effects of AEA on AP and myocyte function.
In conclusion, the results indicate for the first time that AEA interacts directly with Na+- and L-type Ca2+ channels in ventricular myocytes in a manner that is independent of CB1 or CB2 receptors.
Acknowledgments
This study was supported by the National Institute on Drug Abuse/National Institutes of Health, USA and the United Arab Emirates University Research Funds. We thank Mr Anwar Qureshi for his excellent technical help in isolating rat ventricular myocytes.
Glossary
- AEA
anandamide
- AP
action potential
- ATX
sea anemone toxin
- BTX
batrachotoxin
- FAAH
fatty acid amide hydrolase
- IL,Ca
L-type Ca2+ current
- INa
Na+ current
- metAEA
methanandamide
- NAEs
N-acylethanolamines
- PTX
Pertussis toxin
- TTX
tetrodotoxin
- VGCC
voltage-gated calcium channel
- VGSC
voltage-gated sodium channel
Author contributions
P. D., Y. M. S., S. G., C. H. and M. O. all contributed to the design of the experiments, analysis of data and also the writing of the paper.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
http://dx.doi.org/10.1111/bph.12734
Figure S1 Effect of increasing AEA and vehicle (ethanol) concentrations on the voltage-activated sodium currents recorded in rat ventricular myocytes. (A) Effects of AEA (0.1–30 μM) and corresponding vehicle concentrations on the maximal amplitudes of INa currents. Each bar represents mean ± SEM from five to seven cells (B) Corrected concentration response curve for the inhibitory effect of AEA on INa. The amount of inhibition induced by the vehicle was subtracted from the AEA-induced inhibition at corresponding AEA concentrations. Data were fit with logistic equation using Origin data analysis software.
{kind=link}
Figure S2 Effect of PTX pretreatment on BRL-37344 inhibition of voltage-activated Ca2+ currents (ICa) recorded in rat ventricular myocytes. (A) Records of currents presenting the effect of BRL-37344, a β3 adrenoceptor agonist, on ICa in the absence and presence of PTX (2 μg·mL−1 for 3 h in 37°C) pretreatment. Records were obtained by applying a step depolarizing pulse from −50 to +20 mV for a duration of 300 ms (B) Presentation of results on the effect of PTX pretreatment on BRL-37344 inhibition of ICa. Each bar presents mean ± SEM from five to seven cells.* P < 0.05.
{kind=link}
Figure S3 Effects of increasing AEA and vehicle (ethanol) concentrations on the L-type voltage-activated Ca+2 currents (IL,Ca) recorded in rat ventricular myocytes. (A) Effects of AEA (0.1–30 μM) and corresponding vehicle concentrations on the maximal amplitudes of IL,Ca. Each bar represents mean ± SEM from five to seven cells (B) Corrected concentration response curve for the inhibitory effect of AEA on IL,Ca. The amount of inhibition induced by the vehicle was subtracted from the AEA-induced inhibition at corresponding AEA concentrations. Data were fit with logistic equation using Origin data analysis software. Application of AEA inhibited IL,Ca in a concentration-dependent manner with an IC50 = 10.2 ± 0.6 μM, slope factor of 36 ± 0.2, but twofold higher efficacy (i.e. maximal block at saturating concentration A = 72.0 ± 2.9%) compared with INa (see Supporting Information Figure S1B for comparison).
{kind=link}
References
- Abadji V, Lin S, Taha G, Griffin G, Stevenson LA, Pertwee RG, et al. (R)-methanandamide: a chiral novel anandamide possessing higher potency and metabolic stability. J Med Chem. 1994;37:1889–1893. doi: 10.1021/jm00038a020. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The concise guide to PHARMACOLOGY 2013/14: G protein-coupled receptors. Br J Pharmacol. 2013a;170:1459–1581. doi: 10.1111/bph.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Catterall WA, et al. The concise guide to PHARMACOLOGY 2013/14: ion channels. Br J Pharmacol. 2013b;170:1607–1651. doi: 10.1111/bph.12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Kury LT, Voitychuk OI, Ali RM, Galadari S, Yang KH, Howarth FC, et al. Effects of endogenous cannabinoid anandamide on excitation-contraction coupling in rat ventricular myocytes. Cell Calcium. 2014;55:104–118. doi: 10.1016/j.ceca.2013.12.005. [DOI] [PubMed] [Google Scholar]
- Alptekin A, Galadari S, Shuba Y, Petroianu G, Oz M. The effects of anandamide transport inhibitor AM404 on voltage-dependent calcium channels. Eur J Pharmacol. 2010;634:10–15. doi: 10.1016/j.ejphar.2010.02.013. [DOI] [PubMed] [Google Scholar]
- Amoros I, Barana A, Caballero R, Gomez R, Osuna L, Lillo MP, et al. Endocannabinoids and cannabinoid analogues block human cardiac Kv4.3 channels in a receptor-independent manner. J Mol Cell Cardiol. 2010;48:201–210. doi: 10.1016/j.yjmcc.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Andrag E, Curtis MJ. Feasibility of targeting ischaemia-related ventricular arrhythmias by mimicry of endogenous protection by endocannabinoids. Br J Pharmacol. 2013;169:1840–1848. doi: 10.1111/bph.12252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barana A, Amoros I, Caballero R, Gomez R, Osuna L, Lillo MP. Endocannabinoids and cannabinoid analogues block cardiac hKv1.5 channels in a cannabinoid receptor-independent manner. Cardiovasc Res. 2010;85:56–67. doi: 10.1093/cvr/cvp284. [DOI] [PubMed] [Google Scholar]
- Batkai S, Pacher P. Endocannabinoids and cardiac contractile function: pathophysiological implications. Pharmacol Res. 2009;60:99–106. doi: 10.1016/j.phrs.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bebarova M, Matejovic P, Pasek M, Ohlidalova D, Jansova D, Simurdova M, et al. Effect of ethanol on action potential and ionic membrane currents in rat ventricular myocytes. Acta Physiol (Oxf) 2010;200:301–314. doi: 10.1111/j.1748-1716.2010.02162.x. [DOI] [PubMed] [Google Scholar]
- Bendahhou S, Cummins TR, Agnew WS. Mechanism of modulation of the voltage-gated skeletal and cardiac muscle sodium channels by fatty acids. Am J Physiol. 1997;272:C592–C600. doi: 10.1152/ajpcell.1997.272.2.C592. [DOI] [PubMed] [Google Scholar]
- Berger C, Schmid PC, Schabitz WR, Wolf M, Schwab S, Schmid HH. Massive accumulation of N-acylethanolamines after stroke. Cell signalling in acute cerebral ischemia? J Neurochem. 2004;88:1159–1167. doi: 10.1046/j.1471-4159.2003.02244.x. [DOI] [PubMed] [Google Scholar]
- Bonz A, Laser M, Kullmer S, Kniesch S, Babin-Ebell J, Popp V, et al. Cannabinoids acting on CB1 receptors decrease contractile performance in human atrial muscle. J Cardiovasc Pharmacol. 2003;41:657–664. doi: 10.1097/00005344-200304000-00020. [DOI] [PubMed] [Google Scholar]
- Bouchard JF, Lepicier P, Lamontagne D. Contribution of endocannabinoids in the endothelial protection afforded by ischemic preconditioning in the isolated rat heart. Life Sci. 2003;72:1859–1870. doi: 10.1016/s0024-3205(02)02474-8. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005a;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure-function relationships of voltage-gated calcium channels. Pharmacol Rev. 2005b;57:411–425. doi: 10.1124/pr.57.4.5. [DOI] [PubMed] [Google Scholar]
- Chemin J, Nargeot J, Lory P. Chemical determinants involved in anandamide-induced inhibition of T-type calcium channels. J Biol Chem. 2007;282:2314–2323. doi: 10.1074/jbc.M610033200. [DOI] [PubMed] [Google Scholar]
- Danziger RS, Sakai M, Capogrossi MC, Spurgeon HA, Hansford RG, Lakatta EG. Ethanol acutely and reversibly suppresses excitation-contraction coupling in cardiac myocytes. Circ Res. 1991;68:1660–1668. doi: 10.1161/01.res.68.6.1660. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, De Petrocellis L, Bisogno T. The biosynthesis, fate and pharmacological properties of endocannabinoids. Handb Exp Pharmacol. 2005;168:147–185. doi: 10.1007/3-540-26573-2_5. [DOI] [PubMed] [Google Scholar]
- Duan Y, Zheng J, Nicholson RA. Inhibition of [3H]batrachotoxinin A-20α-benzoate binding to sodium channels and sodium channel function by endocannabinoids. Neurochem Int. 2008;52:438–446. doi: 10.1016/j.neuint.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Dunn SM. Voltage-dependent calcium channels in skeletal muscle transverse tubules. Measurements of calcium efflux in membrane vesicles. J Biol Chem. 1989;264:11053–11060. [PubMed] [Google Scholar]
- Epps DE, Schmid PC, Natarajan V, Schmid HH. N-acylethanolamine accumulation in infarcted myocardium. Biochem Biophys Res Commun. 1979;90:628–633. doi: 10.1016/0006-291x(79)91281-6. [DOI] [PubMed] [Google Scholar]
- Epps DE, Mandel F, Schwartz A. The alteration of rabbit skeletal sarcoplasmic reticulum function by N-acylethanolamine, a lipid associated with myocardial infarction. Cell Calcium. 1982;3:531–543. doi: 10.1016/0143-4160(82)90043-4. [DOI] [PubMed] [Google Scholar]
- Farkas A, Curtis MJ. Does QT widening in the Langendorff-perfused rat heart represent the effect of repolarization delay or conduction slowing? J Cardiovasc Pharmacol. 2003;42:612–621. doi: 10.1097/00005344-200311000-00006. [DOI] [PubMed] [Google Scholar]
- Ford WR, Honan SA, White R, Hiley CR. Evidence of a novel site mediating anandamide-induced negative inotropic and coronary vasodilatator responses in rat isolated hearts. Br J Pharmacol. 2002;135:1191–1198. doi: 10.1038/sj.bjp.0704565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulaya NM, Melnik AA, Balkov DI, Volkov GL, Vysotskiy MV, Vaskovsky VE. The effect of long-chain N-acylethanolamines on some membrane-associated functions of neuroblastoma C1300 N18 cells. Biochim Biophys Acta. 1993;1152:280–288. doi: 10.1016/0005-2736(93)90259-3. [DOI] [PubMed] [Google Scholar]
- Hansen HS, Moesgaard B, Hansen HH, Petersen G. N-acylethanolamines and precursor phospholipids – relation to cell injury. Chem Phys Lipids. 2000;108:135–150. doi: 10.1016/s0009-3084(00)00192-4. [DOI] [PubMed] [Google Scholar]
- Hanus LO, Mechoulam R. Novel natural and synthetic ligands of the endocannabinoid system. Curr Med Chem. 2010;17:1341–1359. doi: 10.2174/092986710790980096. [DOI] [PubMed] [Google Scholar]
- Howarth FC, Qureshi MA, White E. Effects of hyperosmotic shrinking on ventricular myocyte shortening and intracellular Ca(2+) in streptozotocin-induced diabetic rats. Pflugers Arch. 2002;444:446–451. doi: 10.1007/s00424-002-0830-0. [DOI] [PubMed] [Google Scholar]
- Ishii I, Chun J. Anandamide-induced neuroblastoma cell rounding via the CB1 cannabinoid receptors. Neuroreport. 2002;13:593–596. doi: 10.1097/00001756-200204160-00011. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Heald SL, Dally RD, Janis RA. Isolation, identification and synthesis of an endogenous arachidonic amide that inhibits calcium channel antagonist 1,4-dihydropyridine binding. Prostaglandins Leukot Essent Fatty Acids. 1993;48:429–437. doi: 10.1016/0952-3278(93)90048-2. [DOI] [PubMed] [Google Scholar]
- Kang JX, Leaf A. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proc Natl Acad Sci U S A. 1996;93:3542–3546. doi: 10.1073/pnas.93.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HI, Kim TH, Shin YK, Lee CS, Park M, Song JH. Anandamide suppression of Na+ currents in rat dorsal root ganglion neurons. Brain Res. 2005;1062:39–47. doi: 10.1016/j.brainres.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Krylatov AV, Uzhachenko RV, Maslov LN, Bernatskaya NA, Makriyannis A, Mechoulam R. Endogenous cannabinoids improve myocardial resistance to arrhythmogenic effects of coronary occlusion and reperfusion: a possible mechanism. Bull Exp Biol Med. 2002;133:122–124. doi: 10.1023/a:1015574100494. [DOI] [PubMed] [Google Scholar]
- Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, et al. Structure–activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J Med Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- Li Q, Ma HJ, Zhang H, Qi Z, Guan Y, Zhang Y. Electrophysiological effects of anandamide on rat myocardium. Br J Pharmacol. 2009;158:2022–2029. doi: 10.1111/j.1476-5381.2009.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozovaya N, Min R, Tsintsadze V, Burnashev N. Dual modulation of CNS voltage-gated calcium channels by cannabinoids: focus on CB1 receptor-independent effects. Cell Calcium. 2009;46:154–162. doi: 10.1016/j.ceca.2009.07.007. [DOI] [PubMed] [Google Scholar]
- Malinowska B, Baranowska-Kuczko M, Schlicker E. Triphasic blood pressure responses to cannabinoids: do we understand the mechanism? Br J Pharmacol. 2012;165:2073–2088. doi: 10.1111/j.1476-5381.2011.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh BT, Hudson B, Yegorova S, Jollimore CA, Kelly ME. Agonist-dependent cannabinoid receptor signalling in human trabecular meshwork cells. Br J Pharmacol. 2007;152:1111–1120. doi: 10.1038/sj.bjp.0707495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal VE, Adler-Graschinsky E. Cannabinoids as therapeutic agents in cardiovascular disease: a tale of passions and illusions. Br J Pharmacol. 2007;151:427–440. doi: 10.1038/sj.bjp.0707261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecucco F, Di Marzo V. At the heart of the matter: the endocannabinoid system in cardiovascular function and dysfunction. Trends Pharmacol Sci. 2012;33:331–340. doi: 10.1016/j.tips.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Nicholson RA, Liao C, Zheng J, David LS, Coyne L, Errington AC. Sodium channel inhibition by anandamide and synthetic cannabimimetics in brain. Brain Res. 2003;978:194–204. doi: 10.1016/s0006-8993(03)02808-7. [DOI] [PubMed] [Google Scholar]
- Oz M. Receptor-independent actions of cannabinoids on cell membranes: focus on endocannabinoids. Pharmacol Ther. 2006;111:114–144. doi: 10.1016/j.pharmthera.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Oz M, Tchugunova YB, Dunn SM. Endogenous cannabinoid anandamide directly inhibits voltage-dependent Ca(2+) fluxes in rabbit T-tubule membranes. Eur J Pharmacol. 2000;404:13–20. doi: 10.1016/s0014-2999(00)00396-4. [DOI] [PubMed] [Google Scholar]
- Oz M, Tchugunova Y, Dinc M. Differential effects of endogenous and synthetic cannabinoids on voltage-dependent calcium fluxes in rabbit T-tubule membranes: comparison with fatty acids. Eur J Pharmacol. 2004;502:47–58. doi: 10.1016/j.ejphar.2004.08.052. [DOI] [PubMed] [Google Scholar]
- Oz M, Alptekin A, Tchugunova Y, Dinc M. Effects of saturated long-chain N-acylethanolamines on voltage-dependent Ca2+ fluxes in rabbit T-tubule membranes. Arch Biochem Biophys. 2005;434:344–351. doi: 10.1016/j.abb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Pacher P, Batkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SP, Di Marzo V, Elphick MR. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB and CB. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Postma SW, Catterall WA. Inhibition of binding of [3H]batrachotoxinin A 20-α-benzoate to sodium channels by local anesthetics. Mol Pharmacol. 1984;25:219–227. [PubMed] [Google Scholar]
- Randall MD, Kendall DA, O'Sullivan S. The complexities of the cardiovascular actions of cannabinoids. Br J Pharmacol. 2004;142:20–26. doi: 10.1038/sj.bjp.0705725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656, and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid HH, Berdyshev EV. Cannabinoid receptor-inactive N-acylethanolamines and other fatty acid amides: metabolism and function. Prostaglandins Leukot Essent Fatty Acids. 2002;66:363–376. doi: 10.1054/plef.2001.0348. [DOI] [PubMed] [Google Scholar]
- Sheldon RS, Cannon NJ, Duff HJ. Binding of [3H]batrachotoxinin A benzoate to specific sites on rat cardiac sodium channels. Mol Pharmacol. 1986;30:617–623. [PubMed] [Google Scholar]
- Sheldon RS, Duff HJ, Thakore E, Hill RJ. Class I antiarrhythmic drugs: allosteric inhibitors of [3H] batrachotoxinin binding to rat cardiac sodium channels. J Pharmacol Exp Ther. 1994;268:187–194. [PubMed] [Google Scholar]
- Soldatov NM, Oz M, O'Brien KA, Abernethy DR, Morad M. Molecular determinants of L-type Ca2+ channel inactivation. Segment exchange analysis of the carboxyl-terminal cytoplasmic motif encoded by exons 40–42 of the human α1C subunit gene. J Biol Chem. 1998;273:957–963. doi: 10.1074/jbc.273.2.957. [DOI] [PubMed] [Google Scholar]
- Straiker AJ, Borden CR, Sullivan JM. G-protein α subunit isoforms couple differentially to receptors that mediate presynaptic inhibition at rat hippocampal synapses. J Neurosci. 2002;22:2460–2468. doi: 10.1523/JNEUROSCI.22-07-02460.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Preusser L, Diaz G, Green J, Liu X, Polakowski J, et al. Negative inotropic effect of a CB2 agonist A-955840 in isolated rabbit ventricular myocytes is independent of CB1 and CB2 receptors. Curr Drug Saf. 2011;6:277–284. doi: 10.2174/157488611798918728. [DOI] [PubMed] [Google Scholar]
- Theile JW, Cummins TR. Inhibition of Navβ4 peptide-mediated resurgent sodium currents in Nav1.7 channels by carbamazepine, riluzole, and anandamide. Mol Pharmacol. 2011;80:724–734. doi: 10.1124/mol.111.072751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugdyzhekova DS, Bernatskaya NA, Stefano JB, Graier VF, Tam SW, Mekhoulam R. Endogenous cannabinoid anandamide increases heart resistance to arrhythmogenic effects of epinephrine: role of CB(1) and CB(2) receptors. Bull Exp Biol Med. 2001;131:251–253. doi: 10.1023/a:1017651432193. [DOI] [PubMed] [Google Scholar]
- Voitychuk OI, Asmolkova VS, Gula NM, Sotkis GV, Galadari S, Howarth FC, et al. Modulation of excitability, membrane currents and survival of cardiac myocytes by N-acylethanolamines. Biochim Biophys Acta. 2012;1821:1167–1176. doi: 10.1016/j.bbalip.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Wagner JA, Jarai Z, Batkai S, Kunos G. Hemodynamic effects of cannabinoids: coronary and cerebral vasodilation mediated by cannabinoid CB(1) receptors. Eur J Pharmacol. 2001;423:203–210. doi: 10.1016/s0014-2999(01)01112-8. [DOI] [PubMed] [Google Scholar]
- Zhang ZS, Cheng HJ, Onishi K, Ohte N, Wannenburg T, Cheng CP. Enhanced inhibition of L-type Ca2+ current by β3-adrenergic stimulation in failing rat heart. J Pharmacol Exp Ther. 2005;315:1203–1211. doi: 10.1124/jpet.105.089672. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Effect of increasing AEA and vehicle (ethanol) concentrations on the voltage-activated sodium currents recorded in rat ventricular myocytes. (A) Effects of AEA (0.1–30 μM) and corresponding vehicle concentrations on the maximal amplitudes of INa currents. Each bar represents mean ± SEM from five to seven cells (B) Corrected concentration response curve for the inhibitory effect of AEA on INa. The amount of inhibition induced by the vehicle was subtracted from the AEA-induced inhibition at corresponding AEA concentrations. Data were fit with logistic equation using Origin data analysis software.
Figure S2 Effect of PTX pretreatment on BRL-37344 inhibition of voltage-activated Ca2+ currents (ICa) recorded in rat ventricular myocytes. (A) Records of currents presenting the effect of BRL-37344, a β3 adrenoceptor agonist, on ICa in the absence and presence of PTX (2 μg·mL−1 for 3 h in 37°C) pretreatment. Records were obtained by applying a step depolarizing pulse from −50 to +20 mV for a duration of 300 ms (B) Presentation of results on the effect of PTX pretreatment on BRL-37344 inhibition of ICa. Each bar presents mean ± SEM from five to seven cells.* P < 0.05.
Figure S3 Effects of increasing AEA and vehicle (ethanol) concentrations on the L-type voltage-activated Ca+2 currents (IL,Ca) recorded in rat ventricular myocytes. (A) Effects of AEA (0.1–30 μM) and corresponding vehicle concentrations on the maximal amplitudes of IL,Ca. Each bar represents mean ± SEM from five to seven cells (B) Corrected concentration response curve for the inhibitory effect of AEA on IL,Ca. The amount of inhibition induced by the vehicle was subtracted from the AEA-induced inhibition at corresponding AEA concentrations. Data were fit with logistic equation using Origin data analysis software. Application of AEA inhibited IL,Ca in a concentration-dependent manner with an IC50 = 10.2 ± 0.6 μM, slope factor of 36 ± 0.2, but twofold higher efficacy (i.e. maximal block at saturating concentration A = 72.0 ± 2.9%) compared with INa (see Supporting Information Figure S1B for comparison).